

Abstract
The micro-neutralization (MN) assay is a standard technique for measuring the infectivity of the influenza virus and the inhibition of virus replication. In this study, we present the protocol of an imaging-based MN assay to quantify the true antigenic relationships between viruses. Unlike typical plaque reduction assays that rely on visible plaques, this assay quantitates the entire infected cell population of each well. The protocol matches the virus type or subtype with the selection of cell lines to achieve maximum infectivity, which enhances sample contrast during imaging and image processing. The introduction of quantitative titration defines the amount of input viruses of neutralization and enables the results from different experiments to be comparable. The imaging setup with a flatbed scanner and free downloadable software makes the approach high throughput, cost effective, user friendly, and easy to deploy in most laboratories. Our study demonstrates that the improved MN assay works well with the current circulating influenza A(H1N1)pdm09, A(H3N2), and B viruses, without being significantly influenced by amino acid substitutions in the neuraminidase (NA) of A(H3N2) viruses. It is particularly useful for the characterization of viruses that either grow to low HA titer and/or undergo an abortive infection resulting in an inability to form plaques in cultured cells.
Keywords: Infection, Issue 118, influenza, micro-neutralization, hemagglutination inhibition, antigenicity, flatbed scanner, high-throughput imaging, virus-infected cell quantitation, plaque assay
Introduction
Micro-neutralization (MN) assays are used in virology for the quantitation of neutralizing antibodies and of antiviral activities. As an alternative of hemagglutination inhibition (HI) assays, MN assays can overcome non-antigenic effects influenced by the affinity changes of receptor-binding in influenza viruses, which can complicate the interpretation of HI results1,2,3. Until recently, most MN assays were based on cytopathic effects (CPE) or enzyme-linked immunosorbent assays (ELISA)4,5. MN assays based on focus and plaque reduction were developed in 19906,7,8. Plaque reduction assays rely on counting visible plaques to quantify infectivity. However, visual counts only cover large plaques that are resolvable by human eyes, even though the majority of plaques are small and invisible for many current circulating viruses. This incomplete coverage can cause significant variation among examiners and between experiments, leading to incomparable results. It is also impossible to use the method when some viruses show abortive infection of single cells or very small plaques.
The poor counting resolution can be improved by the introduction of an imaging-based protocol. With advances in technology, optical microscopy and high-throughput well-plate readers can provide accurate means to count the infected cells9. Under a trans-illuminated microscope, infected cells tagged with certain markers may be visualized by their absorption or fluorescence contrast in sub-cellular resolution. A sample can then be analyzed on a computer screen.
Unfortunately, due to the limitation in the field of view, more than a hundred tiled images are required to cover a single well. Analyzing a plate with 96 wells would require the imaging and processing of about ten thousand images. Such a laborious process is time consuming and expensive, and the resolution gained is in general unnecessary for routine characterization of viral infections. Laboratories with a limited budget may find that the approach built around a flatbed scanner provides a cost-effective, high-throughput alternative.
In this paper, we describe an improved plaque reduction MN assay that is suitable for the antigenic characterization of a large number of viruses and for quantitatively measuring antiviral activities and neutralization antibodies. The assay has several advantages: firstly, it is an imaging-based assay that is able to measure virus infections on the cellular level, regardless of plaque size. Counting the total infected cell population (ICP) within a well greatly increases the detection sensitivity, making it possible to characterize the viruses with low infectivity. Secondly, a more accurate quantitative titration is introduced prior to neutralization to determine the amount of input virus. The quantitative input virus significantly reduces the variation between different experiments and makes the results more comparable between laboratories. Thirdly, neutralization titers can be determined directly by analyzing images, making the quantitation fast and user-friendly. Finally, the protocol provides a cost-effective and high-throughput alternative with the required resolution and accuracy. The quantitation is based on a flatbed scanner and free data processing software. The whole setup has a small footprint and is deployable in most laboratories.
The protocol presented in this paper consists of four major steps, including virus titration, titration quantitation, virus neutralization, and neutralization quantitation. Virus titration is a preparation experiment that determines the amount of input viruses to be used in neutralization. During a titration, a number of viral concentrations are applied to the cell monolayers in a 96-well plate. The infected cells are then quantitated in section 2. The viral dilution that produces 20%-85% ICP is in turn applied as input viruses to the corresponding neutralization insection 3. The titers of the neutralization protocol are quantified using section 4. Experiments that followed the above protocols are presented in the Representative Results. The assay has been tested thoroughly during the last two years with most of the current circulating influenza viruses, such as A(H1N1)pdm09, A(H3N2), and B viruses. The results of the influenza virus characterization were included in the reports for the WHO consultation meeting that gave recommendations on influenza vaccines for use in the Southern Hemisphere in 2016 and the Northern Hemisphere in 2016-2017.
Protocol
NOTE: The biosafety level (BSL) for the following protocol is BSL 2 for seasonal influenza viruses and BSL 3+ for potential pandemic influenza viruses. Viral titration is required in advance of a neutralization experiment to decide the viral concentration of the viral control (VC).
1. Virus Titration
NOTE: Depending on the number of viruses and the number of duplicates, a well plate can be set up with the following flexibilities: (1) The viral dilution can be arranged along either the rows or the columns (Figure 1). Each virus should occupy a separate row/column. (2) The viral dilution increment is flexible, but it should start with the highest viral concentration at the top-left corner. (3) There is no restriction on the number of duplicates.
NOTE: Madin Darby canine kidney (MDCK) and MDCK-SIAT1 (SIAT) cells were kindly provided by Dr. M. Matrosovich, Marburg, Germany10. SIAT cells are MDCK cells stably transfected with the human CMP-N-acetylneuraminate: β-galactoside α-2,6-sialyltransferase gene for enhanced expression of sialic acid (SA) α2-6Gal-terminated oligosaccharides.
Aliquot sufficient MDCK cells or MDCK-SIAT cells8 into 96-well plates (200 µl/well). Incubate the cells at 37 °C with 5% CO2 for 2 or 3 days to reach confluence. Propagate the cells in DMEM supplemented with 10% heat-inactivated fetal calf serum (FCS) and antibiotics (100 U/ml penicillin and 100 µg/ml streptomycin). Incubate the SIAT cells at 37 °C with 5% CO2 and 1 mg/ml G418 sulphate. NOTE: The dilution factor is dependent upon experience and the cell line used. The cell monolayer must be confluent (i.e., no cell wall or visible outline for MDCK cells and a tightly-packed layout for MDCK-SIAT1 cells) at the time of virus inoculation. Examples of dilutions based on the subculture of confluent flasks of MDCK or MDCK-SIAT1 cells are given in Table 1.
Wash the cells 3 times with 200 µl of virus growth medium (VGM) per well. Sterilize the multiwall plate washer with 70% EtOH for 30 min, and then rinse it in sterile phosphate-buffered saline (PBS) or VGM before the wash.
Aspirate the VGM and immediately add 50 µl of VGM to each well.
Add 900 µl of VGM to each of 6 sterile tubes (or fewer, depending upon the number of test viruses and duplicates). Pipette 100 µl of virus into the first tube, mix well, and serially dilute, changing the tips between each dilution. Repeat for each virus to be titrated. NOTE: This will give virus dilutions of 10-1 to 10-6.
Add 50 µl of each virus dilution, starting at the highest dilution (1 x 10-5 in Figure 1), to duplicated wells (Columns 9 and 10 in Figure 1) of a 96-well plate.
Place the plate at 37 °C for 2-3 hr to allow the virus to infect the cells. NOTE: A 2 to 3 hr incubation is necessary to ensure that the viruses in the inoculum have enough time to reach the monolayer.
Prepare the overlay (10 ml/plate) NOTE: The overlay consists of 5 ml of 2x DMEM, Trypsin (2 µg/ml final concentration), 20 µl of 1 mg/ml stock, and 5 ml of cellulose cotton linters (see the Materials List).
Remove the inoculum.
Add the overlay (200 µl to each well). Incubate overnight at 37 °C, UNDISTURBED. NOTE: The virus budding takes place after approximately 4 hr, so it is important that the plate is not disturbed after this time.
Aspirate the overlay from the wells.
Add ice-cold 4% paraformaldehyde in PBS A (200 µl/well). Place them at 4 °C for 30 min or at room temperature for 20 min. NOTE: PBS A is natural-pH phosphate-buffered saline with 10 g of NaCl, 0.25 g of KCl, 1.437 g of Na2HPO4, 0.25 g of KH2PO4, and 1 L of distilled water (see the Materials List). NOTE: Paraformaldehyde is harmful when inhaled and can cause burns if swallowed. There is also limited evidence of a carcinogenic effect, and it may cause sensitization after skin contact.
Aspirate the paraformaldehyde and wash the plates twice with PBS A (200 µl/well).
Store the plates in PBS A at 4 °C for future use, or carry on with the following steps.
Add permeabilization buffer (100 µl/well). Leave it at room temperature for 30 min.
Wash plate twice with PBS A (200 µl/well).
Add 50 µl of the first antibody, mouse MAb against influenza type A (1:1,000 in ELISA Buffer), per well. Incubate it at room temperature for 1 hr (or 4 °C overnight), with shaking.
Wash it 3 times in 100 µl of wash buffer (0.05% Tween 80 in PBS A, v/v) per well. Incubate it at room temperature for 5 min between washes. Alternately, wash it 3 times without incubation using 300 µl per well.
Add 50 µl of the second antibody, goat anti-mouse IgG (H+L) HRP conjugate (1:1,000 in ELISA Buffer), per well. Incubate it at room temperature for 1 hr, with shaking.
Wash it 3 times as described in step 1.17.
Add the substrate (50 µl/well). Incubate it at room temperature for 30 min or until the development of a blue color is clearly visible.
To stop the reaction, wash the plate twice with 200 µl of distilled water per well, incubating at room temperature for 2 -3 min between the washes.
Air-dry the plate and store it in a dark place (e.g., wrap it in aluminum foil).
2. Titration Quantitation
Place a well plate in the scanning area of a flatbed scanner, as shown in Figure 2a. Use the L-shape position limit to ensure an optimum and repeatable imaging location. NOTE: It is possible to image two well plates in one scan.
Scan an image. NOTE: The settings are shown in Table 2.
- Run the "Wellplate Reader" software to calculate the required virus concentration (Figure 3).
- Click the "Load image" button to load an image. Slide the red bar in the histogram of the "Global Threshold" tab to adjust the sampling threshold. Click the "Update" button to examine the effect on the image.
- Tick the "Calculate Neutralization/Titration" box. Click the "Sampling" button to quantify the ICP. Click the "Save" button to save the sampling results when prompted. NOTE: After the sampling process, a new window called "Neutralization & Titration Calculation" appears automatically if the "Calculate Neutralization/Titration" box is ticked.
- Load or input the well-plate definition map. Indicate the threshold (e.g., 30%) in "Titration: Optimal Virus Population (%)". Select the "Titration Process" tab to calculate the titration results. Check the titration results. Click the "Save & Close" button to save the titration results. NOTE: Refer to Supplementary S1 for more detailed instruction on the software. A copy of the bespoke software is available upon request. NOTE: Typically, the best virus dilution for the plaque-reduction assay yields around 50% (20-85%) ICP of total cells within each well11.
3. Virus Neutralization
NOTE: Calculate the number of plates required for the neutralization assay. Each plate can accommodate one virus and a number of antisera.
NOTE: Depending on the number of antisera and the number of duplicates, a well plate can be set up with the following flexibilities: (1) The serum dilution can be arranged along either row or a column (Figure 4). Each serum should occupy a separate row or column. (2) The increment of serum dilution is flexible, but it should start with the highest serum concentration at the top-left corner of a plate. (3) The number of duplicates balances the number of antisera. More duplicates can help to smooth out experimental variations. (4) The number of the VC and the number of the cell control (CC) duplicates are flexible, but they should also be in separate rows or columns. It is expected that the number of VC duplicates is significantly greater than that of the antisera.
Prepare the cell monolayer, as in steps 1.1-1.3, 2 or 3 days in advance.
Add 50 µl of VGM to each well of the plate.
Use Columns 11 and 12 for VC and CC, respectively. Add 50 µl aliquots of 1:20 receptor-destroying enzyme (RDE)-treated serum to the first row (A) of Columns 1-10. NOTE: For each virus and serum combination, the assay is performed in duplicate, so one plate may, for example, contain one virus tested against five antisera.
Perform 2-fold serial dilutions by transferring 50 µl from Row A to Row H (Columns 1-10 in Figure 4) and discarding 50 µl from Row H.
Add 50 µl of diluent to each well of the CC column (Column 12 in Figure 4).
Add 50 µl of the virus to each well of the plate, except for the CC column (Columns 1-11, Rows A-H in Figure 4).
Incubate it at 37 °C for 2-3 hr. Remove the inoculum. Add the overlay (200 µl) to each well. Incubate it overnight at 37 °C, UNDISTURBED. Fix and stain the plates as for the virus titration (steps 1.11-1.22).
4. Neutralization Quantification
Place a well plate in the scanning area of a flatbed scanner, as shown in Figure 2a. Use the L-shape position limit to ensure an optimum and repeatable imaging location. NOTE: It is possible to image two well plates in one scan.
Scan the plate, as described in step 2.2.
- Run the "Wellplate Reader" software to calculate the required viral titers (Figure 3, step 2.3).
- Click the "Load image" button to load an image. Slide the red bar in "Global Threshold" to adjust the sampling threshold. Click the "Update" button to examine the effect.
- Tick the "Calculate Neutralization/Titration" box. Click the "Sampling" button to quantify the ICP. Click the "Save" button to save the sampling results when prompted. NOTE: After the sampling process, a new window called "Neutralization & Titration Calculation" appears automatically if the "Calculate Neutralization/Titration" box is ticked.
- Load or input the well-plate map. Indicate the threshold (e.g., 50%) in "Neutralization: Infection Deduction (%)."
- Select "Neutralization Process" to calculate the titers. Check the titers and click the "Save & Close" button to save the neutralization results. NOTE: Neutralization is reported as the reciprocal of the highest dilution of the serum with the predefined ICP reduction (such as 50% or 80%) against the VC (the mean of Column 11 in Figure 4). A 50% ICP reduction was used in the Representative Results.
Representative Results
Following the above procedures, we present some results from recent antigenic characterization experiments. The titration setup was designed to examine eight input viruses, with double duplicates for each dilution. The viral dilutions were tested between 1.0 x 10-1 and 1.0 x 1.0-6 to cover the variations between the viruses. The test viruses were from the H3N2 subtype, including A/Cameroon/15V-3538/2015, A/Vladivostok/36/2015, A/Moscow/103/2015, A/Tomsk/5/2015/, A/Moscow/101/2015, A/Moscow/100/2015, A/Moscow/133/2015, and A/Bratislava/437/2015. The titration results are illustrated in Figure 5. Figure 5d demonstrates the decrease of ICPs with the increase of virus dilution. The curves were normalized against the ICPs of the same viruses that yielded infections in all cells within a well12 (defined as ICP saturation). If the ICP did not reach saturation with the highest virus concentration, the ICP average from corresponding duplicates was used instead (A/Moscow/103/2015 in Figure 5d). The virus dilutions that produced 30% of ICP saturation were chosen as the input virus dilution for neutralization (Table 3).
Neutralization aimed to have one input virus against five antisera, with double duplicates on each plate. The reference virus shown is H3N2 A/Stockholm/63/2015. The five antisera are A/HK 4801/14 Egg F12/15, A/HK 7295/14 MDCK F02/15, A/South Africa R2665/15 SIAT F50/15, A/Swiss 9715923/13 SIAT NIBF F18/15, and A/Dutch 525/14 SIAT f23/15. The neutralization results are illustrated in Figure 6. Figure 6d shows the infection progress with the increase of serum dilution. The normalized positive population on the vertical axis represents the ratio of ICPs from the corresponding antisera response against the average ICP of the reference virus12. The background ICP from uninfected cell controls was subtracted during the normalization. The neutralization titers were determined as the reciprocals of the antiserum dilutions corresponding to 50% ICP reduction (Table 4). Linear interpolation was used to estimate titers falling between two adjacent serum dilutions.
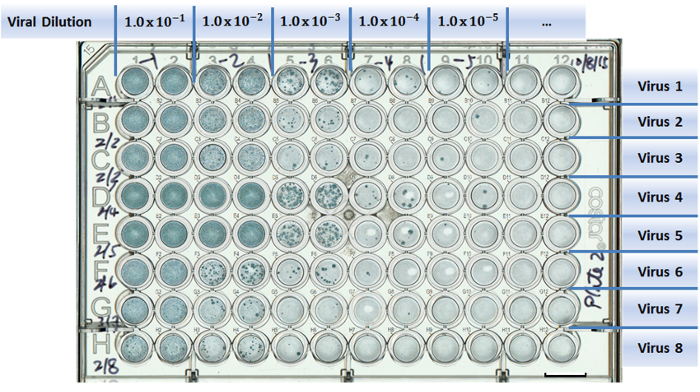 Figure 1:Example of a well-plate setup in a virus titration experiment. Test viruses are assigned to separate rows (A to H). Columns are designed for different viral dilutions (1 to 12). Two columns were used as duplicates for each viral dilution. Scale bar = 10 mm. Please click here to view a larger version of this figure.
Figure 1:Example of a well-plate setup in a virus titration experiment. Test viruses are assigned to separate rows (A to H). Columns are designed for different viral dilutions (1 to 12). Two columns were used as duplicates for each viral dilution. Scale bar = 10 mm. Please click here to view a larger version of this figure.
 Figure 2:Schematics of a sample scanning system. (a) A 96-well plate on the imaging position of a flatbed scanner (republished fromreference11 with permission from Elsevier B.V.), and (b) dimensions of the L-shape position limit shown in (a). Please click here to view a larger version of this figure.
Figure 2:Schematics of a sample scanning system. (a) A 96-well plate on the imaging position of a flatbed scanner (republished fromreference11 with permission from Elsevier B.V.), and (b) dimensions of the L-shape position limit shown in (a). Please click here to view a larger version of this figure.
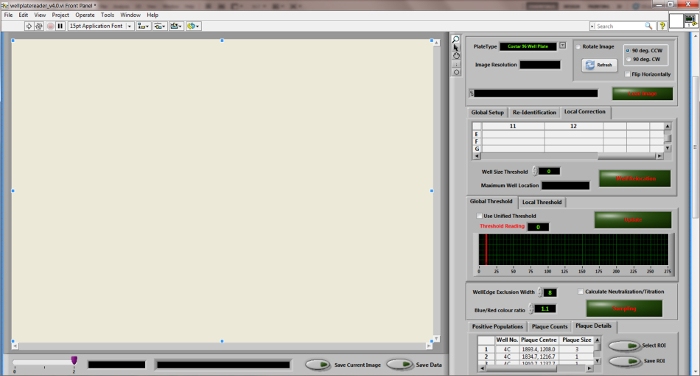 Figure 3:
Desktop diagram of the "Wellplate Reader" quantitation software.
Please click here to view a larger version of this figure.
Figure 3:
Desktop diagram of the "Wellplate Reader" quantitation software.
Please click here to view a larger version of this figure.
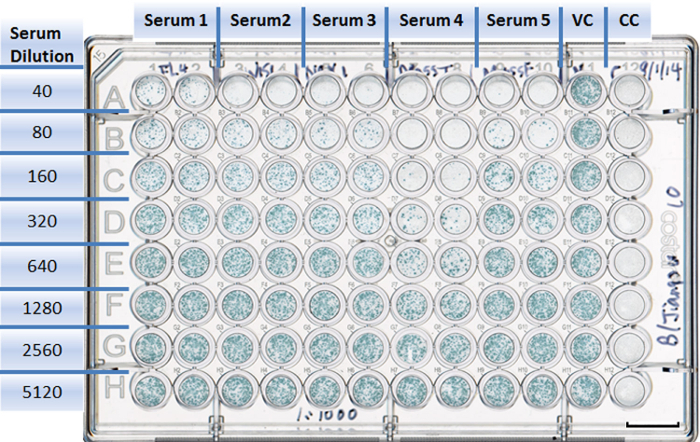 Figure 4:Example of a well-plate setup in a neutralization experiment. Each antiserum is assigned to two columns with duplicates (1 to 10). Rows are designed for different serum dilutions (A to H). The virus control takes Column 11, with eight duplicates (A11 to H11). The cell control is in Column 12, with eight duplicates (A12 to H12). Scale bar = 10 mm. Please click here to view a larger version of this figure.
Figure 4:Example of a well-plate setup in a neutralization experiment. Each antiserum is assigned to two columns with duplicates (1 to 10). Rows are designed for different serum dilutions (A to H). The virus control takes Column 11, with eight duplicates (A11 to H11). The cell control is in Column 12, with eight duplicates (A12 to H12). Scale bar = 10 mm. Please click here to view a larger version of this figure.
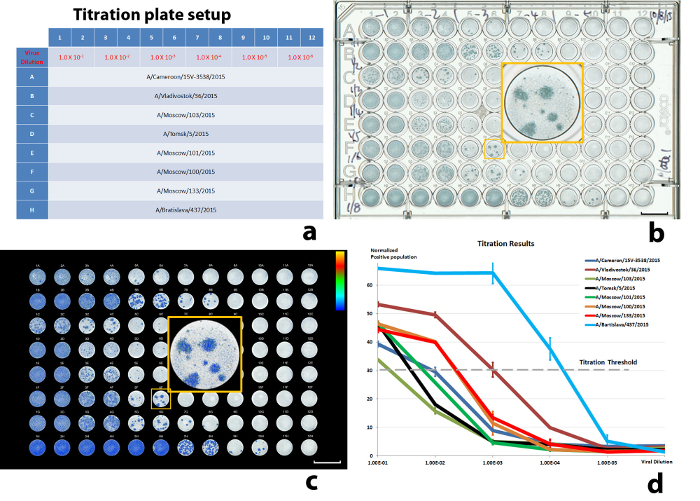 Figure 5: Results of a titration experiment. (a) The well-plate setup, (b) scanned well-plate image, (c) illustration of the color-encoded, quantitated, virus-infected cell population, and (d) normalized virus-infected cell populations against virus dilutions. 30% ICP was used as the threshold. The error bars in (d) are standard deviations (SD) from the sample duplications (± 1 SD). Scale bars in (b) and (c) = 10 mm. Please click here to view a larger version of this figure.
Figure 5: Results of a titration experiment. (a) The well-plate setup, (b) scanned well-plate image, (c) illustration of the color-encoded, quantitated, virus-infected cell population, and (d) normalized virus-infected cell populations against virus dilutions. 30% ICP was used as the threshold. The error bars in (d) are standard deviations (SD) from the sample duplications (± 1 SD). Scale bars in (b) and (c) = 10 mm. Please click here to view a larger version of this figure.
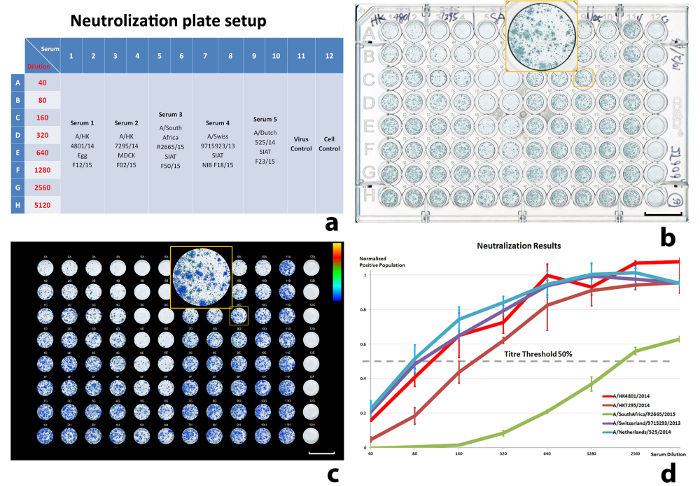 Figure 6:Results of a neutralization experiment. (a) The well-plate setup, (b) scanned well-plate image, (c) illustration of the quantitated, virus-infected cell population, and (d) normalized virus-infected cell population against the serum dilution. The input virus (VC) is A/Stockholm/63/2015. 50% ICP was used to calculate the titers. The error bars in (d) are standard deviations (SD) from the sample duplications (± 1 SD). Scale bars in (b) and (c) = 10 mm. Please click here to view a larger version of this figure.
Figure 6:Results of a neutralization experiment. (a) The well-plate setup, (b) scanned well-plate image, (c) illustration of the quantitated, virus-infected cell population, and (d) normalized virus-infected cell population against the serum dilution. The input virus (VC) is A/Stockholm/63/2015. 50% ICP was used to calculate the titers. The error bars in (d) are standard deviations (SD) from the sample duplications (± 1 SD). Scale bars in (b) and (c) = 10 mm. Please click here to view a larger version of this figure.
Supplementary S1: Please click here to download this file.
| Typical dilution | ||
| MDCK cells | MDCK-SIAT1 cells | |
| 2 days | 1:5 (1 + 4) | 1:10 (1 + 9) |
| 3 days | 1:10 (1 + 9) | 1:20 (1 + 19) |
| Typical number of cells per ml | ||
| MDCK cells | MDCK-SIAT1 cells | |
| 2 days | 2 x 105 | 1 x 105 |
| 3 days | 1 x 105 | 5 x 104 |
Table 1: Typical dilutions on a subculture of confluent flasks of MDCK or MDCK-SIAT1 cells.
| Mode | Professional Mode |
| Document Type | Film (with Film Area Guide) |
| Auto Exposure Type | Photo |
| Image Type | 24-bit Color |
| Resolution | 1,200 dpi |
| Saved image Format | TIFF |
Table 2: Typical setup of a flatbed scanner.
| Row | Virus | Recommended dilution |
| A | A/Cameron/15V-3538/2016 | 1.0 x 10-2 |
| B | A/Vladivostok/36/2015 | 1.0 x 10-3 |
| C | A/Moscow/103/2015 | 1.1 x 10-1 |
| D | A/Tomsk/5/2015 | 5.9 x 10-1 |
| E | A/Moscow/101/2015 | 8.3 x 10-1 |
| F | A/Moscow/100/2015 | 1.4 x 10-2 |
| G | A/Moscow/133/2015 | 1.3 x 10-2 |
| H | A/Bratislava/437/2015 | 1.3 x 10-4 |
Table 3: Viral dilutions calculated from a population of 30% ICP.
| Column | Antisera | Recommended titer |
| 1 - 2 | A/HK4801/2014 | 110 |
| 3 - 4 | A/HK7295/2014 | 213.3 |
| 5 - 6 | A/South Africa/R2665/2015 | 2155.8 |
| 7 - 8 | A/Switzerland/9715293/2013 | 89.4 |
| 9 - 10 | A/Netherlands/525/2014 | 78.6 |
Table 4: Titers at 50% ICP of A/Stockholm/63/2015 against antisera.
Discussion
In this study, we described an imaging-based MN assay to quantify the true antigenic relationships between viruses. Compared with other plaque-reduction assays, the method emphasizes measuring the ICP of an entire well, ensuring the complete coverage of the infective population. Its independence of plaque formation also widens the application of the assay to viruses that can form mainly invisible small plaques or that can only manage single-cell infection. Therefore, the assay is capable of examining more viruses and the effects of a wider range of antibodies than HI, and it helps to more comprehensively reflect the antigenic similarities or differences between viruses12. For example, when oseltamivir carboxylate is included, the MN assay is less affected by NA-dependent binding, reflecting the antigenic differences more accurately. During the development of the assay, great efforts were made to enhance the experiment consistency, speed, and detection sensitivity, including by controlling input viruses through quantitated titration, imaging in high throughput with a flatbed scanner, and implementing a user-friendly data processing platform. Results have generally been consistent with and confirmed those of HI. Notably, the results also revealed antigenic differences between antigenic drift variants of recent type A and B vaccine viruses, as well as antigenic changes caused by culture-selected or sporadic changes, such as the G155E substitution in HA1 of certain A(H1N1)pdm09 viruses12.
The protocol matched the virus type or subtype with the selection of cell lines that gave the strongest infection, which greatly enhanced the imaging signal. Experimental variation was smoothed with the design of duplicates that balanced with the number of tested antisera. Uncertainties can also come from the image quality, which is mainly influenced by the imaging device. A decent color flatbed scanner can significantly improve the image contrast and reduce the noise. The amount of input virus in the neutralization must be quantitatively determined in advance from the corresponding titration while the viruses still maintain a similar status. During neutralization, the antiserum response to an infection is normalized against the difference between the reference virus (VC) and the background level (CC). Accurate measurements of VC and CC are essential to a neutralization experiment. More duplicates are recommended (eight duplicates were used in the Representative Results). Finally, understanding the software is vital to the success of an experiment. The software has been tested for routine virus characterization in the WHO Influenza Centre, UK for more than two years. Detailed instructions for dealing with various situations is provided in the Supplementary S1.
This MN assay is suitable for applications where the samples cover an extremely large field of view but only require moderate imaging resolution. The low cost and simple setup make the system readily available to most virology laboratories. The variation in measurements of the same sample was less than 3%, which roughly defines the uncertainty introduced during the scanning11. Such reproducibility is sufficient in many virological studies and can be further reduced by averaging images from multiple scans.
In conclusion, this study demonstrates a robust MN assay that can be routinely used in antigenic study and to support HI data in detailed analyses of currently-circulating influenza viruses, notably for the biannual selection of viruses for inclusion in human influenza vaccines.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors thank Dr. M. Matrosovich for providing the parent MDCK and MDCK-SIAT1 cell lines and Roche Pharmaceuticals for supplying the oseltamivir carboxylate.We also appreciate Dr. Anne Weston for valuable suggestions on the manuscript. This study was dependent upon the valued collaboration of the WHO National Influenza Centres and the WHO CCs within the WHO GISRS, who provided the influenza viruses used. This work was funded by the Medical Research Council through Programme U117512723.
References
- Walker DL, Horsfall FL. Lack of identity in neutralizing and hemagglutination-inhibiting antibodies against influenza viruses. J Exp Med. 1950;91:65–86. doi: 10.1084/jem.91.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros R, Escriou N, Naffakh N, Manuguerra JC, van der Werf S. Hemagglutinin residues of recent human A(H3N2) influenza viruses that contribute to the inability to agglutinate chicken erythrocytes. Virology. 2001;289:74–85. doi: 10.1006/viro.2001.1121. [DOI] [PubMed] [Google Scholar]
- Lin YP, et al. Neuraminidase receptor binding variants of human influenza A(H3N2) viruses resulting from substitution of aspartic acid 151 in the catalytic site: a role in virus attachment. J Virol. 2010;84:6769–6781. doi: 10.1128/JVI.00458-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmon MW, Rota PA, Walls HH, Kendal AP. Antibody response in humans to influenza virus type B host-cell-derived variants after vaccination with standard (egg-derived) vaccine or natural infection. J Clin Microbiol. 1988;26:333–337. doi: 10.1128/jcm.26.2.333-337.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe T, Abernathy RA, Hu-Primmer J, Thompson WW, Lu X, Lim W, et al. Detection of antibody to avian influenza A (H5N1) virus in human serum by using a combination of serologic assays. J Clin Microbiol.37. 1999;37:937–943. doi: 10.1128/jcm.37.4.937-943.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno Y, Tanaka K, Baba K, Maeda A, Kunita N, Ueda S. Rapid focus reduction neutralization test of influenza A and B viruses in microtiter system. J Clin Microbiol. 1990;28:1308–1313. doi: 10.1128/jcm.28.6.1308-1313.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann MF, Ecabert B, Kopf M. Influenza virus: a novel method to assess viral and neutralizing antibody titers in vitro. J Immunol Methods. 1999;225:105–111. doi: 10.1016/s0022-1759(99)00034-4. [DOI] [PubMed] [Google Scholar]
- Matrosovich M, Matrosovich T, Garten W, Klenk HD. New low-viscosity overlay medium for viral plaque assays. Virol J. 2006;3:63–70. doi: 10.1186/1743-422X-3-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comley J. High content screening - Emerging importance of novel reagents/probes and pathway analysis. Drug Discovery World Summer 2005. 2005. pp. 31–53.
- Matrosovich M, Matrosovich T, Carr J, Roberts NA, Klenk HD. Overexpression of the alpha-2, 6-sialyltransferase in MDCK cells increases influenza virus sensitivity to neuraminidase inhibitors. J. Virol. 2003;77:8418–8425. doi: 10.1128/JVI.77.15.8418-8425.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan K, et al. High throughput virus plaque quantitation using a flatbed scanner. J Virol Methods. 2012;179:81–89. doi: 10.1016/j.jviromet.2011.10.003. [DOI] [PubMed] [Google Scholar]
- Lin YP, et al. Optimization of a micro-neutralization assay and its application in antigenic characterization of influenza viruses. Influenza Other Respir Viruses. 2015;9:331–340. doi: 10.1111/irv.12333. [DOI] [PMC free article] [PubMed] [Google Scholar]