

The function of the dopamine D4 receptor and the product of a clinically significant gene polymorphic variant is revealed.
Keywords: Dopamine D4 receptor, polymorphic variant, glutamate, dopamine, basal ganglia, methamphetamine, attention deficit hyperactivity disorder, substance use disorders
Abstract
Polymorphic variants of the dopamine D4 receptor gene (DRD4) have been repeatedly associated with numerous neuropsychiatric disorders. Yet, the functional role of the D4 receptor and the functional differences of the products of DRD4 polymorphic variants remained enigmatic. Immunohistochemical and optogenetic-microdialysis experiments were performed in knock-in mice expressing a D4 receptor with the long intracellular domain of a human DRD4 polymorphic variant associated with attention deficit hyperactivity disorder (ADHD). When compared with the wild-type mouse D4 receptor, the expanded intracellular domain of the humanized D4 receptor conferred a gain of function, blunting methamphetamine-induced cortical activation and optogenetic and methamphetamine-induced corticostriatal glutamate release. The results demonstrate a key role of the D4 receptor in the modulation of corticostriatal glutamatergic neurotransmission. Furthermore, these data imply that enhanced D4 receptor–mediated dopaminergic control of corticostriatal transmission constitutes a vulnerability factor of ADHD and other neuropsychiatric disorders.
INTRODUCTION
The human D4 receptor gene (DRD4) contains a large number of polymorphisms in its coding sequence (1). The most extensive polymorphism is found in exon 3, a region that codes for the third intracellular loop (3IL) of the receptor. This polymorphism consists of a variable number of tandem repeats (VNTR) in which a 48–base pair sequence exists as 2- to 11-fold repeats (2). The three most common variants contain two, four, and seven TRs and encode for a D4 receptor with two, four, and seven repeats of a proline-rich sequence of 16 amino acids (D4.2, D4.4, and D4.7 receptor). The DRD4 gene with four TRs constitutes the most frequent variant, with a global allelic frequency of 64%, followed by the variants with seven TRs (21%) and two TRs (8%) (3). DRD4 polymorphic variants have been suggested to be associated with numerous behavioral individual differences and neuropsychiatric disorders. The most reported association is the link between the variant with seven repeats and attention deficit hyperactivity disorder (ADHD) (4–6) and substance use disorders (SUDs) (7, 8). However, very little is known about the role of the D4 receptor in the brain and even less about the functional differences between the products of the different polymorphic variants, which should explain their noticeable influence at the behavioral level. In an effort to understand the functional and pharmacological role of the D4 receptor, we recently generated a knock-in mouse with a humanized mouse DRD4 gene containing seven TRs of the human DRD4 in the homologous region of the mouse gene, which codes for the 3IL (D4.7 mouse) (9). Curiously, the rodent gene does not have polymorphisms in the region coding for the 3IL of the D4 receptor, and wild-type control littermates (WT mouse) express a D4 receptor with a short 3IL comparable to the human D4.2 receptor (9). In a previous study, we reported in vitro evidence for blunted mitogen-activated protein kinase signaling in striatal slices from the D4.7 mouse as compared to WT littermates upon coexposure to relatively selective D4 and D2 receptor ligands (9). Parallel studies with the receptors expressed in cultured cells also demonstrated a specific decreased ability of the D4.7 receptor to molecularly interact (oligomerize) with the D2 receptor (9). Therefore, it was suggested, but not proven, that significant differences in the pharmacological and functional properties of human D4DR polymorphic variants should be evident in those brain areas and neuronal elements where both receptors are coexpressed. D4 receptors are predominantly localized in the prefrontal cortex, in GABAergic interneurons and in glutamatergic pyramidal neurons, including their striatal projections (10–12). It is in the pyramidal glutamatergic neurons of the prefrontal cortex and in their striatal terminals where D2 and D4 receptors seem to be particularly colocalized (9–13). The present study was aimed first at finding distinctive differences between D4.7 and WT mice on ex vivo and in vivo pharmacological effects of dopaminergic compounds, which could provide additional clues for the functional role of the D4 receptor in the brain. The results led to the demonstration of a significant role of the D4 receptor in the modulation of corticostriatal glutamatergic neurotransmission.
RESULTS
Blunted response to methamphetamine in D4.7 mice
We first addressed possible differences in the effects of psychostimulants in D4.7 mice as compared with WT littermates and found an unexpected and considerable difference between the dopamine (DA)–releasing and psychomotor-activating effects of cocaine and methamphetamine (METH). Extracellular DA levels were measured with in vivo microdialysis in the nucleus accumbens (NAc) shell of WT and D4.7 mice before and after acute administration of equipotent doses of METH (1 and 3 mg/kg) and cocaine (10 and 30 mg/kg). The basal DA concentration in the dialyzate was not different between genotypes: in means ± SEM, 2.0 ± 1.2 nM (n = 37) and 2.3 ± 1.4 nM (n = 32) for WT and D4.7 mice, respectively (unpaired t test, P = 0.3009). At the dose of 1 mg/kg, METH caused a significantly lower increase of DA in D4.7 mice as compared to WT: in means ± SEM, 323 ± 30% (n = 6) and 452 ± 46% (n = 8), respectively [two-way analysis of variance (ANOVA), genotype factor: F1,152 = 51.40, P < 0.0001]. This difference was more pronounced at the METH dose of 3 mg/kg, which produced a DA increase of 482 ± 136% (n = 5) and 895 ± 165% (n = 4) for D4.7 and WT mice, respectively (two-way ANOVA, genotype factor: F1,84 = 9.99, P = 0.0022) (Fig. 1, A and B). Notably, these differences were specific to METH and were not observed with equipotent doses of cocaine. At the dose of 10 mg/kg, cocaine produced a similar increase of DA in WT mice and D4.7 mice: in means ± SEM, 355 ± 54% (n = 7) and 304 ± 45% (n = 5), respectively (two-way ANOVA, genotype factor: F1,128 = 0.02, P = 0.9018). Similarly, no differences between genotypes were observed with the higher dose of cocaine (30 mg/kg): in means ± SEM, 993 ± 172% (n = 6) and 861 ± 165% (n = 5), respectively (two-way ANOVA, genotype factor: F1,116 = 1.00, P = 0.3192) (Fig. 1, C and D). Correlative results were observed with the analysis of psychomotor activation. D4.7 mice showed significantly lower locomotor activity than WT when treated with METH (1 mg/kg) but not with the equipotent dose of cocaine (10 mg/kg) (unpaired t tests, P = 0.0282 for METH and P = 0.7481 for cocaine) (Fig. 1F). Animals treated with vehicle (saline) did not show differences in locomotor activity (unpaired t test, P = 0.1543) (Fig. 1F). In summary, D4.7 mice showed a specific reduced response to the DA-releasing and the psychomotor-activating effects of METH.
Fig. 1. Blunted response to METH in D4.7 mice.
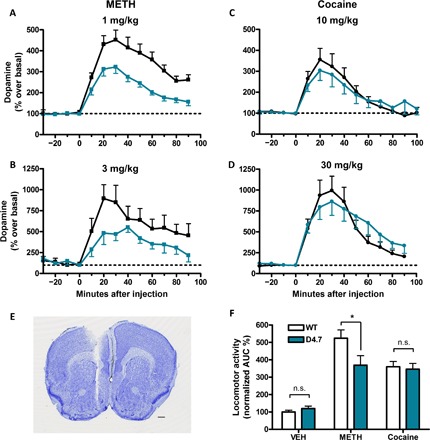
(A to D) Extracellular concentrations of DA in the NAc were measured at 10-min intervals using in vivo microdialysis before and after intraperitoneal injections of METH [1 and 3 mg/kg, (A) and (B), respectively] or cocaine [10 and 30 mg/kg, (C) and (D), respectively] in WT (black symbols/lines) and D4.7 mice (blue symbols/lines). Values (in % over basal) are means ± SEM of four to seven different animals per condition normalized to the mean of the concentration of DA present in the three samples preceding drug administration. (E) Coronal section from a representative experiment showing the microdialysis probe track ending within the medial NAc shell. Scale bar, 5 mm. (F) Locomotor activity quantification from WT (white bars) or D4.7 (blue bars) mice injected with vehicle (VEH), METH (1 mg/kg), or cocaine (10 mg/kg). Values are means ± SEM of 6 to 12 animals and represent the area under the distance × time curve normalized to the average values from WT animals that received the vehicle injection. Statistical significance was calculated by an unpaired t test comparing WT with D4.7 in each condition [*P < 0.05; not significant (n.s.), P > 0.05]. AUC, area under the curve.
Decreased corticostriatal neurotransmission in D4.7 mice
Apart from DA release by direct mechanisms involving the DA transporter, METH also induces striatal glutamate (GLU) release by an indirect mechanism that depends on somatodendritic DA release and activation of DA D1 receptors localized in the terminals of striatonigral GABAergic neurons (14). This leads to disinhibition of thalamic input to the cortex and increased corticostriatal glutamatergic neurotransmission (14, 15). Predominant activation of the nigrothalamocortical loop by METH (3 mg/kg) as compared to an equipotent psychostimulant dose of cocaine (30 mg/kg) was confirmed by analyzing cortical extracellular signal–regulated kinase 1/2 (ERK1/2) phosphorylation by immunohistochemistry in WT and D4.7 mice (Fig. 2). In WT mice, 30 min after an acute injection of METH (3 mg/kg), there was a significantly higher density of cortical cells [medial prefrontal cortex (mPFC)] showing ERK1/2 phosphorylation as compared to that of an equipotent dose of cocaine (30 mg/kg): in means ± SEM, 26 ± 2 cells per field (n = 34) and 11 ± 1 cells per field (n = 57), respectively (one-way ANOVA followed by Bonferroni’s multiple comparison test, P < 0.001). On the other hand, METH produced the same effect as cocaine in D4.7 mice: in means ± SEM, 14 ± 1 cells per field (n = 53) and 12 ± 1 cells per field (n = 32), respectively (one-way ANOVA followed by Bonferroni’s multiple comparison test, P > 0.05) (Fig. 2I). Then, we compared the acute effects of the systemic administrations of METH (3 mg/kg) on the extracellular concentration of GLU in the NAc shell of WT and D4.7 with in vivo microdialysis. No significant differences were observed in the basal GLU levels between WT and D4.7 mice: in means ± SEM, 1.8 ± 0.2 μM (n = 12) and 2.0 ± 0.3 μM (n = 12), respectively (unpaired t test, P = 0.6077). METH (3 mg/kg) caused a significantly lower GLU increase in D4.7 as compared to WT mice: in means ± SEM, 224 ± 65% (n = 6) and 123 ± 13% (n = 8), respectively (two-way ANOVA, genotype factor: F1,122 = 3.99, P = 0.0481) (Fig. 3A). Nonetheless, GLU extracellular levels were only slightly increased after an equipotent dose of cocaine (30 mg/kg), and no differences were observed between WT and D4.7 mice: in means ± SEM, 110 ± 14% (n = 7) and 109 ± 11% (n = 7), respectively (two-way ANOVA, genotype factor: F1,117 = 0.32, P = 0.5745) (Fig. 3B).
Fig. 2. Preferential ERK phosphorylation in the mPFC after systemic administration of METH versus cocaine.
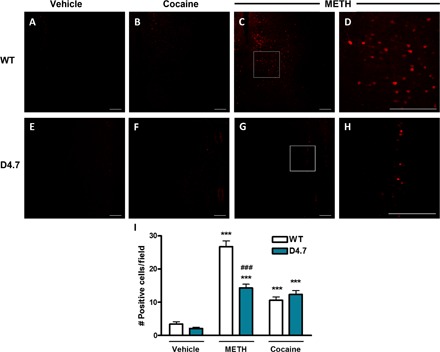
(A) to (C) and (E) to (G) Immunohistochemical analysis of phosphorylated ERK1/2 in the mPFC of WT (upper row) or D4.7 (lower row) mice treated with vehicle (A and E), cocaine (30 mg/kg; B and F), or METH (3 mg/kg; C and G). (D and H) Magnification of the framed area in (C) and (G), respectively. Scale bars, 200 μm. (I) Quantification of positive cells from 400-μm × 400-μm fields in the mPFC. Values are means ± SEM of 32 to 57 different fields obtained from three different animals per condition. Statistical significance was calculated by one-way ANOVA followed by Bonferroni’s post hoc tests (***P < 0.001, compared with the respective vehicle, and ###P < 0.001 compared with WT).
Fig. 3. Decreased corticostriatal GLU release in D4.7 mice.
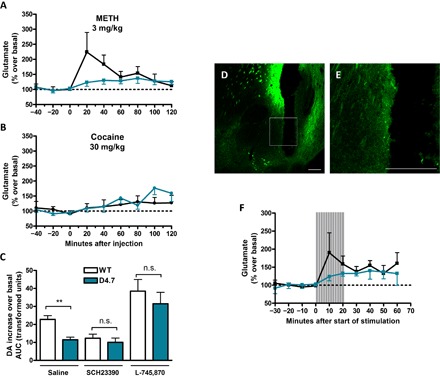
(A and B) Extracellular concentrations of GLU in the NAc were measured at 20-min intervals using in vivo microdialysis before and after an intraperitoneal injection of METH (3 mg/kg) or cocaine (30 mg/kg) in WT (black symbols/lines) or D4.7 mice (blue symbols/lines). Values (in % over basal) are means ± SEM of seven to eight different animals per condition normalized to the mean of the concentration of GLU present in the three samples preceding drug injection. (C) Area under the increase × time curve of extracellular DA measured at 10-min intervals after intraperitoneal injections of METH (1 mg/kg) in WT (white) or D4.7 (blue) mice pretreated (10 min before METH injection) or not with the D1R antagonist SCH23390 (1 mg/kg) or the D4 receptor antagonist L-745,870 (1 mg/kg). Values are means ± SEM of six to eight different animals per condition. Statistical significance was calculated by an unpaired t test comparing WT with D4.7 in each condition (**P < 0.01; n.s., P > 0.05). (D) Representative image of the light-sensitive corticostriatal terminals (monitored as EYFP expression) and optogenetic-microdialysis probe track within the medial NAc shell. (E) Magnification of the framed area in (C). Scale bars, 200 μm. (F) Extracellular GLU concentration in the NAc shell as measured by in vivo microdialysis before, during (shaded area), and after 20 min of photostimulation (16 2-ms pulses at 100 Hz at one burst per second). Values (in % over basal) are means ± SEM of seven to eight different animals per condition normalized to the mean of the concentration of GLU present in the three samples preceding stimulation.
Because striatal GLU release is known to facilitate DA release by acting on N-methyl-D-aspartate (NMDA) receptors localized in the dopaminergic terminals (16–18), the differential effect of METH on striatal GLU release between WT and D4.7 mice could explain their differences in DA release and psychomotor activation. D4.7 receptors localized in the dendritic region of mPFC pyramidal neurons and in their striatal terminals could then promote a stronger inhibitory control of corticostriatal glutamatergic transmission by DA, thereby reducing the GLU-mediated component of METH-induced DA release. In complete agreement, systemic administration of the D4 receptor antagonist L-745,870 eliminated the differences in the DA-releasing effects of METH between both genotypes. Both genotypes responded equally: in means ± SEM, 802 ± 156% (n = 6) for WT, and 661 ± 137% (n = 7) for D4.7 (two-way ANOVA, genotype factor: F1,129 = 3.32, P = 0.0709) and significantly higher than the WT mice in the absence of the D4 receptor antagonist (Fig. 3C). Therefore, these results reveal the relevance of the GLU-mediated component of METH-induced DA release (Fig. 3C). Likewise, in agreement with the involvement of somatodendritic DA release and activation of mesencephalic D1 receptors in METH-induced striatal GLU release, systemic administration of the D1 receptor antagonist SCH23390 also eliminated the differences in the DA-releasing effects of METH between both genotypes: in means ± SEM, 336 ± 62% (n = 7) for WT, and 306 ± 49% (n = 7) for D4.7 (two-way ANOVA, genotype factor: F1,162 = 0.70, P = 0.4056) (Fig. 3).
A previous study of the effect of relatively selective D4 receptor ligands on rat striatal slice preparations suggested that activation of D4 receptors localized in striatal glutamatergic terminals specifically inhibits depolarization-induced GLU release, without a direct influence on DA release (9). Using our recently introduced optogenetic-microdialysis probe (19), we evaluated the existence of differences between the two D4DR genotypes on the local influence of basal extracellular DA levels on optogenetically induced striatal GLU release from corticostriatal terminals. The design of this probe allows laser illumination and consequent neuronal depolarization of channelrhodopsin 2 (ChR2)–containing terminals around the dialysis membrane, within the same area sampled for extracellular GLU (see Methods). Four weeks after microinjection in the mPFC of an adeno-associated virus (AAV) expressing the light-activated cation channel ChR2 fused to the enhanced yellow fluorescent protein (EYFP), its expression (fluorescence) could be detected at the site of injection, in the mPFC, and in the medial NAc shell (Fig. 3, D and E). EYFP was also detected in other projecting areas, such as septal, amygdalar, thalamic, and hypothalamic areas, which were not the object of this study. The optogenetic-microdialysis probe was implanted in the medial NAc shell. Optogenetic stimulation induced a significantly higher increase in extracellular GLU levels in WT mice as compared with their D4.7 littermates: in means ± SEM, 176 ± 49% (n = 8) and 124 ± 12% (n = 7), respectively (two-way ANOVA, genotype factor: F1,55 = 6.47, P = 0.0138) (Fig. 3F). Therefore, these results demonstrate that striatal D4 receptors play a key modulatory role in the local striatal dopaminergic control of corticostriatal GLU release and that the degree of this modulation is significantly increased with a humanized D4.7 receptor–like variant.
Quantitative polymerase chain reaction (PCR) and radioligand binding measurements were performed to discard a possible influence of differences in the expression or density of D4 receptors on the gain of function observed in D4.7 animals as compared to WT. There were no substantial differences in D4DR transcript mRNA copies in the mPFC. D4DR/GADPH mRNA ratios for WT and D4.7 were, in means ± SEM, 3.6 × 10−5 ± 0.2 × 10−5 (n = 6) and 6.2 × 10−5 ± 0.6 (n = 6), respectively. There were no significant differences in the estimated amount of D4 receptor binding sites in membrane preparations from the mPFC of WT and D4.7 mice: in means ± SEM, 78 ± 7 fmol/mg of protein (n = 6) and 80 ± 20 fmol/mg of protein (n = 6), respectively (unpaired t test, P = 0.8585).
DISCUSSION
The specific and significant effects obtained with the D4.7 mouse, with an expanded long intracellular domain corresponding to the human D4DR gene polymorphism, allowed us to establish a main role of the D4 receptor in the brain: the dopaminergic control of corticostriatal neurotransmission, both under basal conditions and upon administration of METH. This can explain the previously documented increases of striatal extracellular GLU levels in DRD4 knockout mice (20). In addition, the present results demonstrate the significant contribution of corticostriatal glutamatergic neurotransmission in the ability of METH to increase the striatal extracellular levels of DA. Two main mechanisms that control striatal DA release have been described: a firing-dependent vesicular release and a firing-independent, NMDA receptor–modulated mechanism (18). It is through this lesser known and characterized mechanism that GLU controls DA release, by acting on NMDA receptors localized in dopaminergic terminals adjacent to glutamatergic terminals, in a microcircuit or “local module” centered in the dendritic spine of the striatal GABAergic efferent neuron (17, 21).
The analysis of cortical ERK1/2 phosphorylation and the measurement of striatal GLU with the optogenetic-microdialysis technique allowed us to demonstrate that D4 receptors mediate a significant inhibitory role of DA in corticostriatal neuronal function, both at the dendritic level (mPFC) and at the terminal level (NAc shell). The results from our ex vivo study in the mPFC agree with a recent in vitro electrophysiological study in cortical slices from DRD4 knockout mice, which reported an increased ability of a putative selective D4 receptor agonist to suppress glutamatergic excitatory network bursts upon viral infection with human D4.7 as compared with D4.4 receptor cDNA or as compared with the suppression observed in slices from WT mice (22). We have recently shown that the optogenetic-microdialysis method allows the study of the functional role of a G protein–coupled receptor (GPCR) localized in striatal glutamatergic terminals in the modulation of GLU release and the consequent local GLU-dependent modulation of striatal extracellular levels of DA (19). It could be demonstrated that adenosine modulates striatal DA release by its ability to modulate glutamatergic neurotransmission acting on adenosine receptors localized in the corticostriatal glutamatergic terminals (19). The existence of D2-like receptors (classified into the subtypes D2, with isoforms D2S and D2L, D3, and D4) in striatal glutamatergic terminals that mediate a dopaminergic inhibitory control of glutamatergic transmission had been previously reported by Maura et al. (23). Our results imply that the D4 receptor is a major D2-like receptor subtype involved in glutamatergic transmission and that the addition in its 3IL of seven repeats of 16 amino acids, corresponding to the product of the clinically significant DRD4 polymorphic variant with seven TRs, leads to a profound gain of function. Together with the recent results from in vitro electrophysiological studies in cortical slices from DRD4 knockout mice, our results imply that the previous view on the relative functional differences among the products of DRD4 polymorphic variants should be revisited. Thus, initial and still canonically cited studies seemed to indicate that the D4.7 receptor signals with less efficiency than D4.2 or D4.4 receptors (24). Nevertheless, a recent study found no significant differences in the ability of D4.2, D4.4, and D4.7 receptor variants to promote DA-induced activation with any of the five different Gi/o protein subtypes (25). This would support the premise that functional differences between the products of DRD4 polymorphic variants depend on their molecular interactions with other GPCRs, such as the D2 receptor (9, 26). The D4.7 receptor forms less heteromers with the D2 receptor than the more common D4.4 receptor, and it was previously suggested that this heteromerization provides a gain of function of the D2 receptor (9, 26). This would apparently predict opposite results from those obtained in the present study and in the recently reported electrophysiological study in cortical slices from DRD4 knockout mice (22). However, those experiments were performed with synthetic DA receptor agonists. We recently reported that DA is significantly more potent at activating any D4 receptor (D4.2, D4.4, and D4.7) than D2 receptors (D2S or D2L) (25), and we are obtaining results that indicate that DA is also more potent at activating D4 receptors (including D4.7) than D2-D4 receptor heteromers (in preparation). Therefore, the inability of D4.7 receptors to heteromerize should lead to a gain of function.
To our knowledge, these are the first clear ex vivo and in vivo experimental data showing a significant specific neuronal function that depends on the DRD4 VNTR: corticostriatal glutamatergic neurotransmission. Our study provides a neuronal correlate of recent results from imaging studies showing that subjects carrying the DRD4 polymorphism with seven TRs show decreased function and connectivity of brain regions involved in inhibitory control during the execution of impulse control tasks, particularly the right inferior frontal gyrus cortex (27, 28). A blunted corticostriatal transmission should affect the activity of both the “Go” and “NoGo” GABAergic striatal efferent pathways, decreasing their respective ability to increase the reactivity to reward-related stimuli and to suppress the reactivity to nonrewarded- or aversive-related stimuli (29). The outcome should be an increased “interest” for irrelevant stimuli and a reduced inhibition of irrelevant responses, which could be important in explaining the attentional deficit and impulsivity of ADHD. Thus, DRD4 polymorphism with seven TRs in the third exon sequence that codes for the 3IL has been repeatedly associated with ADHD (4–6) and, more specifically, with the two major dimensions of impulsivity, action and choice impulsivity (30, 31), traits that constitute endophenotypes of ADHD and SUD (8, 32). Therefore, the present results provide a specific functional correlate of a vulnerability factor for these neuropsychiatric disorders featuring an enhanced D4 receptor–mediated dopaminergic control of corticostriatal glutamatergic neurotransmission.
MATERIALS AND METHODS
Animals
Transgenic D4.7 mice, with a humanized mouse DRD4 gene containing seven TRs of the human DRD4 in the homologous region of the mouse gene, which codes for the 3IL (9), were used. Homozygous (D4.7) and WT littermates were obtained from a breeding colony of D4.7 heterozygous mice (in a C57Bl/6J background) kept in the National Institute on Drug Abuse Intramural Research Program (NIDA IRP) breeding facility. Male mice, weighting 30 to 40 g, were used in all microdialysis, optogenetic-microdialysis, and locomotor activity experiments. Male mice weighting 25 to 30 g were used for virus injections. Animals were housed (four per cage) and kept on a 12-hour light/12-hour dark cycle with food and water available ad libitum. All animals used in the study were maintained in accordance with the guidelines of the National Institutes of Health (NIH) Animal Care, and the animal research conducted to perform this study was approved by the NIDA IRP Animal Care and Use Committee (protocols #12-BNRB-73, #15-BNRB-73, and #12-MTMD-2).
Surgical procedures
For all surgical procedures, animals were anesthetized with a mixture of ketamine (60 mg/kg, intraperitoneally) and xylazine (12 mg/kg, intraperitoneally). For optogenetic-microdialysis experiments, animals received a unilateral injection of an AAV encoding ChR2 (ChR2/H134R) fused to EYFP under control of the CaMKIIa neuronal promoter [AAV-CaMKIIa-hChR2(H134R)-EYFP; University of North Carolina core vector facility, Chapel Hill, NC] in the mPFC. Coordinates of injection were 1.9-mm anterior, 0.5-mm lateral, and 2.9-mm ventral from bregma. Viral particles (0.5 μl of purified and concentrated AAV; 1 × 1012 infectious units/ml) were injected using a 105-μm-thick silica tubing injector coupled to a 1-μl syringe driven by an infusion pump. Virus suspension was injected over a 10-min period at a rate of 50 nl/min, and the injector was left in place for an additional 10 min to allow diffusion of the suspension. Four weeks after virus injection, a modified optogenetic-microdialysis probe with an embedded light-guiding optic fiber, described in the study of Quiroz et al. (19), was implanted into the medial NAc shell; coordinates were 1.2-mm anterior, 0.6-mm lateral, and 5.2-mm ventral from bregma. The probe was fixed to the skull with successive layers of glass ionomer cement (CX-Plus, Shofu Dental) and dental acrylic resin (Lang Dental Manufacturing Co. Inc.). For microdialysis experiments, microdialysis probes (see below) were implanted into the medial NAc shell (coordinates were 1.2- to 1.5-mm anterior, 0.6-mm lateral, and 5.2-mm ventral from bregma) following the same procedures described for optogenetic-microdialysis experiments.
Probe designs and preparations
Optogenetic-microdialysis probes were prepared as described in the study of Quiroz et al. (19) for rats, with some modifications to be used in mice. Briefly, a 125–μm–core diameter optic fiber (Thorlabs) was stripped and inserted into an AN-69 fiber (Hospal Industrie) together with two silica capillary tubes to allow liquid perfusion. The tip of the optical fiber was given a conical shape to ensure local light dispersion around the working portion of the dialysing membrane. The dialysing surface of the probes was 1 mm long. Conventional microdialysis probes were prepared in a similar procedure without the insertion of the optic fiber.
In vivo microdialysis experiments
Experiments were performed 36 hours after probe implantation in freely moving mice. Ringer’s solution (147 mM NaCl, 4 mM KCl, and 2.2 mM CaCl2) was pumped through the probes at a constant rate of 1 μl/min. After 30 min of equilibration, samples were collected every 10 min (DA analysis) or 20 min (GLU analysis) for 80 min. The indicated doses of (+)-METH-HCl (NIDA Pharmacy) or (−)-cocaine-HCl (NIDA Pharmacy) were administered intraperitoneally using saline (10 ml/kg) (0.9% NaCl) as a vehicle. After the systemic administration, samples were collected every 10 min (DA analysis) or 20 min (GLU analysis) for 90 or 120 min, respectively. Where indicated, vehicle (saline), the D1 receptor antagonist SCH23390 (1 mg/kg; Tocris), or the D4 receptor antagonist L-745,870 (1 mg/kg; Tocris) was systemically administered (10 ml/kg, intraperitoneally) 10 min before METH or cocaine treatment. DA content in the dialyzate fractions was measured by high-performance liquid chromatography (HPLC) coupled to a coulometric detector (5200a Coulochem III, ESA). GLU was measured by HPLC coupled to a GLU oxidase enzyme reactor and electrochemical detector (Eicom Corporation).
In vivo optogenetic-microdialysis experiments
Experiments were performed 36 hours after probe implant on freely moving mice. Ringer’s solution (147 mM NaCl, 4 mM KCl, and 2.2 mM CaCl2) was pumped through the probes at a constant rate of 1.25 μl/min. After 30 min of equilibration, samples were collected every 10 min for 40 to 60 min before optogenetic stimulation. Optogenetic stimulation was applied for 20 min, and 10-min samples were taken for 60 additional minutes after the beginning of the stimulation. Optical stimulation was delivered by coupling the light-guiding port of the probe to a 473-nm solid-state laser module driven by an electrical stimulator (model S88, Grass Technologies). Light was applied over a 20-min period in 160-ms trains of 2-ms pulses at 100 Hz. Light intensity was adjusted at 5 to 8 mW at the probe tip, calibrated before the implant using an integrating sphere silicon photodiode power sensor designed for optical power measurements independent of beam shape and divergence (model S144C, Thorlabs). GLU was measured as described above.
Histology
At the end of the optogenetic-microdialysis or microdialysis experiments, animals were anesthetized with equithesin (3 ml/kg) (4.44 g of chloral hydrate, 0.972 g of sodium pentobarbital, 2.124 g of MgSO4, 44.4 ml of propylene glycol, 12 ml of ethanol, and distilled water up to 100 ml of the final solution; NIDA Pharmacy) and perfused transcardially with 0.1 M phosphate-buffered saline (PBS) followed by ice-cold 4% paraformaldehyde in PBS. Brains were kept in the same fixative for 18 hours and then stored in a 30% sucrose solution for at least 48 hours. Coronal sections (40 μm thick) were cut in a cryostat (model CM3050S, Leica) and collected in PBS. Sections were then evaluated for localization of implanted probes and ChR2-EYFP expression. Confocal fluorescence microscopy images were acquired with a confocal microscope (Examiner Z1, Zeiss) with a laser scanning module (LSM-710, Zeiss). Experiments where the probe was misplaced or where EYFP expression was not detected in the injection and target areas were discarded.
Locomotor activity
Mice (WT and D4.7) received an intraperitoneal injection of METH (1 mg/kg in saline), cocaine (10 mg/kg in saline), or saline and were placed immediately in the motility chambers (50 cm × 50 cm; Coulbourn Instruments), and locomotion was recorded by counting the number of breaks in the infrared beams of the chambers for 2 hours in 10-min bins. The area under the curve (AUC) of the distance versus time curve was calculated and normalized by the average value obtained in WT animals that received a saline injection.
ERK1/2 phosphorylation assay
Drug-naïve animals (WT and D4.7) were brought to the experimental room, weighed, individually housed, and allowed to habituate to their new home cages for 2 hours. Then they were administered (intraperitoneally) METH (3 mg/kg), cocaine (30 mg/kg), or saline and placed back in the home cages. After 30 min, animals were anesthetized with equithesin (see above) and perfused transcardially (see above). After fixation and sectioning (see above), 40-μm-thick coronal slices of the brains were permeabilized with PBS with Triton X-100 (0.1%; washing buffer), blocked with bovine serum albumin 5% in washing buffer [2 hours, room temperature (RT)], incubated overnight at 4°C with a polyclonal anti–phospho-ERK1/2 primary antibody (1:1000, Cell Signaling), washed again, and incubated with Alexa 635 goat anti-rabbit secondary antibody (1:200; 2 hours, RT; Invitrogen) and washed again. Coronal sections were mounted, and confocal images were taken as described above. Images were analyzed using ImageJ 1.48v software (NIH). Briefly, images were thresholded (20 to 255), and particles larger than 50 μm2 were counted in fields of 400 μm × 400 μm from the mPFC.
Quantitative PCR and binding experiments
WT and D4.7 mice were euthanized by overdose of equithesin (10 ml/kg) and decapitated, the brains were quickly removed and placed on ice, and the mPFC was dissected out. For mRNA quantification, the tissue was homogenized by sonication, and total RNA was extracted using RNeasy Plus Mini kit (QIAGEN GmbH). Total RNA was converted to cDNA by RNA to cDNA EcoDry (Clontech) using Oligo-dT primer. Quantitative real-time reverse transcription PCR was performed and analyzed using LightCycler 480 II (Roche). PCR reactions were carried out in a total volume of 20 μl in a PCR mix containing 10 μl of LightCycler probe master, 500 nM reference gene primer (GAPDH), 500 nM D4 receptor sense and antisense primer, 100 nM target gene probe and reference gene probe, and 2 μl of 100-ng cDNA. To normalize the cDNA content of the samples, we used the comparative threshold (DDCt) cycle method, which consists of the normalization of the number of target gene copies versus the endogenous reference gene, GAPDH. For binding experiments, the dissected tissue was homogenized in 50 mM tris-HCl (pH 7.4) containing a protease inhibitor cocktail (1:1000; Sigma-Aldrich Co.). Membranes were obtained by centrifugation at 48,000g (50 min, 4°C) and washed two times in the same conditions. Protein was quantified by the bicinchoninic acid method (Pierce). Pellets were resuspended, and membrane suspensions (50 μg of protein/ml) were incubated in 50 mM tris-HCl (pH 7.4) with 120 mM NaCl, 5 mM KCl, 10 mM MgCl2, and increasing concentrations (0.03 to 1 nM) of the nonselective D2-like antagonist [3H]N-methylspiperone (77.7 Ci/mmol, PerkinElmer) in the absence or the presence of the selective D4 receptor antagonist L-745,870 (1 μM) to prevent the radioligand binding to the D4 receptor. Nonspecific binding was determined in the presence of 10 μM nonlabeled N-methylspiperone. Free and membrane-bound ligands were separated by rapid filtration on 500-μl aliquots in a 96-well plate harvester (Brandel) and washed with 2 ml of ice-cold tris-HCl. MicroScint 20 scintillation liquid (65 μl per well; PerkinElmer) was added to the filter plates, the plates were incubated overnight at RT, and radioactivity counts were determined in a MicroBeta2 plate counter (PerkinElmer) with an efficiency of 41%. Saturation curves were fitted to one-site binding hyperbolic curves using Prism 4 (GraphPad Software), and the D4 receptor binding sites were estimated by subtraction.
Acknowledgments
We thank F. Vautier and his team from the NIDA Breeding Facility for their work in maintaining the D4.7 colony. Funding: This work was supported by the National Agency for Scientific and Technical Research, Argentina University of Buenos Aires (grant no. PICT2012-0893) and intramural funds of the National Institute on Drug Abuse. Author contributions: J.B., G.T., and S.F. designed and supervised the study. J.B. and N.-S.C. performed the experiments and analyzed the data. C.Q. and J.B. designed and adapted the microdialysis-optogenetic technology. M.R. contributed to the generation of the D4.7 mouse line. J.B. and S.F. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data availability: All data needed to evaluate the conclusions in the paper are present in the paper. Additional data related to this paper may be requested from the authors.
REFERENCES AND NOTES
- 1.LaHoste G. J., Swanson J. M., Wigal S. B., Glabe C., Wigal T., King N., Kennedy J. L., Dopamine D4 receptor gene polymorphism is associated with attention deficit hyperactivity disorder. Mol. Psychiatry 1, 121–124 (1996). [PubMed] [Google Scholar]
- 2.Wang E., Ding Y.-C., Flodman P., Kidd J. R., Kidd K. K., Grady D. L., Ryder O. A., Spence M. A., Swanson J. M., Moyzis R. K., The genetic architecture of selection at the human dopamine receptor D4 (DRD4) gene locus. Am. J. Hum. Genet. 74, 931–944 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang F.-M., Kidd J. R., Livak K. J., Pakstis A. J., Kidd K. K., The world-wide distribution of allele frequencies at the human dopamine D4 receptor locus. Hum. Genet. 98, 91–101 (1996). [DOI] [PubMed] [Google Scholar]
- 4.Faraone S. V., Perlis R. H., Doyle A. E., Smoller J. W., Goralnick J. J., Holmgren M. A., Sklar P., Molecular genetics of attention-deficit/hyperactivity disorder. Biol. Psychiatry 57, 1313–1323 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Li D., Sham P. C., Owen M. J., He L., Meta-analysis shows significant association between dopamine system genes and attention deficit hyperactivity disorder (ADHD). Hum. Mol. Genet. 15, 2276–2284 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Gizer I. R., Ficks C., Waldman I. D., Candidate gene studies of ADHD: A meta-analytic review. Hum. Genet. 126, 51–90 (2009). [DOI] [PubMed] [Google Scholar]
- 7.McGeary J., The DRD4 exon 3 VNTR polymorphism and addiction-related phenotypes: A review. Pharmacol. Biochem. Behav. 93, 222–229 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belcher A. M., Volkow N. D., Moeller F. G., Ferré S., Personality traits and vulnerability or resilience to substance use disorders. Trends Cogn. Sci. 18, 211–217 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.González S., Rangel-Barajas C., Peper M., Lorenzo R., Moreno E., Ciruela F., Borycz J., Ortiz J., Lluís C., Franco R., McCormick P. J., Volkow N. D., Rubinstein M., Floran B., Ferré S., Dopamine D4 receptor, but not the ADHD-associated D4.7 variant, forms functional heteromers with the dopamine D2S receptor in the brain. Mol. Psychiatry 17, 650–662 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lauzon N. M., Laviolette S. R., Dopamine D4-receptor modulation of cortical neuronal network activity and emotional processing: Implications for neuropsychiatric disorders. Behav. Brain Res. 208, 12–22 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Tarazi F. I., Campbell A., Yeghiayan S. K., Baldessarini R. J., Localization of dopamine receptor subtypes in corpus striatum and nucleus accumbens septi of rat brain: Comparison of D1-, D2-, and D4-like receptors. Neuroscience 83, 169–176 (1998). [DOI] [PubMed] [Google Scholar]
- 12.Svingos A. L., Periasamy S., Pickel V. M., Presynaptic dopamine D4 receptor localization in the rat nucleus accumbens shell. Synapse 36, 222–232 (2000). [DOI] [PubMed] [Google Scholar]
- 13.Gaspar P., Bloch B., Le Moine C., D1 and D2 receptor gene expression in the rat frontal cortex: Cellular localization in different classes of efferent neurons. Eur. J. Neurosci. 7, 1050–1063 (1995). [DOI] [PubMed] [Google Scholar]
- 14.Mark K. A., Soghomonian J.-J., Yamamoto B. K., High-dose methamphetamine acutely activates the striatonigral pathway to increase striatal glutamate and mediate long-term dopamine toxicity. J. Neurosci. 24, 11449–11456 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gross N. B., Duncker P. C., Marshall J. F., Cortical ionotropic glutamate receptor antagonism protects against methamphetamine-induced striatal neurotoxicity. Neuroscience 199, 272–283 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borland L. M., Michael A. C., Voltammetric study of the control of striatal dopamine release by glutamate. J. Neurochem. 91, 220–229 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Quarta D., Borycz J., Solinas M., Patkar K., Hockemeyer J., Ciruela F., Lluis C., Franco R., Woods A. S., Goldberg S. R., Ferré S., Adenosine receptor-mediated modulation of dopamine release in the nucleus accumbens depends on glutamate neurotransmission and N-methyl-d-aspartate receptor stimulation. J. Neurochem. 91, 873–880 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Grace A. A., Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat. Rev. Neurosci. 17, 524–532 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quiroz C., Orrú M., Rea W., Ciudad-Roberts A., Yepes G., Britt J. P., Ferré S., Local control of extracellular dopamine levels in the medial nucleus accumbens by a glutamatergic projection from the infralimbic cortex. J. Neurosci. 36, 851–859 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thomas T. C., Grandy D. K., Gerhardt G. A., Glaser P. E. A., Decreased dopamine D4 receptor expression increases extracellular glutamate and alters its regulation in mouse striatum. Neuropsychopharmacology 34, 436–445 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferré S., Agnati L. F., Ciruela F., Lluis C., Woods A. S., Fuxe K., Franco R., Neurotransmitter receptor heteromers and their integrative role in `local modules’: The striatal spine module. Brain Res. Rev. 55, 55–67 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhong P., Liu W., Yan Z., Aberrant regulation of synchronous network activity by the attention-deficit/hyperactivity disorder-associated human dopamine D4 receptor variant D4.7 in the prefrontal cortex. J. Physiol. 594, 135–147 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maura G., Giardi A., Raiteri M., Release-regulating D-2 dopamine receptors are located on striatal glutamatergic nerve terminals. J. Pharmacol. Exp. Ther. 247, 680–684 (1988). [PubMed] [Google Scholar]
- 24.Asghari V., Sanyal S., Buchwaldt S., Paterson A., Jovanovic V., Van Tol H. H. M., Modulation of intracellular cyclic AMP levels by different human dopamine D4 receptor variants. J. Neurochem. 65, 1157–1165 (1995). [DOI] [PubMed] [Google Scholar]
- 25.Sánchez-Soto M., Bonifazi A., Cai N. S., Ellenberger M. P., Newman A. H., Ferré S., Yano H., Evidence for noncanonical neurotransmitter activation: Norepinephrine as a dopamine D2-like receptor agonist. Mol. Pharmacol. 89, 457–466 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Borroto-Escuela D. O., Van Craenenbroeck K., Romero-Fernandez W., Guidolin D., Woods A. S., Rivera A., Haegeman G., Agnati L. F., Tarakanov A. O., Fuxe K., Dopamine D2 and D4 receptor heteromerization and its allosteric receptor–receptor interactions. Biochem. Biophys. Res. Commun. 404, 928–934 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gilsbach S., Neufang S., Scherag S., Vloet T. D., Fink G. R., Herpertz-Dahlmann B., Konrad K., Effects of the DRD4 genotype on neural networks associated with executive functions in children and adolescents. Dev. Cogn. Neurosci. 2, 417–427 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mulligan R. C., Kristjansson S. D., Reiersen A. M., Parra A. S., Anokhin A. P., Neural correlates of inhibitory control and functional genetic variation in the dopamine D4 receptor gene. Neuropsychologia 62, 306–318 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bromberg-Martin E. S., Matsumoto M., Hikosaka O., Dopamine in motivational control: Rewarding, aversive, and alerting. Neuron 68, 815–834 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Congdon E., Lesch K. P., Canli T., Analysis of DRD4 and DAT polymorphisms and behavioral inhibition in healthy adults: Implications for impulsivity. Am. J. Med. Genet. Part B 147B, 27–32 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Sweitzer M. M., Halder I., Flory J. D., Craig A. E., Gianaros P. J., Ferrell R. E., Manuck S. B., Polymorphic variation in the dopamine D4 receptor predicts delay discounting as a function of childhood socioeconomic status: Evidence for differential susceptibility. Soc. Cogn. Affect. Neurosci. 8, 499–508 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gray J. C., MacKillop J., Impulsive delayed reward discounting as a genetically-influenced target for drug abuse prevention: A critical evaluation. Front. Psychol. 6, 1104 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]