

Abstract
Structural interactions between the endoplasmic reticular (ER) and mitochondrial membranes, in domains known as mitochondria-associated membranes (MAM), are crucial hubs for cellular signaling and cell fate. Particularly, these inter-organelle contact sites allow the transfer of calcium from the ER to mitochondria through the voltage-dependent anion channel (VDAC)/glucose-regulated protein 75 (GRP75)/inositol 1,4,5-triphosphate receptor (IP3R) calcium channeling complex. While this subcellular compartment is under intense investigation in both physiological and pathological conditions, no simple and sensitive method exists to quantify the endogenous amount of ER-mitochondria contact in cells. Similarly, MAMs are highly dynamic structures, and there is no suitable approach to follow modifications of ER-mitochondria interactions without protein overexpression. Here, we report an optimized protocol based on the use of an in situ proximity ligation assay to visualize and quantify endogenous ER-mitochondria interactions in fixed cells by using the close proximity between proteins of the outer mitochondrial membrane (VDAC1) and of the ER membrane (IP3R1) at the MAM interface. Similar in situ proximity ligation experiments can also be performed with the GRP75/IP3R1 and cyclophilin D/IP3R1 pairs of antibodies. This assay provides several advantages over other imaging procedures, as it is highly specific, sensitive, and suitable to multiple-condition testing. Therefore, the use of this in situ proximity ligation assay should be helpful to better understand the physiological regulations of ER-mitochondria interactions, as well as their role in pathological contexts.
Keywords: Molecular Biology, Issue 118, mitochondria-associated membranes, mitochondria, endoplasmic reticulum, in situ proximity ligation assay, immunofluorescence, VDAC, IP3R.
Introduction
Mitochondria and endoplasmic reticulum (ER) are not independent organelles in the cell, but they interact structurally and functionally at contact sites defined as mitochondria-associated endoplasmic reticulum membranes (MAM). In fact, MAMs correspond to regions where the membranes of the ER and mitochondria are closely apposed, allowing interactions between proteins from both sides. Nonetheless, the membranes of these organelles do not fuse within these regions, so they maintain their separate entities. The MAMs play a crucial role in calcium (Ca2+) and phospholipid transfer from ER to mitochondria, impacting energy metabolism and cell survival1-3.
The association between the ER and mitochondria was first visualized in the 1970s with electron microscopy. Since then, transmission electron microscopy4,5, electron tomography6,7 or immuno-localization of ER and mitochondria-specific fluorophores/fluorescent proteins8 were classically used to study ER-mitochondria interactions. Another useful tool for the analysis of MAM is based on the use of subcellular fractionation. It allows the isolation of MAM fractions by differential ultracentrifugation coupled to a Percoll gradient9. However, the final product contains enriched MAM fractions, rather than pure fractions. Altogether, these strategies are not particularly sensitive and/or quantitative, and they are not easily amenable to large screening. Alternatively, genetic approaches using drug-inducible fluorescent inter-organelle linkers have emerged, but they do not allow the analysis of organelle interactions at the endogenous expression levels of proteins10.
Based on Szabadkai's discovery of the IP3R/GRP75/VDAC complex at the MAM11, we developed a quantitative method to analyze ER-mitochondria interactions. We used the in situ proximity ligation assay to detect and quantify interactions between VDAC1 and IP3R1, two organelle-surface proteins involved in the Ca2+-channeling complex at the MAM interface in fixed cells12. Briefly, we probed VDAC1 at the outer mitochondrial membrane (mouse anti-VDAC1 primary antibody) and IP3R1 at the ER membrane (rabbit anti-IP3R1 primary antibody) (Figure 1, panel a). Then, according to the assay, we added both anti-mouse and anti-rabbit IgG (mouse and rabbit proximity ligation assay probes), which are conjugated to complementary oligonucleotide extensions. If the two targeted proteins are at a distance below 40 nm, the oligonucleotides can hybridize with the subsequently added connector oligos to allow the formation of a circular DNA template (Figure 1, panel b). This circular DNA molecule is ligated and amplified, creating a single-stranded DNA product covalently attached to one of the proximity probes (Figure 1, panel c). Since the distance between the ER and mitochondria at the MAM interface ranges from 10 nm to 25 nm6, proximity ligation and amplification can be done, leading to subsequent detection due to the hybridization of Texas red-labeled oligonucleotides probes (Figure 1, panel d). Each fluorescent dot represents interactions between VDAC1/IP3R1, thus allowing the quantification of in situ ER-mitochondria interactions in individual cells.
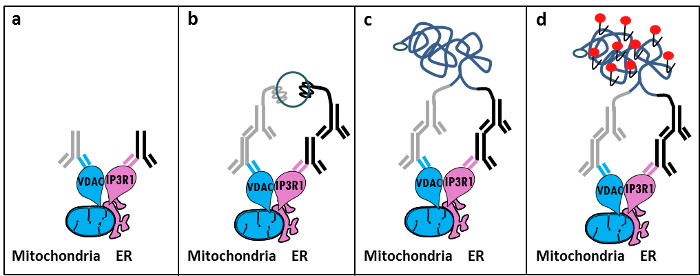 Figure 1: Schematic Illustration of the Detection of the Endoplasmic Reticulum-mitochondria Interactions by In Situ Proximity Ligation Assay.a) A mouse primary antibody directed against VDAC1 and a rabbit primary antibody directed against IP3R1 can bind to their epitopes in proximity at the MAM interface, b) The addition of a pair of proximity ligation probes directed against mouse and rabbit IgG. These probes have attached DNA strands that can form templates for the ligation of connector oligos. c) The circular DNA strand formed after ligation can be amplified and d) visualized by microscopy as a fluorescent dot by using Texas red-labeled oligonucleotides. Please click here to view a larger version of this figure.
Figure 1: Schematic Illustration of the Detection of the Endoplasmic Reticulum-mitochondria Interactions by In Situ Proximity Ligation Assay.a) A mouse primary antibody directed against VDAC1 and a rabbit primary antibody directed against IP3R1 can bind to their epitopes in proximity at the MAM interface, b) The addition of a pair of proximity ligation probes directed against mouse and rabbit IgG. These probes have attached DNA strands that can form templates for the ligation of connector oligos. c) The circular DNA strand formed after ligation can be amplified and d) visualized by microscopy as a fluorescent dot by using Texas red-labeled oligonucleotides. Please click here to view a larger version of this figure.
Similar in situ proximity ligation assay experiments can be performed with the GRP75/IP3R1 pair of antibodies, as well as cyclophilin D (CypD)/IP3R1 antibodies, considering that CypD was shown to interact with the IP3R/GRP75/VDAC complex at the MAM interface12-14.
Protocol
1. Preparation of Solutions
Prepare 10% formaldehyde in PBS (low salt) by diluting 5.5 ml of 37% formaldehyde in 14.5 ml of PBS. Prepare 1 M glycine, pH 8.0, by dissolving 3.8 g of glycine in 50 ml of PBS; dilute this solution to obtain 100 mM glycine in 1x PBS.
Prepare 0.1% Triton-X100 in 1x PBS. Prepare 20x Saline Sodium Citrate (SSC) by using 3.0 M sodium chloride and 0.30 M trisodium citrate prepared in a deionized water buffer with pH 7.0 at 25 °C. Dilute this buffer to 1x and 0.01x using deionized water.
2. Fixation of Cells
NOTE: We used the HuH7 hepatocarcinoma cell line in this study, but this method is applicable to other adherent cell cultures.
Plate HuH7 cells (cultured in DMEM 1 g/L glucose, supplemented with 10% fetal calf serum and 0.01% penicillin-streptomycin stock) at 150,000 cells per uncoated 35-mm glass-bottom dish. When working on primary cell cultures, use collagen-coated dishes.
The next day, remove the culture medium. Wash the cells with 1 ml of PBS and aspirate. Fix the cells by adding 1 ml of formaldehyde 10% and incubate for 10 min at room temperature (RT) under agitation.
Stop the reaction with 1 ml of 1 M glycine and mix by rotation. Remove the stop reaction solution and wash the cells by adding 1 ml of 1x PBS; agitate by rotation and aspirate. Add 1 ml of 100 mM glycine to the cells and incubate them for 15 min at RT under agitation, and then aspirate. NOTE: The protocol can be stopped here and the following steps can be postponed to another day. In that case, add 1 ml of 100 mM glycine and keep at 4 °C, if necessary.
3. Permeabilization of Cells
Add 1 ml of 0.1% Triton-X100 in 1x PBS, incubate for 15 min at RT under agitation, and then aspirate. This incubation time could be increased up to 15 - 20 min when working on primary cell cultures (e.g., primary mice hepatocytes). Wash the cells by adding 1 ml of 1x PBS; aspirate.
4. Blocking
Add 40 µl of blocking solution to each sample (provided by the kit); this volume can be increased in order to cover the sample. Incubate the dishes for 30 min at 37 °C in a humidity chamber.
Tap the blocking solution off of the dishes. Try to obtain equal residual volumes for each slide, as this will affect reproducibility. Do not let the samples dry!
5. Primary Antibodies
Dilute the primary antibodies in 1x PBS and add the solution to the dishes (VDAC1 mouse antibody: 1/100, IP3R1 rabbit antibody: 1/500). Alternatively, GRP75 or CypD antibodies (both mouse antibodies used at 1/500) can be used instead of VDAC1.
Incubate overnight in a humidity chamber at 4 °C. Wash the slides two times using Tris Buffered Saline with 0.01% Tween (TBS-T).
6. Proximity Ligation Assay Probes
Proximity ligation assay probes are provided by the kit. Choose probes according to the species of primary antibodies.
Prepare the two proximity ligation assay probes 1:5 in antibody diluent. Allow the mixture to sit for 20 min at RT. Add the diluted proximity ligation assay probe solution. Incubate the dishes in a pre-heated humidity chamber for 1 hr at 37 °C. Wash the dishes two times with TBS-T.
7. Ligation
Use the in situ detection reagent Texas red kit.
Dilute the 5x ligation stock (provided by the kit) 1:5 in high-purity water and mix well. Dilute the ligase in the 1x ligation solution (provided by the kit) 1:40 and vortex. Wait to add the ligase until immediately before addition to the samples.
Add this solution to each sample (40 µl for 35-mm glass-bottom dishes) and incubate the slides in a pre-heated humidity chamber for 30 min at 37 °C. Wash the slides two times with TBS-T.
8. Amplification
NOTE: Be careful, light sensitive reagents.
Dilute the 5x amplification stock (provided by the kit) 1:5 in high-purity water. Remove the polymerase from the freezer using the freezing block (-20 °C). Dilute the polymerase (provided by the kit) 1:80 in the 1x amplification solution and vortex. Add the polymerase immediately before using the mixture.
Add this solution to each sample (40 µl for 35-mm glass-bottom dishes). Incubate the slides in a pre-heated humidity chamber for 100 min at 37 °C. Tap the Amplification-Polymerase solution off of the slides.
9. Final Washing
Wash the slides in 1x SSC washing buffer for 2 min. Wash the slides in 0.01x SSC washing buffer for 2 min. Let the dishes dry at RT in the dark.
10. Preparation for Imaging
Mount the slides using a minimal volume of mounting medium containing DAPI (aqueous and does not solidify), ensuring that no air bubbles get caught under the cover slip. Nail polish can be used to seal the edges. Wait for approximately 15 min before analyzing using a fluorescence or confocal microscope (excitation: 594 nm, emission: 624 nm, magnification: 63X).
After imaging, store the slides at -20 °C in the dark.
Representative Results
Based on our experience using this protocol, we can safely recommend this method for the visualization and quantification of ER-mitochondria interactions in fixed cells. Representative images of in situ proximity ligation assay-visualized ER-mitochondria interactions in the HuH7 hepatocarcinoma cell line, using several pairs of antibodies, are shown. As shown in Figure 2 by fluorescent microscopy, each red dot represents an interaction between the two targeted proteins, and nuclei appear in blue. As a negative control, the use of only one primary antibody led to no signal (Figure 2, panel a), validating the dual recognition requirement for in situ proximity ligation assay signal generation. Classically, we analyze ER-mitochondria interactions using the VDAC1/IP3R1 pair of antibodies (Figure 2, panel b). Similar in situ proximity ligation assay experiments could also be performed with the GRP75/IP3R1 or CypD/IP3R1 pairs of antibodies (Figure 2, panels c and d, respectively), since GRP75 is a chaperone coupling VDAC and IP3R at the MAM interface11 and CypD was demonstrated to interact with the VDAC/GRP75/IP3R Ca2+-channeling complex12-14. Quantification of fluorescent dots can be performed using either Blob-finder15 or ImageJ software. As the nuclei of cells are labeled in blue, quantifications of the ER-mitochondria interactions per cell are suitable to perform. We thoroughly validated this assay12, and we can conclude that these targeted proteins are good candidates to study ER-mitochondria interactions.
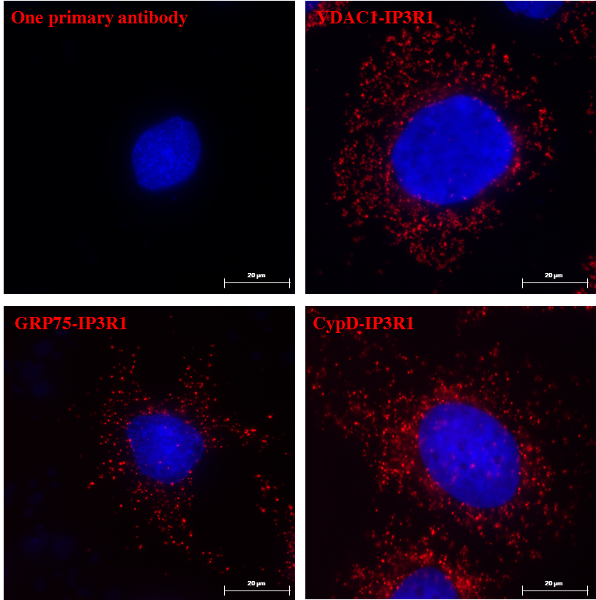 Figure 2: Visualization of the VDAC1/IP3R1, GRP75/IP3R1 and CypD/IP3R1 Interactions by In Situ Proximity Ligation Assay in HuH7 Cells. Cell nuclei appear in blue, and interactions between the two targeted proteins are depicted in red (63X magnification; scale bar = 20 µm). We show that VDAC1, GRP75, and CypD interact with IP3R1. As a negative control, we demonstrate that only one primary antibody does not lead to red fluorescence. To validate the assay, several other controls have been performed to demonstrate that VDAC1, GRP75, and CypD do not interact with other ER proteins and that IP3R1 does not interact with other mitochondrial proteins (data not shown, see ref. 9 and 13). Please click here to view a larger version of this figure.
Figure 2: Visualization of the VDAC1/IP3R1, GRP75/IP3R1 and CypD/IP3R1 Interactions by In Situ Proximity Ligation Assay in HuH7 Cells. Cell nuclei appear in blue, and interactions between the two targeted proteins are depicted in red (63X magnification; scale bar = 20 µm). We show that VDAC1, GRP75, and CypD interact with IP3R1. As a negative control, we demonstrate that only one primary antibody does not lead to red fluorescence. To validate the assay, several other controls have been performed to demonstrate that VDAC1, GRP75, and CypD do not interact with other ER proteins and that IP3R1 does not interact with other mitochondrial proteins (data not shown, see ref. 9 and 13). Please click here to view a larger version of this figure.
Discussion
Collectively, our studies indicate that the in situ proximity ligation assay is truly a relevant strategy to follow and quantify endogenous ER-mitochondria interactions in fixed cells, without the need for using organelle-specific fluorophores or fluorescent proteins. The specific use of VDAC1/IP3R1 antibodies has been adapted to study ER-mitochondria interactions in HuH7 cells. However, alternative isoforms of VDAC and IP3R may be used, depending on the cell type. In this case, antibodies need to be validated by immunofluorescence before the proximity ligation assay experiments in order to ensure specificity and to find the correct working dilutions. We also note that the dilution of antibodies needs to be systematically adjusted when using a new proximity ligation assay kit or a new antibody batch. We chose to use in situ detection Texas red reagents, but it is also possible to use reagents in other wavelengths or to use the HRP/Novared kit for bright-field detection. We used this option to analyze ER-mitochondria interactions in paraffin-embedded liver samples16, enabling the visualization of ER-mitochondria interactions as red-brownish dots, thus overcoming the difficulty of working on liver tissue that presents marked autofluorescence.
Compared to conventional imaging approaches6,7,17, the in situ proximity ligation assay18 avoids using organelle-specific fluorophores or fluorescent proteins that could induce some artefacts. Furthermore, it offers improved selectivity because it requires dual recognition and increased sensitivity. It includes DNA amplification as a component for detecting the target protein by using fluorescence microscope. Finally, the procedure is fast, suitable for multiple-condition testing, reproducible, and consistent. Indeed, we systematically found very reproducible results when targeting several pairs of proteins (VDAC/IP3R, GRP75/IP3R, CypD/IP3R) at the MAM interface or when using alternative strategies (electron microscopy analysis, subcellular fractionation, or co-immunoprecipitation)12,13,16, strongly supporting the robustness of the technique and its ability to monitor organelle coupling. Nevertheless, as some targeted proteins (e.g., VDAC and GRP75) have been reported to be present in other intracellular structures19,20, we cannot completely exclude unspecific signals if IP3R is not far away from these structures. However, co-localization studies of each targeted proteins with specific labeling of respective organelles have been performed in HuH7 cells12, and the majority of the signal was merged in each case, suggesting that another localization of proteins, if present, is minor. Therefore, we recommend that this possibility is tested in each investigated cell or tissue and that the proximity ligation assay is performed with the three pairs of antibodies to totally confirm the results. In addition, the expression level of each targeted proteins should be investigated in each case to determine whether the modulation of organelle proximity is linked to a structural modification or to variation in the expression levels of the targeted proteins.
Until now, the lack of adapted analytical methods has prevented dynamic monitoring of ER-mitochondria contact in health and disease. Now, we think that the use of this in situ proximity ligation assay should allow a better understanding of the physiological regulation of ER-mitochondria interactions, as well as of their role in pathological contexts.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
We thank all the people in our laboratory who contributed to optimize and validate the protocol. This work was supported by INSERM and the national research agency (ANR-09-JCJC-0116 AND ANR-11-BSV1-033-02). E.T. was supported during her PhD by a research fellowship from the French ministry of higher education and research.
References
- Bravo-Sagua R, et al. Organelle communication: signaling crossroads between homeostasis and disease. The international journal of biochemistry & cell biology. 2014;50:55–59. doi: 10.1016/j.biocel.2014.01.019. [DOI] [PubMed] [Google Scholar]
- Giorgi C, et al. Mitochondria-associated membranes: composition, molecular mechanisms, and physiopathological implications. Antioxidants & redox signaling. 2015;22:995–1019. doi: 10.1089/ars.2014.6223. [DOI] [PubMed] [Google Scholar]
- Phillips MJ, Voeltz GK. Structure and function of ER membrane contact sites with other organelles. Nature reviews. Molecular cell biology. 2016;17:69–82. doi: 10.1038/nrm.2015.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosson P, et al. The RTM resistance to potyviruses in Arabidopsis thaliana: natural variation of the RTM genes and evidence for the implication of additional genes. PLoS One. 2012;7:39169. doi: 10.1371/journal.pone.0039169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannella CA. Structure and dynamics of the mitochondrial inner membrane cristae. Biochim Biophys Acta. 2006;1763:542–548. doi: 10.1016/j.bbamcr.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Csordas G, et al. Structural and functional features and significance of the physical linkage between ER and mitochondria. The Journal of cell biology. 2006;174:915–921. doi: 10.1083/jcb.200604016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannella CA, Buttle K, Rath BK, Marko M. Electron microscopic tomography of rat-liver mitochondria and their interaction with the endoplasmic reticulum. Biofactors. 1998;8:225–228. doi: 10.1002/biof.5520080309. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J, Pinton P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat Protoc. 2009;4:1582–1590. doi: 10.1038/nprot.2009.151. [DOI] [PubMed] [Google Scholar]
- Csordas G, et al. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol Cell. 2010;39:121–132. doi: 10.1016/j.molcel.2010.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadkai G, et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol. 2006;175:901–911. doi: 10.1083/jcb.200608073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubbs E, et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes. 2014;63:3279–3294. doi: 10.2337/db13-1751. [DOI] [PubMed] [Google Scholar]
- Paillard M, et al. Depressing Mitochondria-Reticulum Interactions Protects Cardiomyocytes From Lethal Hypoxia-Reoxygenation Injury. Circulation. 2013;128:1555–1565. doi: 10.1161/CIRCULATIONAHA.113.001225. [DOI] [PubMed] [Google Scholar]
- Rieusset J, et al. Disruption of calcium transfer from ER to mitochondria links alterations of mitochondria-associated ER membrane integrity to hepatic insulin resistance. Diabetologia. 2016;59:614–623. doi: 10.1007/s00125-015-3829-8. [DOI] [PubMed] [Google Scholar]
- Allalou A, Wahlby C. BlobFinder, a tool for fluorescence microscopy image cytometry. Computer methods and programs in biomedicine. 2009;94:58–65. doi: 10.1016/j.cmpb.2008.08.006. [DOI] [PubMed] [Google Scholar]
- Theurey P, et al. Mitochondria-associated endoplasmic reticulum membranes allow adaptation of mitochondrial metabolism to glucose availability in the liver. Journal of molecular cell biology. 2016. [DOI] [PubMed]
- de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- Soderberg O, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nature methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- De Pinto V, Messina A, Lane DJ, Lawen A. Voltage-dependent anion-selective channel (VDAC) in the plasma membrane. FEBS letters. 2010;584:1793–1799. doi: 10.1016/j.febslet.2010.02.049. [DOI] [PubMed] [Google Scholar]
- Kaul SC, Taira K, Pereira-Smith OM, Wadhwa R. Mortalin: present and prospective. Experimental gerontology. 2002;37:1157–1164. doi: 10.1016/s0531-5565(02)00135-3. [DOI] [PubMed] [Google Scholar]