

Abstract
The gene expression platform assay allows for robust and highly reproducible quantification of the expression of up to 800 transcripts (mRNA or miRNAs) in a single reaction. The miRNA assay counts transcripts by directly imaging and digitally counting miRNA molecules that are labeled with color-coded fluorescent barcoded probe sets (a reporter probe and a capture probe). Barcodes are hybridized directly to mature miRNAs that have been elongated by ligating a unique oligonucleotide tag (miRtag) to the 3' end. Reverse transcription and amplification of the transcripts are not required. Reporter probes contain a sequence of six color positions populated using a combination of four fluorescent colors. The four colors over six positions are used to construct a gene-specific color barcode sequence. Post-hybridization processing is automated on a robotic prep station. After hybridization, the excess probes are washed away, and the tripartite structures (capture probe-miRNA-reporter probe) are fixed to a streptavidin-coated slide via the biotin-labeled capture probe. Imaging and barcode counting is done using a digital analyzer. The immobilized barcoded miRNAs are visualized and imaged using a microscope and camera, and the unique barcodes are decoded and counted. Data quality control (QC), normalization, and analysis are facilitated by a custom-designed data processing and analysis software that accompanies the assay software. The assay demonstrates high linearity over a broad range of expression, as well as high sensitivity. Sample and assay preparation does not involve enzymatic reactions, reverse transcription, or amplification; has few steps; and is largely automated, reducing investigator effects and resulting in high consistency and technical reproducibility. Here, we describe the application of this technology to identifying circulating miRNA perturbations in irritable bowel syndrome.
Keywords: Genetics, Issue 117, Irritable Bowel Syndrome, MicroRNA, Multiplex Analysis, Gene Expression, Circulating, MicroArray, High-Throughput Screening, Molecular Barcode
Introduction
Irritable Bowel Syndrome (IBS) is the most common outpatient diagnosis in Gastroenterology, afflicting 10 - 15% of the general population and representing a significant burden on health services. Currently, there are no generally accepted diagnostic biomarkers for IBS (i.e., the diagnosis is based on clinical symptoms and ruling out other organic disorders, such as GI malignancy, Inflammatory Bowel Diseases, and gastrointestinal (GI) infections). When studying gene expression profiles in common conditions with subclinical histopathology, such as IBS, high sensitivity and accuracy (reproducibility) are needed, as perturbations in gene expression profiles are often subtle1-3. miRNAs have been identified as both diagnostic and functional targets in IBS4,5, and we are particularly interested in assessing their use as circulating biomarkers of IBS-associated biological perturbations3,4,6,7. The clinical utilization of circulating biomarkers is of current interest due to the ease and minimally invasive nature of the collection of the biomarkers' source (e.g., peripheral blood) from patients.
Gene expression platform assays, because of their unique chemistry and streamlined, mostly automated workflow, provide a microarray platform that provides wide and customizable coverage (800 targets in a single reaction) of the transcriptome for targeted discovery. They also yield high precision and linearity over a wide spectrum of expression levels in the quantification of transcriptomic targets. The assay does not require the reverse transcription of RNA, the amplification of resulting cDNA, or any other enzymatic reactions. Thus, potential errors introduced by the high cycle numbers of amplification from RNA species with low abundance or rare splice variants are reduced by the process of digital counting. This means that a wide variety of sample types, such as purified total RNA (i.e., mRNA + miRNA), RNA from formalin-fixed paraffin-embedded (FFPE) specimens, as well as crude tissue and cell lysates, can be used with the assays.
RNAs of interest are detected using a pair of probes (capture and reporter probes), each containing a target-specific sequence that recognizes a corresponding region on the target RNA. In order to ensure the detection of short RNAs (i.e., miRNAs), the mature miRNA sequence is elongated by annealing a unique oligonucleotide tag (miRtag) to the 3' end of the molecule. A bridging oligonucleotide, which is complimentary to a portion of both the mature target miRNA and the miRNA-specific miRtag, is used to ensure sequence specificity. Capture and reporter probes ligate to the 3' and 5' ends of the mature miRNA-miRtag complex, respectively. The capture probes have a biotin label at the 3' end, which allow them to attach to the surface of a streptavidin-coated glass slide, fixing the capture probe-miRNA-miRtag-reporter probe complex to the slide. An electric current is then used to orient the complex on the slide surface, allowing fluorescent barcodes carried by the reporter probe on the 5' end to be visualized using a microscope and charge-coupled device (CCD) camera. Barcodes are counted and decoded, yielding a count of the specific target miRNAs. The fluorescent barcode consists of six positions that can be occupied by one of four fluorescent colors, which can be used to construct over 4,000 color-barcode combinations, each coding for a specific RNA.
Sample preparation (i.e., RNA purification or cell/tissue lysate preparation), as well as hybridization of the capture and reporter probes, are done at the bench. Twelve specimens can be multiplexed on a single slide. After the overnight hybridization, all further specimen preparation is performed by a robotic prep station. The loaded slides are then transferred to a digital analyzer for imaging, counting, decoding, and data processing. The resulting data are imported to the accompanying software, where they are further processed with a high degree of user control. The unique chemistry, simple preparation, automated nature, simple data type (counts), and overall streamlined workflow of the assay facilitate high-precision gene expression quantification, making it a useful tool in studying circulating miRNA expression profiles and signatures in functional conditions such as IBS.
Protocol
The research described here was approved by the Institutional Review Board of the National Institutes of Health (Clinicaltrial.gov # NCT00824941).
1. Collection of Whole Blood Samples
Collect whole blood samples (2.5 ml) from study participants via venipuncture in blood collection tubes containing an RNA stabilizing solution (allowing for long-term storage), and store them at -80 °C until RNA purification.
2. Total RNA Purification
Thaw and incubate the blood tubes for 2 hr, and then centrifuge them using a swing-out rotor for 10 min at 5,000 x g. Remove the supernatant by carefully pouring or pipetting it off into a waste tube, being careful not to disturb the pellet.
Wash the pellet by adding 4 ml of RNase-free water, replace the cap securely, and vortex until the pellet is visibly dissolved. Centrifuge using a swing-bucket rotor at 5,000 x g for 10 min and remove the supernatant, as described in step 2.1.
Add 350 µl of BM1 buffer (provided) to the pellet. Vortex the pellet until it is visibly dissolved and transfer it to a 2-ml processing tube for automated total RNA extraction.
- Perform automated purification of intracellular RNA, including miRNA, from the whole blood using a commercially available RNA extraction kit, which can be automated on a robotic RNA extraction system. NOTE: The automated robotic system we used can process twelve samples at a time, and it takes 3 hr for the RNA purification protocol to complete.
- Load the pre-loaded pipette tips, centrifuge tubes, buffers, and reagents provided in the RNA purification kit into the robotic RNA extraction system.
- Select the "Blood miRNA Part A" protocol from the protocol selection menu and start the run. NOTE: See Supplementary Materials for a description of the automated procedures.
- Once the robot has completed Part A of the automated miRNA purification protocol, empty the waste receptacle and remove the used plastics and reagent containers from the robotic system.
- Close the lids on all the micro-centrifuge tubes containing eluted RNA and place them in the shaker adaptor. Select "Blood miRNA Part B" from the protocol selection menu to incubate the samples at 65 oC for 5 min.
- Once the robot has completed running Part B of the miRNA purification protocol, immediately remove the samples and chill them on ice.
Quantitate and evaluate the purified RNA quality using a spectrophotometer. NOTE: As per the protocol recommendations for the miRNA assay, a minimum concentration of 33 ng/µl is required, and all samples should meet or exceed a 280/260 ratio of 1.9 and a minimum 260/230 ratio of 1.8 to ensure the absence of significant organic contamination, which can affect assay performance (refer to Reference 17).
Store the samples at -80 oC.
3. miRNA Sample Preparation
Normalize 12 RNA samples to 33 ng/µl using nuclease-free water.
Prepare a 1:500 dilution of the miRNA controls provided in the assay kit using nuclease-free water. Keep on ice.
Prepare the annealing master mix: combine 13 µl of annealing buffer (provided), 26 µl of miRtag reagent (provided), and 6.5 µl of miRNA controls (prepared in step 3.2) using 10- and 20-µl pipettes.
Using a 10-µl pipette, add 3.5 µl of the annealing master mix and 3 µl of the RNA sample (i.e., 100 ng) to each of twelve 0.2-ml strip tubes. Flick to mix, spin down using a benchtop mini microcentrifuge (2,000 x g), and place in the thermocycler (94 oC for 1 min, 65 oC for 2 min, and 45 oC for 10 min; hold at 48 oC).
- Prepare the ligation master mix by combining 3 µl of the ligation buffer (provided) and 19.5 µl of the Polyethylene glycol (PEG; provided) using a 20-µl pipette. Add 2.5 µl of the ligation master mix to each of the strip tubes from step 3.4 using a 10-µl pipette.
- Mix gently, spin down using a benchtop mini microcentrifuge (2,000 x g), and return to the thermocycler at 48 oC for 5 min. After 5 min, using a 10-µl pipette, add 1 µl of ligase directly to each tube in the thermocycler and incubate them at 48 oC for 3 min, 47 oC for 3 min, 46 oC for 3 min, 45 oC for 5 min, and 65 oC 10 min; hold at 4 oC.
Remove the tubes from the thermocycler, mix them gently, spin them down using a benchtop mini microcentrifuge (2,000 x g), and add 1 µl of ligation clean-up enzyme (provided). Incubate the tubes in the thermocycler (37 oC for 1 hr and 70 oC for 10 min; hold at 4 oC). Use a 10-µl pipette.
Remove the tubes and, using a 100-µl pipette, add 40 µl of nuclease-free water, mix, and spin down using a benchtop mini microcentrifuge (2,000 x g).
4. miRNA Hybridization
Thaw the reporter and capture probe sets (provided) on ice and spin them down using a benchtop mini microcentrifuge (2,000 x g).
Using a 200-µl pipette, add 130 µl of hybridization buffer to the reporter code set tube (130 µl) to create the hybridization master mix. Mix and spin down using a benchtop mini microcentrifuge (2,000 x g). NOTE: Reporter and captures probes are specific and standard for each miRNA included in the panel. Custom panels and user-designed capture and probe sets can be designed.
In 12 new 0.2-ml strip tubes, add 20 µl of the hybridization master mix using a 20-µl pipette.
Denature the miRNA samples from step 3.7 at 85 oC for 5 min and cool them on ice. Add 5 µl to each of the tubes from step 4.3 using a 10-µl pipette.
Program the thermocycler to 65 oC at a 30-µl volume-calculated temperature and heated lid at the forever time setting (do not program it to ramp down to 4 oC at the end of the run).
Using a 10-µl pipette, add 5 µl of the capture probe set to each tube, mix, spin down using a benchtop mini microcentrifuge (2,000 x g), and immediately place in the thermocycler at 65 oC. Incubate for no less than 12 hr and no more than 30 hr.
5. Prep Station and Digital Analyzer
- Load the prep station.
- Open the station door and load the reagent plates (provided), cartridge (provided), pipette tips (provided), prepared samples in twelve 0.2-ml strip tubes, and twelve empty 0.2-ml strip tubes (provided). Remove the strip tube caps and reagent plate cover. Close the prep-station door and run the appropriate assay from the control panel. NOTE: Post-hybridization processing is the same, regardless of the assay being run. All reagents and plastic wares are prepackaged and require no preparation, other than bringing them to room temperature and spinning down the reagent-containing 96-well plates at 2,000 x g for 2 min. All components are loaded in clearly defined positions in the prep station. See Supplementary Materials for a description of the automated procedures.
- Visualize and count the barcodes of the immobilized and oriented tripartite complexes.
- Remove the cartridge from the prep station, seal it with the cover slips provided, and place it in the digital analyzer in the slot above the camera optic.
- Select the appropriate assay (miRNA panel assay) and assay version indicated on the assay kit and run the program. NOTE: The digital analyzer then takes field of view images for each sample position at four excitation wavelengths, allowing the fluorescent barcodes to be read and decoded. Specific barcodes, corresponding to a specific miRNA, are counted.
- Export the data files to a USB or directly to the server via an internet link.
6. Data Processing and Analysis
Click on the "Raw Data" tab and select "Import RCC Files". Select the folder containing RCC files (raw data files) from the USB.
Click "Next", select all QC boxes, and click "Import".
Click on the "New Study" tab. Name and describe the study and save.
In the new study, select the "New Experiment" tab and name and describe the new experiment. Click "Next" and select the RLF folder from the left pane that contains the relevant raw data files that have been uploaded. Once it has been selected, click on "Keep Selected", and then click "Next".
Add annotations if desired and click "Next".
Select the background subtraction method; either negative control, blank lane subtraction, or a user-defined subtraction can be selected. Once selected, click "Next". NOTE: Determine the most appropriate background correction approach based on the technical parameters and biological context of the study. NOTE: Counts are less precise for low-abundance targets. For the purposes of the study we describe here, the data was first processed without background correction in order to examine QC parameters and to examine variation in counts for miRNA of different abundances across technical replicates. The data was then reprocessed using a 25-count background threshold.
Select the "Top 100" normalization, and then click "Next".
To obtain the ratio data, select the ratio parameters, and then click "Next".
Click "Finished".
Click on the named experiment in left pane to reveal a drop down menu containing "Raw", "Normalized", "Grouped", "Ratio Data", and "Analysis Data".
- Click on "Raw Data" and observe a QC report in the right pane. Observe a flag next to the samples that do not pass the default QC parameters. Build a new "Study" which does not include the flagged samples and reprocess as described, or simply exclude flagged QC samples from the data analysis step by selecting "Exclude samples by QC/normalization flag".
- Export "Raw", "Normalized", or "Grouped" data files in CVS or another compatible file format for analysis in another statistical or microarray software.
Select "Normalized Data" from the left pane and select the samples to be included in the analysis in the right pane.
Click on "Analysis" and select the desired analysis type (e.g., "Heat Map", "Violin Plot", "Box Plot", "Scatter Plot", or "Histogram Plot"). Select the desired exclusion criteria boxes (e.g., exclude the outlier samples or genes, exclude the samples by QC flags, exclude the control genes, and/or exclude the normalization probes from data analysis).
Select individual samples to be excluded from the analysis as desired.
Select individual genes to be excluded from or included in the analysis as desired.
Select the parameters for the selected statistical or data visualization manipulation and click on "Finish".
Representative Results
Transcriptomic perturbation in functional disorders may be subtle, and in order to reliably detect those subtle changes in expression, quantification of gene expression needs to be precise. The gene expression platform assay system reduces technical noise through its highly automated nature, and the unique chemistry it uses produces high-precision gene expression data. Technical replicates shown in Figure 1 demonstrate the high reproducibility in both endogenous (blue dots) and viral miRNA (orange dots; housekeeping genes are represented by yellow dots) expression quantification that is possible using this method. Using this method, viral (Figure 2a) and endogenous (Figure 2b) miRNAs that show deviation from expected expression, as defined by healthy study participants, can be identified as targets of interest in IBS. It should be noted that it is unclear whether the viral miRNAs detected here derive from viral genes integrated into the human genome or from viral load. As the assay only counts mature miRNAs with single-nucleotide specificity, cross hybridization with similar endogenous human miRNAs is unlikely. Nonetheless, differential steady-state levels of these molecules are of interest as biomarkers. The precision of the method allows for perturbations among low expression targets to be detected, as accurate detection of these transcripts are not swamped out by more abundant transcripts. Subtle perturbations can also be reliably observed. Figures 3 and 4 show some of these perturbations in circulating miRNAs in IBS and IBS-subtypes compared to healthy controls, and Figures 5 and 6 (both adapted from 3) show the two most significant differentially elevated miRNAs (Figure 5: miRNA 342-3p, Figure 6: miRNA 150) in IBS participants. Figure 6 is an example of the graphical output of results generated by the associated software.
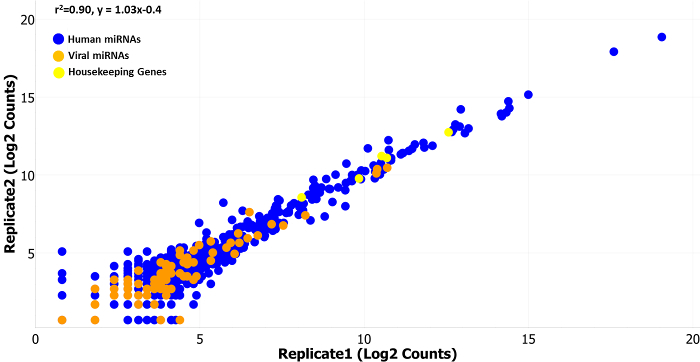 Figure 1:Normalized Counts from two Technical Replicates. Technical replicates run on two separate cartridges show high linear correlation (r2 = 0.90, y = 1.03x - 0.4) over the range of the normalized (normalized to the top 100 expressed miRNAs) miRNA counts (log2 counts on the x- and y-axis). The scatter plot includes data for endogenous human miRNAs in blue (654 miRNAs), endogenous viral miRNAs in orange (80 miRNAs), and housekeeping genes in yellow (8 genes). Endogenous viral miRNAs were available on earlier versions of the assay (version 1.4 was used here), but are no longer included on the standard assay, although they are available as custom additions. Please click here to view a larger version of this figure.
Figure 1:Normalized Counts from two Technical Replicates. Technical replicates run on two separate cartridges show high linear correlation (r2 = 0.90, y = 1.03x - 0.4) over the range of the normalized (normalized to the top 100 expressed miRNAs) miRNA counts (log2 counts on the x- and y-axis). The scatter plot includes data for endogenous human miRNAs in blue (654 miRNAs), endogenous viral miRNAs in orange (80 miRNAs), and housekeeping genes in yellow (8 genes). Endogenous viral miRNAs were available on earlier versions of the assay (version 1.4 was used here), but are no longer included on the standard assay, although they are available as custom additions. Please click here to view a larger version of this figure.
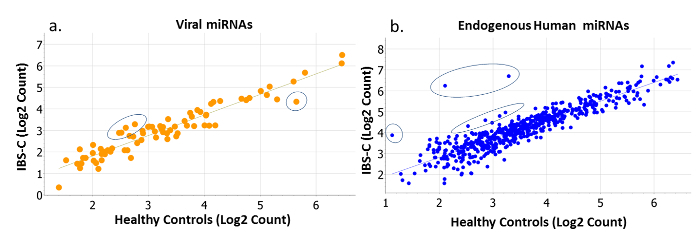 Figure 2: Normalized Counts of Viral and Endogenous Human miRNAs in IBS-constipation (IBS-C) versus Healthy Controls. Perturbations in (a) viral (orange) and (b) endogenous human miRNAs (blue) are evident in this example, where miRNA counts (log2 transformed) of IBS-C participants (y-axis) are regressed against the miRNA counts (log2 transformed) of healthy controls (x-axis). The majority of miRNAs express very similarly in both populations, but some notably deviate from the correlation. These deviations are evident among both rare and abundantly expressed targets. Please click here to view a larger version of this figure.
Figure 2: Normalized Counts of Viral and Endogenous Human miRNAs in IBS-constipation (IBS-C) versus Healthy Controls. Perturbations in (a) viral (orange) and (b) endogenous human miRNAs (blue) are evident in this example, where miRNA counts (log2 transformed) of IBS-C participants (y-axis) are regressed against the miRNA counts (log2 transformed) of healthy controls (x-axis). The majority of miRNAs express very similarly in both populations, but some notably deviate from the correlation. These deviations are evident among both rare and abundantly expressed targets. Please click here to view a larger version of this figure.
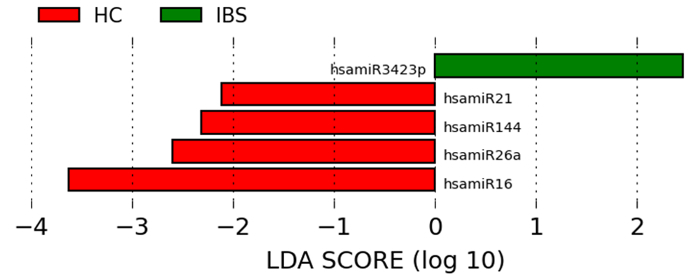 Figure 3:miRNAs are Significantly and Uniformly Differentially Expressed in IBS Compared to Healthy Controls. Differentially expressed miRNAs in IBS are revealed using the linear discriminant analysis effect size method, which combines tests of statistical significance with checks for consistency within categories. Please click here to view a larger version of this figure.
Figure 3:miRNAs are Significantly and Uniformly Differentially Expressed in IBS Compared to Healthy Controls. Differentially expressed miRNAs in IBS are revealed using the linear discriminant analysis effect size method, which combines tests of statistical significance with checks for consistency within categories. Please click here to view a larger version of this figure.
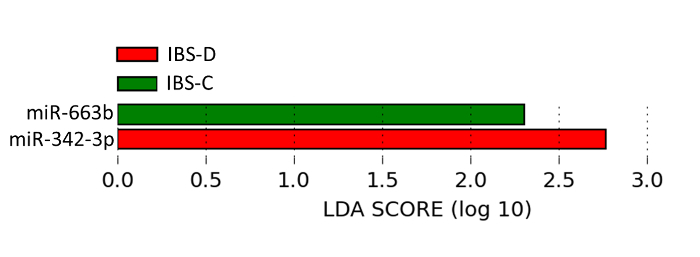 Figure 4:miRNAs are Significantly and Uniformly Differentially Expressed in IBS Subtypes Compared to Healthy Controls. miRNAs were differentially expressed in IBS-subtypes (-D: diarrhea, -C: constipation) compared to healthy controls using the linear discriminant analysis effect size method, which combines tests of statistical significance with checks for consistency within categories. Please click here to view a larger version of this figure.
Figure 4:miRNAs are Significantly and Uniformly Differentially Expressed in IBS Subtypes Compared to Healthy Controls. miRNAs were differentially expressed in IBS-subtypes (-D: diarrhea, -C: constipation) compared to healthy controls using the linear discriminant analysis effect size method, which combines tests of statistical significance with checks for consistency within categories. Please click here to view a larger version of this figure.
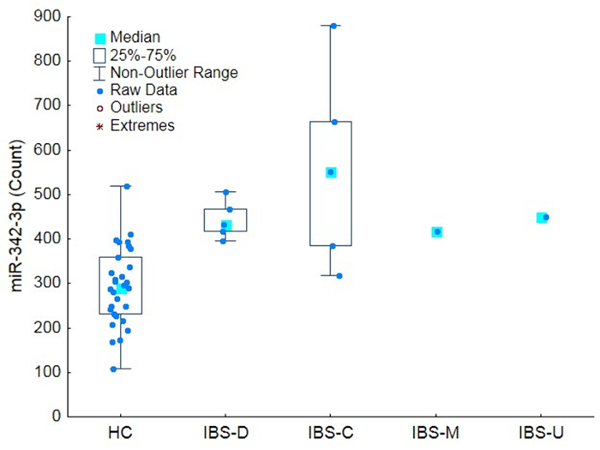 Figure 5:Differential Expression of miRNA-342-3p. Chronic abdominal pain of unknown etiology is a principal symptom of IBS. The box plot shows that miR-342-3p (normalized counts on the y-axis), a miRNA associated with chronic bladder pain of unknown etiology, was found to be present in circulation at higher counts across all IBS subtypes compared to healthy controls. The bars represent the non-outlier range, the boxes the inter-quartile range and the blue square the median count. Modified from 3. Please click here to view a larger version of this figure.
Figure 5:Differential Expression of miRNA-342-3p. Chronic abdominal pain of unknown etiology is a principal symptom of IBS. The box plot shows that miR-342-3p (normalized counts on the y-axis), a miRNA associated with chronic bladder pain of unknown etiology, was found to be present in circulation at higher counts across all IBS subtypes compared to healthy controls. The bars represent the non-outlier range, the boxes the inter-quartile range and the blue square the median count. Modified from 3. Please click here to view a larger version of this figure.
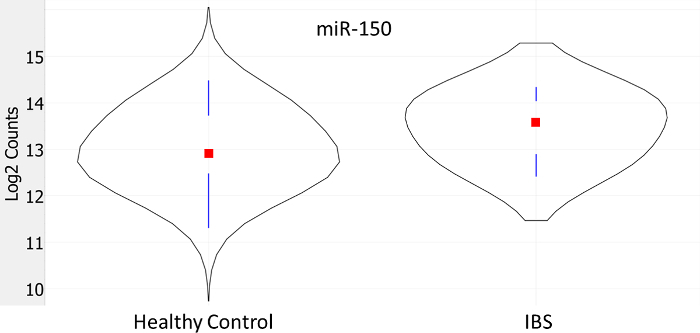 Figure 6:Differential Expression of miRNA-150. A violin plot showing that IBS participants had significantly higher circulating miR-150 counts compared to healthy controls (log2 transformed counts on the y-axis). The curves indicate the frequency of counts in the category, the vertical bars represent the 1st and 3rd quartiles, and the red square indicates the median count. Modified from 3. Please click here to view a larger version of this figure.
Figure 6:Differential Expression of miRNA-150. A violin plot showing that IBS participants had significantly higher circulating miR-150 counts compared to healthy controls (log2 transformed counts on the y-axis). The curves indicate the frequency of counts in the category, the vertical bars represent the 1st and 3rd quartiles, and the red square indicates the median count. Modified from 3. Please click here to view a larger version of this figure.
Discussion
Critical Steps within the Protocol
For the miRNA assay in particular, it is critical not to use unpurified lysates (these can be used in mRNA and protein assays), as the reagents have not been optimized for use with lysates, and sample preparation reactions are likely to be inhibited. Additionally, contaminants carried over from lysis and RNA purification steps (e.g., guanidinium compounds, ethanol, and phenols) can inhibit ligation and sample purification reactions. Good-quality RNA is therefore critical to the sensitivity and accuracy of the miRNA assay.
Using a thermocycler with a heated lid is critical to ensure proper temperature control to optimize the performance of all reaction steps. During step 3.5.1, it is important not to remove the strips from the thermocycler in order to maintain the temperature during the addition of the ligase. Removing the strips to add the ligase will compromise the reaction. When performing the hybridization steps, it is critical to avoid vigorous shaking, pipetting, or flicking when mixing reagents in the tubes, as this may shear the reporter probes. To ensure optimal performance and consistency, it is important to thoroughly mix reagents in the tubes by gentle flicking. Always spin down reactions after mixing, and use a new pipette tip each time a reagent is dispensed to ensure accurate pipetting and to avoid accidental cross-contamination.
Modifications and Troubleshooting
The code sets can be customized to include only targets of interest. In this way, costs can be balanced to allow for the testing of large sample sets when further investigating or validating a bio-signature. Custom probes can be designed for genes or targets not available from the a la carte menu. Sensitivity for less-abundant targets or low-concentration RNA can be manipulated by allowing for a longer hybridization time and/or by using higher-resolution digital counting.
In our study we used the predefined 800 target miRNA panel with total RNA from whole blood; however, the assays can be customized to include fewer miRNAs if a more targeted approach is needed. A total of 24 samples can be prepared on a single day, staggered in two sets of 12, because two cartridges can comfortably be passed through post-hybridization processing on the robotic prep station and imaged on the digital analyzer the next day. Staggered processing of the samples in batches of 12 is important to standardize the hybridization time. Cartridges that have been imaged can be saved and stored at 4 oC to be rescanned if data analyses suggest that a higher resolution scan may improve the detection of rarer miRNAs. Detection of rarer molecules may also be improved by hybridizing samples for longer; however, prolonged hybridization may result in oversaturation and may only improve results if the starting RNA concentrations are low (as in extracellular vesicle-derived RNA). The platform's sensitivity and precision allow relatively low-abundance miRNAs to be reliably counted.
Limitations of the Technique
The described gene expression assay platform only accommodates up to 800 targets per array. It is therefore not suited for whole miRNA-transcriptome/whole transcriptome studies. Only known, described, and specified targets can be detected using the platform, so it is not suited to the discovery of new molecular species. Other methods, such as RNASeq or other traditional microarray methods, are more suited to whole transcriptome exploratory studies.
Significance of the Technique with Respect to Existing/Alternative Methods
IBS is characterized by a combination of symptoms, principally including abdominal pain; visceral hypersensitivity; and changes in bowel habits, such as frequent diarrhea, constipation, or a combination of both9,10. IBS symptoms have no known organic cause, and patients do not present with clinically significant inflammation, histopathological perturbations of gastrointestinal tissues, or systemic markers9-12. Research suggests that biological dysregulation in IBS is subtle, subclinical, and heterogeneous, with common themes emerging around inflammation and immune function10. Therefore, we needed to control experimenter-introduced variation and use a platform that was capable of reliably detecting subtle perturbations in gene expression. The unique attributes of the assay and system standardize sample processing and reduce experimenter handling through automation, reducing technical variance to produce highly reproducible data. These features allow for focused investigations and produce highly precise and replicable data. This allows for the detection of subtle perturbations and perturbations among low-expression targets that may be overlooked or swamped by the signals of more abundant targets when using intensity- or amplification-based methods. PCR-based microarrays and RNAseq methods are viable alternatives to using the described platform and may be attractive as alternative when higher throughputs are required or when the aim is to characterize the whole transcriptome or to look for novel molecules. However, alternatives may not perform as well in accurately and sensitively detecting and quantitating rare targets and subtle perturbations.
Future Applications or Directions after Mastering this Technique
Circulating miRNA expression in IBS participants showed a number of perturbations that were found to be both significant and consistent within categories. The identified miRNAs were associated with relevant pathways and pathological conditions, including a functional pain condition (miR-342-3p: chronic bladder pain)8 and inflammation (miR-150)13. The example study was based on an exploratory cohort used to define targets and systems of interest. A more targeted, smaller miRNA array was then constructed, lowering the per sample cost and allowing for much larger cohorts to be analyzed in a cost-effective manner in order to validate and refine signatures and biomarkers. The gene expression platform assay system strikes a balance between the traditional exploratory nature of the microarray and more targeted, hypothesis-driven data collection to refine and test molecular signatures and biomarkers14-16. The method will be used to examine molecular and protein perturbations in in vivo and in vitro models where the described target miRNAs are experimentally over-expressed or inhibited. New assays allow for the simultaneous quantification of mRNAs and proteins directly from cell and tissue lysates. Furthermore, by optimizing the purification of specific fractions of peripheral blood and excreta (e.g., urine) and by customizing and optimizing certain assay parameters, molecular profiles and signatures of low molecular concentration fractions (e.g., exosomal fractions) will be examined.
Disclosures
W.A.H., S.K.A, N.H.F., and R.M.P. hold a US patent (no. PCT/US14/38638) for a circulating miRNA signature of IBS emanating from the research described herein.
Acknowledgments
The authors acknowledge funding from the U.S. Department of Health and Human Services, the National Institutes of Health, the National Institute of Nursing Research, and the Division of Intramural Research to Wendy A. Henderson, 1ZIANR000018-01-06; the Intramural Research Training Awards to Nicolaas H. Fourie, Ralph M. Peace, Sarah K. Abey, and Leeanne B. Sherwin; and special support from the Burroughs Wellcome Fund to Howard Hughes-NIH Research Scholar Ralph M. Peace.
The opinions expressed herein and the interpretation and reporting of these data are the responsibility of the author(s) and should not be seen as an official recommendation, interpretation, or policy of the National Institutes of Health or the United States Government.
References
- Aerssens J, et al. Alterations in Mucosal Immunity Identified in the Colon of Patients With Irritable Bowel Syndrome. Clin Gastroenterol Hepatol. 2008;6:194–205. doi: 10.1016/j.cgh.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbar A, et al. Increased capsaicin receptor TRPV1-expressing sensory fibres in irritable bowel syndrome and their correlation with abdominal pain. Gut. 2008;57:923–929. doi: 10.1136/gut.2007.138982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourie NH, et al. Elevated circulating miR-150 and miR-342-3p in patients with irritable bowel syndrome. Exp Mol Pathol. 2014;96:422–425. doi: 10.1016/j.yexmp.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Souba WW, Croce CM, Verne GN. MicroRNA-29a Regulates Intestinal Membrane Permeability in Patients with Irritable Bowel Syndrome. Gut. 2010;59:775–784. doi: 10.1136/gut.2009.181834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Verne GN. miRNA-based therapies for the Irritable Bowel Syndrome. Expert Opin Biol Ther. 2011;11:991–995. doi: 10.1517/14712598.2011.577060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlova IA, et al. MicroRNA modulation in complex regional pain syndrome. J Transl Med. 2011;9:1–11. doi: 10.1186/1479-5876-9-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C-K, et al. Extracellular MicroRNAs Activate Nociceptor Neurons to Elicit Pain via TLR7 and TRPA1. Neuron. 2014;82:47–54. doi: 10.1016/j.neuron.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gheinani AH, Burkhard FC, Monastyrskaya K. Deciphering microRNA code in pain and inflammation: lessons from bladder pain syndrome. Cell Mol Life Sci. 2013;70:3773–3789. doi: 10.1007/s00018-013-1275-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drossman DA, et al. Rome III: The Functional Gastrointestinal Disorders. 3rd edn. Degnon Associates, Inc; 2006. [Google Scholar]
- Chey WD, Kurlander J, Eswaran S. Irritable bowel syndrome: A clinical review. JAMA. 2015;313:949–958. doi: 10.1001/jama.2015.0954. [DOI] [PubMed] [Google Scholar]
- Del Valle-Pinero AY, Sherwin LB, Anderson EM, Caudle RM, Henderson WA. Altered vasoactive intestinal peptides expression in irritable bowel syndrome patients and rats with trinitrobenzene sulfonic acid-induced colitis. World J Gastroenterol. 2015;21:155–163. doi: 10.3748/wjg.v21.i1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson WA, et al. Inverse relationship of interleukin-6 and mast cells in children with inflammatory and non-inflammatory abdominal pain phenotypes. World J Gastrointest Pathophysiol. 2012;3:102–108. doi: 10.4291/wjgp.v3.i6.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekow JR, Kwon JH. MicroRNAs in inflammatory bowel disease. Inflamm Bowel Dis. 2012;18:187–193. doi: 10.1002/ibd.21691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldman-Jones MH, et al. Flexible Molecular Subtyping of Clinical DLBCL Samples Using the NanoString nCounter System. Clin Cancer Res. 2015;21:2367–2378. doi: 10.1158/1078-0432.CCR-14-0357. [DOI] [PubMed] [Google Scholar]
- Lee J, et al. Nanostring-Based Multigene Assay to Predict Recurrence for Gastric Cancer Patients after Surgery. PLoS ONE. 2014;9:e90133. doi: 10.1371/journal.pone.0090133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohavanichbutr P, et al. A 13-gene signature prognostic of HPV-negative OSCC: discovery and external validation. Clin Cancer Res. 2013;19:1197–1203. doi: 10.1158/1078-0432.CCR-12-2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- nCountermiRNA Expression Assay User Manual (MAN-C0009-05) NanoString Technologies, Inc; 2013. Available from: http://www.nanostring.com/media/pdf/MAN_nCounter_miRNA_Expression_Assay.pdf. [Google Scholar]