

Abstract
Background:
Prenatal exposure to air pollution is considered to be associated with adverse effects on child health. This may partly be mediated by mechanisms related to DNA methylation.
Objectives:
We investigated associations between exposure to air pollution, using nitrogen dioxide (NO2) as marker, and epigenome-wide cord blood DNA methylation.
Methods:
We meta-analyzed the associations between NO2 exposure at residential addresses during pregnancy and cord blood DNA methylation (Illumina 450K) in four European and North American studies (n = 1,508) with subsequent look-up analyses in children ages 4 (n = 733) and 8 (n = 786) years. Additionally, we applied a literature-based candidate approach for antioxidant and anti-inflammatory genes. To assess influence of exposure at the transcriptomics level, we related mRNA expression in blood cells to NO2 exposure in 4- (n = 111) and 16-year-olds (n = 239).
Results:
We found epigenome-wide significant associations [false discovery rate (FDR) p < 0.05] between maternal NO2 exposure during pregnancy and DNA methylation in newborns for 3 CpG sites in mitochondria-related genes: cg12283362 (LONP1), cg24172570 (3.8 kbp upstream of HIBADH), and cg08973675 (SLC25A28). The associations with cg08973675 methylation were also significant in the older children. Further analysis of antioxidant and anti-inflammatory genes revealed differentially methylated CpGs in CAT and TPO in newborns (FDR p < 0.05). NO2 exposure at the time of biosampling in childhood had a significant impact on CAT and TPO expression.
Conclusions:
NO2 exposure during pregnancy was associated with differential offspring DNA methylation in mitochondria-related genes. Exposure to NO2 was also linked to differential methylation as well as expression of genes involved in antioxidant defense pathways.
Citation:
Gruzieva O, Xu CJ, Breton CV, Annesi-Maesano I, Antó JM, Auffray C, Ballereau S, Bellander T, Bousquet J, Bustamante M, Charles MA, de Kluizenaar Y, den Dekker HT, Duijts L, Felix JF, Gehring U, Guxens M, Jaddoe VV, Jankipersadsing SA, Merid SK, Kere J, Kumar A, Lemonnier N, Lepeule J, Nystad W, Page CM, Panasevich S, Postma D, Slama R, Sunyer J, Söderhäll C, Yao J, London SJ, Pershagen G, Koppelman GH, Melén E. 2017. Epigenome-wide meta-analysis of methylation in children related to prenatal NO2 air pollution exposure. Environ Health Perspect 125:104–110; http://dx.doi.org/10.1289/EHP36
Introduction
Air pollution exposure has been associated with different types of health effects, such as adverse pregnancy outcomes (Pedersen et al. 2013), childhood airway disease (Minelli et al. 2011), and neurodevelopmental disorders (Calderón-Garcidueñas et al. 2014). Oxidative stress and inflammatory responses have been suggested to be among key pathophysiological mechanisms linking air pollution exposure to the health end points. Even though the molecular processes are not fully understood, there is evidence that air pollution may act partly through epigenetic mechanisms (Gruzieva et al. 2014). Some studies show that DNA methylation, one of the key epigenetic mechanisms, is altered in children exposed to air pollution (Perera et al. 2009; Rossnerova et al. 2013; Tang et al. 2012). A few candidate gene studies have reported differential methylation in genes involved in oxidative stress and chronic inflammation in relation to prenatal (Perera et al. 2009; Tang et al. 2012) and postnatal (Hew et al. 2015; Nadeau et al. 2010; Salam et al. 2012) air pollution exposure. These findings were further supported by animal studies showing that methylation changes within inflammatory genes after exposure to diesel exhaust particles (Liu et al. 2008). Some of these epigenetic modifications were also linked to differential protein expression (Hew et al. 2015). However, genome-wide methylation analyses allowing a hypothesis-free assessment of epigenetic modifications in relation to air pollution exposure are sparse (Jiang et al. 2014; Rossnerova et al. 2013).
Both animal and human studies suggest that exposures affecting epigenetic markers may have a substantial impact if occurring in utero (de Planell-Saguer et al. 2014), particularly in light of extensive epigenetic reprogramming during embryogenesis (Cortessis et al. 2012; Wright and Brunst 2013). This has been demonstrated in epigenome-wide studies of methylation in offspring related to maternal smoking during pregnancy (Joubert et al. 2016; Richmond et al. 2015). To our knowledge, no study has evaluated the role of prenatal air pollution exposure on methylation levels across the genome in newborns.
For the present study, we used a large collection of genome-wide DNA methylation data to investigate associations between prenatal exposure to nitrogen dioxide (NO2), as an indicator of traffic-related air pollution, and cord blood DNA methylation. In addition, we applied a literature-based candidate approach to evaluate the importance of prenatal NO2 exposure for DNA methylation within a set of antioxidant and anti-inflammatory genes. Furthermore, the continuance of associations between maternal exposure to NO2 and cord blood DNA methylation changes at key cytosine-guanine dinucleotide sites (CpGs) was examined in a sample of 4- and 8-year-old children, as well as differences in gene expression of selected genes in relation to air pollution exposure.
Methods
Study Population
Four studies participating in the Pregnancy and Childhood Epigenetics consortium (PACE) were included in the meta-analysis of NO2 exposure during pregnancy and cord blood DNA methylation. These are Mechanisms of the Development of ALLergy (MeDALL), the Generation R Study (the Netherlands), the Children’s Health Study (CHS; USA), and the Mother and Child Cohort Study (MoBa; Norway). MeDALL represents a pooled sample of four cohorts with uniform methylation measurements in paired samples either in cord blood and 4–5 years: Infancia y Medio Ambiente (INMA; Spain) and Etudes des Déterminants pré et postnatals précoces du développement et de la santé de l’ENfant (EDEN; France); or at 4 and 8 years: Children’s Allergy Environment Stockholm Epidemiology study (BAMSE; Sweden) and Prevention and Incidence of Asthma and Mite Allergy (PIAMA; the Netherlands). Two of the MEDALL cohorts with cord blood methylation data (INMA and EDEN) contributed to the meta-analysis on newborns. Methylation data in older children in MeDALL (age 4–5 years for INMA, EDEN, BAMSE, and PIAMA, and age 8 years for BAMSE and PIAMA), as well as an independent methylation data set from the BAMSE cohort at age 8 years (BAMSE EpiGene), the latter consisting of asthma cases and healthy controls (Melén et al. 2013), were used for the subsequent look-up of the findings in cord blood meta-analysis. Information about study design, recruitment, and procedures for data collection in each cohort are provided in the “Materials and Methods” section in the Supplemental Material. Consent for blood sampling was obtained from all parents. Ethics approval for each study was obtained from local authorized review boards.
Air Pollution Exposure Assessment
In the MeDALL cohorts, the Generation R Study, and BAMSE EpiGene mean concentrations of NO2 during pregnancy were estimated at maternal home addresses through land-use regression (LUR) models developed for each study area within the ESCAPE (European Study of Cohorts for Air Pollution Effects) project (Pedersen et al. 2013). LUR models for MoBa were developed following the ESCAPE methodology. In the CHS, air quality monitoring data (Peters et al. 1999) and the U.S. Environmental Protection Agency (EPA) Air Quality System (https://www.epa.gov/aqs) were used to assign estimates of prenatal exposure for NO2. Detailed descriptions of exposure assessment are provided in the “Materials and Methods” section in the Supplemental Material.
Profiling of DNA Methylation
Each cohort independently conducted laboratory measurements and quality control (QC) as described in the “Materials and Methods” section in the Supplemental Material. The samples for each cohort underwent bisulfite treatment using the EZ-96 DNA Methylation kit (Zymo Research Corporation, Irvine, CA, USA), and were subsequently processed with the Illumina Infinium HumanMethylation450 BeadChip (Illumina Inc., San Diego, CA, USA).
Details on QC of samples are provided in the “Materials and Methods” section in the Supplemental Material. Cohorts used validated, published statistical methods for normalizing their methylation data on the untransformed methylation beta values (ranging from 0 to 1), such as “DASEN” (Pidsley et al. 2013), “DASES” (Touleimat and Tost 2012), and BMIQ (Teschendorff et al. 2013). Furthermore, we excluded from the meta-analysis probes that mapped to the X (n = 11,232) or Y (n = 416) chromosomes, leaving a total of 472,299 CpGs included in the meta-analysis.
Data on mRNA gene expression were available in the BAMSE (239 children 16 years of age) and the INMA (111 children 4 years of age) cohorts through the MeDALL project (Bousquet et al. 2011). Whole blood was collected in PAXGene tubes, and RNA was extracted using PAXgene Blood RNA kit (QIAGEN, Courtaboeuf, France) and assessed for quality. Gene expression data were obtained using Affymetrix HTA 2.0 Genechips (Affymetrix, Inc., Santa Clara, CA, USA). Additional information is provided in the “Materials and Methods” section in the Supplemental Material.
Statistical Analyses
First, we examined the association between exposure to NO2 and methylation levels across the genome using robust linear regression to account for any potential outliers and heteroskedasticity in the data (Fox and Weisberg 2011). Untransformed normalized methylation β-values were used. All included samples were analyzed on a cohort level, except for the pooled MeDALL study with coordinated methylation measurements as well as air pollution exposure assessment according to a harmonized protocol. All analyses were adjusted for an a priori selected panel of covariates: sex, maternal smoking during pregnancy, municipality at birth (in BAMSE), cohort-specific batch indicator(s), cohort indicator (in the pooled MeDALL sample set), and ancestry (in CHS). In addition, age at biosampling was included in the analyses of the older children. As a sensitivity analysis we also adjusted for asthma status in the older children analyses. Cohort-specific results of the cord blood EWAS (epigenome-wide association studies) were subsequently included in a fixed-effects meta-analysis (I 2 random-effects tests for heterogeneity did not display heterogeneity across cohorts) by combining p-values across studies, taking into account study-specific weights based on the inverse of the corresponding standard errors (Willer et al. 2010).
DNA methylation sites were annotated based on data provided by Illumina (Bibikova et al. 2011). Because DNA methylation patterns within genetic regions are correlated, we used the false discovery rate (FDR) procedure to account for multiple testing (Strimmer 2008), rather than the more stringent Bonferroni adjustment that assumes independent effects of all CpG sites. CpG sites with FDR < 0.05 threshold were labeled significant.
It has been demonstrated that differences in DNA methylation can arise from variability of cell composition in whole blood (Reinius et al. 2012). To adjust for this, we estimated the fraction of CD8T, CD4T, NK cells, B cells, monocytes, and granulocytes in each sample through the reference-based Houseman method (Houseman et al. 2012) using the estimateCellCounts function in the minfi Bioconductor package in R (Jaffe and Irizarry 2014). We adjusted for cell composition by including the six estimated cell type fractions as covariates in the multivariate linear regression. Additionally, as a sensitivity analysis we applied a new method of cell proportion estimation for cord blood samples in the MeDALL study (Bakulski et al. 2016).
Second, we investigated whether associations between NO2 exposure and methylation levels in the top 25 CpGs (corresponding to p < 2.59 × 10–5) in the cord blood analyses persisted in older children, employing a single CpG look-up approach in available samples of 4-year-olds (pooled MeDALL sample), as well as in 8-year-olds (meta-analyzed pooled MeDALL sample and the BAMSE EpiGene). For these look-up analyses, a CpG with a nominal p-value < 0.05 was considered to be statistically significant.
Third, using a candidate-gene approach based on a literature search for air pollution associated genes we investigated separately a set of 739 CpGs in 38 antioxidant and inflammatory genes (TGFB1, ARG1, ARG2, GSTM1, GSTP1, NQO1, SOD2, GPX1, HMOX-1, CAT, GSTT1, EPHX1, NOS2, TNF, NFE2L2, GSS, GPX7, GPX2, GSTZ1, ALB, SRXN1, NOX5, ALOX12, NCF2, AOX1, MPV17, SIRT2, MBL2, OXSR1, OXR1, NUDT1, DUOX2, EPX, PXDNL, PXDN, MPO, LPO, TPO) (Carlsten and Melén 2012; Chen et al. 2015; Minelli et al. 2011; Nagiah et al. 2015), by extracting the results from meta-analysis (excluding cg01957222, available only in one cohort). Fourth, to assess functional effects related to methylation profiles, we investigated whether genes annotated to the identified CpGs were differentially expressed in relation to air pollution exposure during pregnancy and at the time of biosampling by means of linear regression analysis. Finally, pathways associated with differentially methylated sites (p < 0.0001) were interrogated using ConsensusPathDB database (http://cpdb.molgen.mpg.de) (Kamburov et al. 2013).
Air pollution concentrations were entered as continuous variables without transformation. The results are presented as change in methylation β-value per 10 μg/m3 of increase in NO2. All study-specific statistical analyses were performed using R (version 3.0.1; R Project for Statistical Computing) and Bioconductor packages (Gentleman et al. 2004), and the meta-analysis was performed using METAL software (Willer et al. 2010). For the most significant results we used the web-based plotting tool CoMet to graphically display additional information about all available CpGs within the same gene including physical location, correlation, and statistical significance (Martin et al. 2015). Cord blood methylation data from the MeDALL samples (n = 280) were used to compute the correlations between the CpG sites within selected genes.
Results
The baseline characteristics of the study population of the original cohorts, and of subjects included in the present analyses are presented in Table S1 and Table 1, respectively. Exposure contrasts, indicated by the interquartile ranges were smallest for the MoBa (5.4 μg/m3) and Generation R (5.8 μg/m3) cohorts, and highest for the pooled MeDALL sample (28.1 μg/m3). In total, 1,508 children were included in the discovery meta-analysis of prenatal NO2 exposure and cord blood methylation. Plotted –log10 (p-values) from the combined analysis of 472,299 CpGs across the genome in cord blood samples of participants of the MeDALL, Generation R, CHS, and MoBa studies are presented in Figure 1. The quantile–quantile plot did not reveal any significant inflation in the distribution of observed p-values (lambda = 1.08). We found epigenome-wide significant associations (FDR p-value < 0.05) between NO2 exposure and DNA methylation for 3 CpGs, one mapped to lon peptidase 1 (LONP1, cg12283362, chromosome 19), one located 3.8 kbp upstream of the 3-hydroxyisobutyrate dehydrogenase (HIBADH, cg24172570, chromosome 7), and a third mapped to solute carrier family 25 (SLC25A28, cg08973675, chromosome 10) (Table 2). We also observed that methylation levels of these top 3 CpGs significantly changed with NO2 exposure levels in a dose-dependent manner with negative trend for cg24172570 (3.8 kbp upstream of HIBADH) and cg12283362 (LONP1), and positive for cg08973675 (SLC25A28) as indicated by the trend test (see Figure S1), although threshold effects or nonlinear associations cannot be completely ruled out. The top hits were largely unaltered by adjustment for predicted cell type components, although cg08973675 (SLC25A28 chromosome 10) was no longer significant at the FDR significance level (see Figure S2 and Table S2). Interestingly, cg01610636 in PLVAP (chromosome 19) encoding plasmalemma vesicle associated protein, as well as cg21022949 located 19.7 kbp downstream of G-protein-coupled receptor 55 (GPR55, chromosome 2) appeared to be FDR-significant after cell-type correction (p = 7.0 × 10–7 and p = 8.9 × 10–7 corrected and p = 0.002 and 1.5 × 10–5 uncorrected, respectively). A sensitivity analysis based on the MeDALL sample set applying a novel adjustment approach for cord blood cells according to Bakulski et al. (2016) showed very good agreement between the results of analyses with and without cell-count adjustment (epigenome-wide correlation of β-coefficients = 0.95, and p-values = 0.82; see Table S3). In addition, we checked the potential influence of outliers on our top hits results in the MeDALL sample set by trimming outliers defined by > 3 interquartile ranges below the first quartile or above the fourth quartile (NIST 2013). After outlying CpGs have been excluded by the trim, we re-ran the analyses and got essentially unchanged results (data not shown).
Table 1.
Characteristics for individuals of the included cohorts.
| Characteristic | Birth | 4–5 years | 8 years | ||||
|---|---|---|---|---|---|---|---|
| MeDALL pooleda EDEN (France), INMA (Spain) (n = 280) | Generation R (the Netherlands) (n = 809) | CHS (USA) (n = 226) | MoBa (Norway) (n = 193) | MeDALL pooleda BAMSE (Sweden), EDEN (France), INMA (Spain), PIAMA (the Netherlands) (n = 733) | MeDALL pooleda BAMSE (Sweden), PIAMA (the Netherlands) (n = 444) | BAMSE EpiGene (Sweden) (n = 342) | |
| NO2 during pregnancy (μg/m3): percentiles 25th, 50th, 75th (minimum–maximum) | 19.0, 37.5, 47.1 (9.0–89.9) | 36.0, 38.7, 41.8 (28.6–55.9) | 23.0, 32.1, 36.9 (7.5–51.0) | 7.5, 10.3, 12.9 (0.01–27.6) | 20.2, 31.2, 40.4 (9.0–89.9) | 19.7, 27.3, 35.4 (9.9–59.8) | 17.9, 23.3, 33.3 (9.3–58.7) |
| Annual NO2 at the current address at the time of biosampling (μg/m3): percentiles 25th, 50th, 75th (minimum–maximum) | — | — | — | — | 11.4, 21.1, 23.7 (2.6–95.6) | 9.1, 14.1, 22.5 (6.1–39.7) | 8.1, 9.4, 13.1 (6.0–29.1) |
| Male sex [n (%)] | 155 (55.4) | 427 (52.8) | 93 (41.2) | 101 (52.3) | 398 (54.3) | 230 (51.8) | 181 (52.9) |
| Age at biosampling (years): mean ± SD (minimum–maximum) | — | — | — | — | 4.4 ± 0.5 (3.3–6.0) | 8.2 ± 0.4 (7.3–9.7) | 8.3 ± 0.5 (7.4–10.5) |
| Maternal smoking during pregnancy [n (%)] | 48 (17.1) | 200 (24.7) | 14 (6.2) | 15 (7.8) | 101 (13.8) | 52 (11.7) | 41 (12.0) |
| aIn the MeDALL sample, methylation data measured in cord blood are available in EDEN (n = 93) and INMA (n = 187); at 4–5 years available in EDEN (n = 82), INMA (n = 195), BAMSE (n = 232), and PIAMA (n = 224); at 8 years available in BAMSE (n = 243) and PIAMA (n = 201). | |||||||
Figure 1.
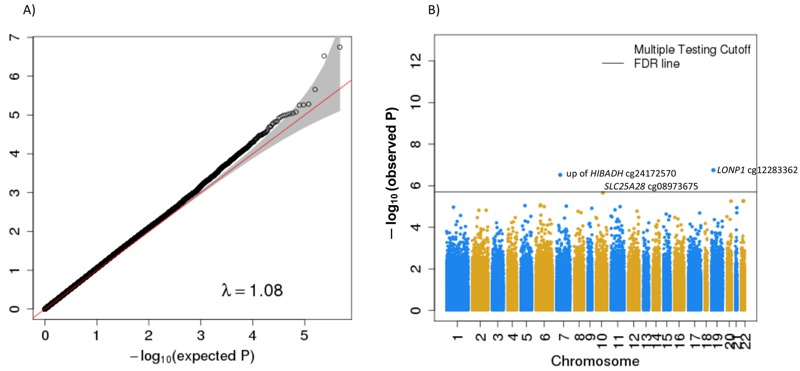
Quantile–quantile plot (A) and Manhattan plot (B) for epigenome-wide meta-analysis of the association between prenatal NO2 exposure and cord blood DNA methylation (n = 1,508). (B) Three CpGs were considered statistically significant using FDR correction (solid horizontal line): cg12283362 in LONP1, cg24172570 3.8 kbp upstream of HIBADH, and cg08973675 in SLC25A28.
Table 2.
Top 25 CpGs from the epigenome-wide meta-analysis of the association between prenatal NO2 exposure and newborn cord blood DNA methylation (n = 1,508 newborns from MeDALL, Generation R, CHS and MoBa cohorts).
| Chr | Position (build 37) | CpG | Mapped gene | Gene group | Coef | SE | p-Value | Direction |
|---|---|---|---|---|---|---|---|---|
| 19 | 5709149 | cg12283362 | LONP1FDR,a | Body | –0.007 | 0.0014 | 1.78 × 10–7 | –??–b |
| 7 | 27561178 | cg24172570 | HIBADHFDR,a,c | –0.004 | 0.0008 | 3.01 × 10–7 | ––?–b | |
| 10 | 101380289 | cg08973675 | SLC25A28FDR,a | TSS200 | 0.005 | 0.0011 | 2.20 × 10–6 | ++++ |
| 22 | 40355732 | cg17988310 | GRAP2 | Body | 0.004 | 0.0009 | 5.25 × 10–6 | ++++ |
| 20 | 61427684 | cg14582546 | C20orf20 | TSS200 | 0.005 | 0.0011 | 5.50 × 10–6 | ++++ |
| 22 | 39323510 | cg12276768 | APOBEC3Ac | 0.003 | 0.0006 | 5.60 × 10–6 | ++++ | |
| 6 | 30688588 | cg21660604 | TUBB | Body | 0.002 | 0.0003 | 8.36 × 10–6 | ++++ |
| 5 | 77284206 | cg26815688 | AP3B1c | –0.002 | 0.0005 | 9.03 × 10–6 | –––– | |
| 6 | 30524763 | cg03860665 | PRR3;GNL1 | 5’UTR; 1stExon | 0.002 | 0.0005 | 9.17 × 10–6 | ++++ |
| 7 | 117824040 | cg08301459 | NAA38 | TSS200 | 0.002 | 0.0003 | 9.58 × 10–6 | ++?+ |
| 6 | 33359817 | cg04757012 | KIFC1 | Body | 0.001 | 0.0003 | 1.01 × 10–5 | ++++ |
| 11 | 74871202 | cg12537437 | SLCO2B1 | Body; 5’UTR | –0.004 | 0.0008 | 1.02 × 10–5 | –––+ |
| 1 | 35226135 | cg01828548 | GJB4 | 5’UTR | –0.005 | 0.0011 | 1.08 × 10–5 | –––+ |
| 21 | 46032086 | cg26386968 | C21orf29;KRTAP10-8 | Body; 1stExon | –0.007 | 0.0015 | 1.15 × 10–5 | ––?– |
| 9 | 139607421 | cg12657416 | FAM69B | Body | 0.103 | 0.0236 | 1.22 × 10–5 | ?+?+ |
| 11 | 34460856 | cg03728580 | CAT | Body | 0.003 | 0.0007 | 1.43 × 10–5 | ++++ |
| 2 | 231809697 | cg21022949 | GPR55c | 0.001 | 0.0002 | 1.51 × 10–5 | ++++ | |
| 2 | 98409069 | cg06840305 | TMEM131 | Body | –0.002 | 0.0004 | 1.51 × 10–5 | –––– |
| 12 | 120967065 | cg11075121 | COQ5 | TSS200 | 0.002 | 0.0004 | 1.66 × 10–5 | ++++ |
| 8 | 48099615 | cg03271173 | IGLV8OR8-1c | –0.003 | 0.0006 | 1.70 × 10–5 | ––+– | |
| 8 | 110346503 | cg25407888 | ENY2;NUDCD1 | TSS200 | 0.003 | 0.0006 | 1.98 × 10–5 | ++++ |
| 21 | 45753677 | cg24316255 | C21orf2 | Body | –0.003 | 0.0006 | 2.00 × 10–5 | –––+ |
| 17 | 78851213 | cg08314949 | RPTOR | Body; Body | 0.013 | 0.0031 | 2.06 × 10–5 | +–?+ |
| 15 | 59063272 | cg01889112 | FAM63B | TSS200; TSS200 | 0.002 | 0.0004 | 2.29 × 10–5 | ++++ |
| 6 | 31382102 | cg26504614 | MICA | Body | –0.005 | 0.0011 | 2.59 × 10–5 | –?–– |
| Shown are top 25 CpGs ordered by p-value. Results presented per 10 μg/m3 increase in prenatal NO2 exposure. Column heads: Chr: chromosome; Position: chromosomal position based on NCBI human reference genome assembly build 37; Mapped Gene: UCSC annotated gene; Gene group: UCSC gene region feature category; Coef: regression coefficient; SE: standard error for regression coefficient; Direction: direction of effect across cohorts included in the statistical model (MeDALL, Generation R, CHS, and MoBa): NO2 exposure during pregnancy was associated with increased (+) or decreased (–) methylation, or missing (?) result. aGenome-wide significance threshold (FDR p < 0.05). bData on methylation for cg12283362 were available in 473 individuals, and for cg24172570 in 1,282 individuals. ccg24172570 is located 3.8 kbp upstream of HIBADH; cg12276768 is located 25.2 kbp upstream of APOBEC3A; cg26815688 is located 21.1 kbp upstream of AP3B1; cg21022949 is located 19.7 kbp downstream of GPR55; cg03271173 is located 14.5 kbp upstream of IGLV8OR8-1. | ||||||||
We further investigated whether these three associations (cg24172570 3.8 kbp upstream of HIBADH, cg12283362 LONP1 and cg08973675 SLC25A28) between air pollution exposure and methylation at birth persisted later in childhood. We observed similar significant change in methylation level of cg08973675 (SLC25A28) in all available samples of the 4-year-old children of the MeDALL study (p = 0.03), as well as of the 8-year-olds of the meta-analyzed MeDALL and BAMSE EpiGene samples (p = 0.04), in relation to prenatal NO2 exposure (see Table S4). None of the other two top hits could be replicated in the older children. In addition, because the MeDALL sample set of 4-year-olds included two cohorts with paired samples (cord blood and 4 years), EDEN and INMA, we re-ran the look-up analysis in 4-year-olds separately in these two cohorts. A significant change in methylation of cg08973675 (SLC25A28) associated with NO2 exposure during pregnancy was seen in the combined EDEN and INMA samples (p = 0.005), providing further evidence of the persistence of the association between air pollution exposure and methylation at birth into older age (see Table S5). The results remained unchanged after additional adjustment for asthma status (data not shown).
Among CpGs of selected antioxidant defense genes previously linked to air pollution exposure, 2 CpGs in catalase gene (CAT cg03728580 and cg17034036, chromosome 11), as well as 1 in thyroid peroxidase gene (TPO cg01385533, chromosome 2) were differentially methylated (FDR p < 0.05) (see 10 top significant CpGs in Table 3 and all nominally significant CpGs in Table S6). In addition, 4 of 15 available CpGs in the CAT, as well as 9 of 87 CpGs in the TPO were differentially methylated at the nominal significance level (p < 0.05) (see Tables S7 and S8). In the analyses in older children, methylation changes in cg01385533 (TPO) in 4-year-olds were found to be of similar direction as in the newborns in relation to annual NO2 exposure at the time of biosampling (p = 0.04), as well as in 8-year-olds in relation to prenatal exposure (p = 0.04) (see Table S9). Associations did not persist to older ages for the CAT probes. We found some evidence for localized clustering around the top FDR-significant CpGs in CAT and TPO, with moderate co-methylation within the CAT region but weak in the TPO (see Figure S3).
Table 3.
Top 10 significant CpGs within oxidative stress genes extracted from the epigenome-wide meta-analysis of the association between prenatal NO2 exposure and newborn cord blood DNA methylation (n = 1,508 newborns from MeDALL, Generation R, CHS, and MoBa cohorts).
| Chr | Position (build 37) | CpG | Mapped gene | Gene group | Coef | SE | p-Value | Direction |
|---|---|---|---|---|---|---|---|---|
| 11 | 34460856 | cg03728580 | CATFDR | Body | 0.003 | 0.001 | 0.00001 | ++++ |
| 11 | 34461028 | cg17034036 | CATFDR | Body | 0.002 | 0.001 | 0.0001 | ++++ |
| 2 | 1482597 | cg01385533 | TPOFDR | Body | –0.003 | 0.001 | 0.0004 | –?–– |
| 1 | 226023590 | cg05935800 | EPHX1 | Body | –0.002 | 0.001 | 0.002 | –––– |
| 20 | 33539306 | cg13607138 | GSS | Body | –0.003 | 0.001 | 0.003 | ––?– |
| 8 | 107642385 | cg17526936 | OXR1 | Body | –0.002 | 0.001 | 0.004 | ––?– |
| 2 | 1544120 | cg19407717 | TPO | Body | –0.002 | 0.001 | 0.004 | –––– |
| 2 | 1479523 | cg13703866 | TPO | Body | –0.001 | 0.000 | 0.005 | –––– |
| 11 | 34460336 | cg07768201 | CAT | TSS200 | 0.003 | 0.001 | 0.006 | ++++ |
| 1 | 226012507 | cg03337430 | EPHX1 | TSS1500;5’UTR | 0.001 | 0.000 | 0.006 | +–++ |
| Shown are the top 10 CpGs ordered by p-value. Three CpGs were statistically significant using genome-wide significance threshold (FDR p < 0.05). Results presented per 10 μg/m3 increase in prenatal NO2 exposure. Column heads: Chr: chromosome; Position: chromosomal position based on NCBI human reference genome assembly Build 37. Mapped Gene: UCSC annotated gene; Gene group: UCSC gene region feature category; Coef: regression coefficient; SE: standard error for regression coefficient; Direction: direction of effect across cohorts included in the statistical model (MeDALL, Generation R, CHS, and MoBa): NO2 exposure during pregnancy associated with increased (+) or decreased (–) methylation, or missing (?) result. | ||||||||
In functional analysis of available expression data from the 16-year-olds in the BAMSE cohort and the 4-year-olds in the INMA cohort, no significant association of in utero NO2 exposure with gene expression was detected for any of the studied probes (data not shown). However, current NO2 exposure at 16 years was significantly associated with LONP1, CAT, and TPO expression levels in peripheral blood cells of the BAMSE children (Table 4). The results were robust to additional adjustment for measured cell counts. In the INMA cohort, TPO and GPR55 were also significantly differentially expressed in relation to current NO2 exposure at 4 years after adjustment for cell counts (p < 0.05), although the direction of change differed compared to that in BAMSE.
Table 4.
Associations between current NO2 exposure and gene expression levels in the children of the BAMSE (n = 239) and INMA (n = 111) cohorts.
| Gene | Cohort | LogFCa | p-Value |
|---|---|---|---|
| TPO | BAMSE 16 years | 0.038 | 0.032 |
| INMA 4 years | –0.028 | 0.004 | |
| CAT | BAMSE 16 years | –0.098 | 0.042 |
| INMA 4 years | 0.014 | 0.660 | |
| LONP1 | BAMSE 16 years | 0.034 | 0.008 |
| INMA 4 years | –0.007 | 0.372 | |
| SLC25A28 | BAMSE 16 years | 0.003 | 0.829 |
| INMA 4 years | –0.0003 | 0.968 | |
| PLVAP | BAMSE 16 years | –0.071 | 0.142 |
| INMA 4 years | 0.002 | 0.954 | |
| GPR55 | BAMSE 16 years | 0.027 | 0.180 |
| INMA 4 years | –0.033 | 0.003 | |
| Results presented per 10-μg/m3 increase in NO2 exposure current with biosampling in the BAMSE and INMA cohorts. LogFC, logarithm fold-change (1 unit of the logFCs translates to a 2-fold change in expression). In the INMA cohort, cell count estimation using expression data was performed using R package CellMix and Abbas data set. Actual cell counts in BAMSE were used. aAdjusted for sex, age, municipality at birth (only in BAMSE), maternal smoking during pregnancy, and cell composition. | |||
Finally, to identify plausible pathways associated with air pollution exposure, we also performed gene set enrichment analysis based on CpGs significantly associated with prenatal NO2 in the meta-analysis using an arbitrary cut-off of p < 0.0001. A total of 71 unique gene identifiers were entered in the ConsensusPathDB database of which 58 matched. Using FDR p < 0.05, a few enriched pathways were identified including “negative regulation of cellular process” (GO term GO:0048523, FDR p = 0.04), “negative regulation of biological process” (GO:0048519, FDR p = 0.04), and the “integrin-linked kinase signaling” pathway (FDR p = 0.02).
Discussion
This study represents a large-scale epigenome-wide meta-analysis evaluating the association between prenatal air pollution exposure and DNA methylation in newborns. The combined results show suggestive evidence for associations of NO2 exposure during pregnancy with methylation differences in several genes, involved in mitochondria function, providing a potential epigenetic biomarker of in utero exposure that persisted in early childhood. Using a hypothesis-based approach, we also identified a link between prenatal NO2 exposure and methylation of CpG loci in antioxidant enzyme genes, such as CAT and TPO. Furthermore, we observed differential expression of these two genes in relation to recent exposure to NO2.
The three differentially methylated CpG sites—cg12283362 in LONP1, cg24172570 3.8 kbp upstream of HIBADH, and cg08973675 in SLC25A28—represent novel associations in the context of air pollution exposure. The top significant cg12283362 localizes to the gene LONP1 encoding a protein that belongs to the Lon family of ATP-dependent proteases and mediates the selective degradation of misfolded, unassembled or oxidatively damaged polypeptides in the mitochondrial matrix (Pinti et al. 2015). However, cg12283362 did not pass the QC filter in two of the cohorts with cord blood samples (n = 1,035), and the results should therefore be interpreted with caution. The second significant site, cg24172570, was located 3.8 kbp upstream of HIBADH that encodes a protein playing a critical role in the catabolism of l-valine. The third one, cg08973675, is annotated to the SLC25A28 coding for a mitochondrial iron transporter protein that mediates iron uptake. SLC25A28 was the only top gene with persistent prenatal NO2–methylation associations in older children. Interestingly, all three genes are involved in mitochondria function, and mitochondria are known to play an important role in several key pathways of cellular responses to environmental stressors, including response to reactive oxygen species (ROS), nutrient and ATP sensing, and DNA damage response (Shaughnessy et al. 2014).
Recent studies demonstrated that air pollution exposure during pregnancy is associated with changes in global DNA methylation in cord blood cells and placental tissue sampled from the fetal side (Herbstman et al. 2012; Janssen et al. 2013). Global methylation, however, represents the overall methylation state of the genome without indicating which genomic locations are methylated. A study conducted in schoolchildren suggested an impact of air pollution exposure on the DNA methylation patterns in genes related to the immune system, DNA–protein binding, and metabolism of xenobiotics as measured by the Illumina 27K platform (Rossnerova et al. 2013).
We also compared the methylation status at a candidate gene level for genes previously implicated in biologic response to air pollution using a hypothesis-based approach. Oxidative stress and inflammation have been hypothesized as the main mechanisms through which ambient air pollution can affect human health (Esposito et al. 2014). Both experimental and observational studies demonstrate the capacity of NO2 along with other air pollutants to activate oxidant pathways through formation of ROS, triggering inflammation and cell death (Lodovici and Bigagli 2011). Studies in human bronchial epithelial cells showed differential expression of genes involved in response to oxidative stress following air pollution exposure (Rossner et al. 2015; Zhou et al. 2015). In our study we observed differential methylation in CAT and TPO. CAT encodes catalase, an antioxidant that catalyzes degradation of hydrogen peroxide and plays a crucial role in protecting cells against ROS. However, long-term exposure to ROS may downregulate CAT expression via hypermethylation of a CpG island (Min et al. 2010), which would be in line with our results. Even though DNA methylation and gene expression were measured at different ages, we observed increased methylation in CpGs of the CAT gene in newborns together with decreased gene expression in adolescents of the BAMSE study in relation to current NO2 exposure at 16 years. This observed pattern of increased methylation and decreased gene expression by NO2 exposure is in the expected direction (i.e., the higher the methylation, the lower the gene expression). Furthermore, additional pathway analysis demonstrated that CAT was significantly enriched in several gene ontology terms. Thyroid peroxidase, originally described as thyroid specific enzyme, has also been identified in human airway epithelial cells as the only peroxidase differentially expressed in severe asthmatics, thus distinguishing them from healthy controls and milder asthma cases (Voraphani et al. 2014). A recent functional study indicated significantly higher expression of TPO in peripheral lymphocytes in pregnant women residing in a highly industrialized area (Nagiah et al. 2015). We also observed decreased methylation in the TPO gene in newborns as well as in older children together with differential TPO expression in both the BAMSE and the INMA cohorts, although with the opposite direction. Relatively small sample sizes, differences in age, as well as in other exposures might have contributed to the observed difference in the direction of effects; therefore, these results should be interpreted with caution. Furthermore, the present analysis does not involve the possible various isoforms of the genes. Thus, future studies need to assess whether different isoforms are expressed in response to air pollution.
One challenge of genome-wide DNA methylation analyses in blood samples with a mixed cell composition is the difference in methylation patterns between different cell types. In the present analyses we used the reference data for adult peripheral blood to correct for cell type proportions in the cord blood analyses (Reinius et al. 2012). A sensitivity analysis in one of the included studies that applied a new cell type referenced by Bakulski et al. (2016), which takes cord blood cell composition into account, further supported robustness of the results. Although no major differences were detected in the top results with cell-type correction, cg01610636 in PLVAP and cg21022949 located 19.7 kbp downstream of GPR55 appeared to be FDR-significant after cell-type adjustment (according to the Houseman method). Interestingly, PLVAP is known to be involved in leukocyte transendothelial cell migration (Keuschnigg et al. 2009). GPR55 has been implicated as cannabinoid receptor (Ryberg et al. 2007). Further functional analysis did not reveal any difference in expression profiles of PLVAP in relation to NO2 exposure, but weak associations with GPR55 expression were observed in the INMA study. Tissue specificity is another potential limitation that may complicate the assessment of epigenetic patterns relevant for air pollution exposure (Bakulski and Fallin 2014). Therefore, using other biological samples, such as airway epithelium or placenta, in future studies may identify important methylation differences in the primary tissues.
The comprehensive evaluation of genome-wide DNA methylation using the Illumina 450K BeadChip together with air pollution exposure information on individual level, as well as availability of samples at multiple ages, are major strengths of this study. All cohort-specific analyses were conducted according to the same analytical protocol. However, the between-cohort differences in statistical methods applied for the quality control, normalization, and adjustment for technical variation may to some extent contribute to diluting of possible associations. A recently published EWAS meta-analysis including the same cohorts reported very robust results in relation to different data processing methods used across the cohorts for normalization and corrections for technical variables such as batch (Joubert et al. 2016). It is also important to note that our analyses were based mainly on Caucasian populations, and it remains to be investigated whether the findings can be extrapolated to other ethnic groups.
We used NO2 as a marker of traffic-derived combustion pollutants. Road traffic is considered to be the principal outdoor source of nitrogen dioxide (WHO 2010). Previous measurement studies around roadways have shown that traffic-related pollutants are characterized well by NO2, as indicated by high correlations (r ~ 0.7–0.96) between measurements of NO2 and PM2.5 (particulate matter ≤ 2.5 μm), ultrafine particles, and black carbon (Beckerman et al. 2008), including increases in benzene and polycyclic aromatic hydrocarbons (Karner et al. 2010). A potential limitation of the exposure assessment is that the modeled individual concentrations account only for outdoor air pollution at residential addresses and therefore are not equivalent to personal exposure. Indoor exposure and time–activity patterns may introduce some bias, although this will most likely be nondifferential and thus would generally tend to attenuate the associations. Furthermore, several measurement studies conducted in different areas have demonstrated that indoor and outdoor NO2 levels are strongly correlated (R 2 = 0.7–0.9), pointing to indoor NO2 concentrations being largely affected by outdoor sources (El-Hougeiri and El Fadel 2004; Verriele et al. 2015; Wichmann et al. 2010).
Conclusions
Our epigenome-wide meta-analysis provides evidence of cord blood methylation differences in several mitochondria-related genes, in relation to air pollution exposure during pregnancy. Our study also contributes to further understanding of potential underlying mechanisms of the negative health effects of air pollution by highlighting the implications of DNA methylation in several candidate genes involved in antioxidant defense pathways, such as CAT and TPO.
Supplemental Material
Acknowledgments
For all studies, information on acknowledgments and funding can be found in the Supplemental Material, “Acknowledgments.”
Footnotes
The authors declare they have no actual or potential competing financial interests.
References
- Bakulski KM, Fallin MD. Epigenetic epidemiology: promises for public health research. Environ Mol Mutagen. 2014;55:171–183. doi: 10.1002/em.21850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakulski KM, Feinberg JI, Andrews SV, Yang J, Brown S, McKenney SL, et al. DNA methylation of cord blood cell types: applications for mixed cell birth studies. Epigenetics. 2016;11:354–362. doi: 10.1080/15592294.2016.1161875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckerman B, Jerrett M, Brook JR, Vermae DK, Araine MA, Finkelstein MM. Correlation of nitrogen dioxide with other traffic pollutants near a major expressway. Atmos Environ. 2008;42:275–290. [Google Scholar]
- Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–295. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Bousquet J, Anto J, Auffray C, Akdis M, Cambon-Thomsen A, Keil T, et al. MeDALL (Mechanisms of the Development of ALLergy): an integrated approach from phenotypes to systems medicine. Allergy. 2011;66:596–604. doi: 10.1111/j.1398-9995.2010.02534.x. [DOI] [PubMed] [Google Scholar]
- Calderón-Garcidueñas L, Torres-Jardón R, Kulesza RJ, Park SB, D’Angiulli A. 2014. Air pollution and detrimental effects on children’s brain. The need for a multidisciplinary approach to the issue complexity and challenges. Front Hum Neurosci 8 613, doi: 10.3389/fnhum.2014.00613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsten C, Melén E. Air pollution, genetics, and allergy: an update. Curr Opin Allergy Clin Immunol. 2012;12:455–461. doi: 10.1097/ACI.0b013e328357cc55. [DOI] [PubMed] [Google Scholar]
- Chen Z, Salam MT, Eckel SP, Breton CV, Gilliland FD. Chronic effects of air pollution on respiratory health in Southern California children: findings from the Southern California Children’s Health Study. J Thorac Dis. 2015;7:46–58. doi: 10.3978/j.issn.2072-1439.2014.12.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortessis VK, Thomas DC, Levine AJ, Breton CV, Mack TM, Siegmund KD, et al. Environmental epigenetics: prospects for studying epigenetic mediation of exposure–response relationships. Hum Genet. 2012;131:1565–1589. doi: 10.1007/s00439-012-1189-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Planell-Saguer M, Lovinsky-Desir S, Miller RL. Epigenetic regulation: the interface between prenatal and early-life exposure and asthma susceptibility. Environ Mol Mutagen. 2014;55:231–243. doi: 10.1002/em.21836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hougeiri N, El Fadel M. Correlation of indoor-outdoor air quality in urban areas. Indoor Built Environ. 2004;13:421–431. [Google Scholar]
- Esposito S, Tenconi R, Lelii M, Preti V, Nazzari E, Consolo S, et al. 2014. Possible molecular mechanisms linking air pollution and asthma in children. BMC Pulm Med 14 31, doi: 10.1186/1471-2466-14-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox J, Weisberg S. Thousand Oaks, CA: SAGE; 2011. Robust regression in R. In: An R Companion to Applied Regression. 2nd ed. pp. 149–228. [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5 R80, doi: 10.1186/gb-2004-5-10-r80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruzieva O, Merid SK, Melén E. An update on epigenetics and childhood respiratory diseases. Paediatr Respir Rev. 2014;15:348–354. doi: 10.1016/j.prrv.2014.07.003. [DOI] [PubMed] [Google Scholar]
- Herbstman JB, Tang D, Zhu D, Qu L, Sjödin A, Li Z, et al. 2012. Prenatal exposure to polycyclic aromatic hydrocarbons, benzo[a]pyrene-DNA adducts, and genomic DNA methylation in cord blood. Environ Health Perspect 120 733 738, doi: 10.1289/ehp.1104056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hew KM, Walker AI, Kohli A, Garcia M, Syed A, McDonald-Hyman C, et al. Childhood exposure to ambient polycyclic aromatic hydrocarbons is linked to epigenetic modifications and impaired systemic immunity in T cells. Clin Exp Allergy. 2015;45:238–248. doi: 10.1111/cea.12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. 2012. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 13 86, doi: 10.1186/1471-2105-13-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AE, Irizarry RA. 2014. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol 15 R31, doi: 10.1186/gb-2014-15-2-r31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen BG, Godderis L, Pieters N, Poels K, Kiciński M, Cuypers A, et al. 2013. Placental DNA hypomethylation in association with particulate air pollution in early life. Part Fibre Toxicol 10 22, doi: 10.1186/1743-8977-10-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R, Jones MJ, Sava F, Kobor MS, Carlsten C. 2014. Short-term diesel exhaust inhalation in a controlled human crossover study is associated with changes in DNA methylation of circulating mononuclear cells in asthmatics. Part Fibre Toxicol 11 71, doi: 10.1186/s12989-014-0071-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joubert BR, Felix JF, Yousefi P, Bakulski KM, Just AC, Breton C, et al. DNA methylation in newborns and maternal smoking in pregnancy: genome-wide consortium meta-analysis. Am J Hum Genet. 2016;98:680–696. doi: 10.1016/j.ajhg.2016.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamburov A, Stelzl U, Lehrach H, Herwig R. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 2013;41(database issue):D793–D800. doi: 10.1093/nar/gks1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karner AA, Eisinger DS, Niemeier DA. Near-roadway air quality: synthesizing the findings from real-world data. Environ Sci Technol. 2010;44:5334–5344. doi: 10.1021/es100008x. [DOI] [PubMed] [Google Scholar]
- Keuschnigg J, Henttinen T, Auvinen K, Karikoski M, Salmi M, Jalkanen S. The prototype endothelial marker PAL-E is a leukocyte trafficking molecule. Blood. 2009;114:478–484. doi: 10.1182/blood-2008-11-188763. [DOI] [PubMed] [Google Scholar]
- Liu J, Ballaney M, Al-alem U, Quan C, Jin X, Perera F, et al. Combined inhaled diesel exhaust particles and allergen exposure alter methylation of T helper genes and IgE production in vivo. Toxicol Sci. 2008;102:76–81. doi: 10.1093/toxsci/kfm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodovici M, Bigagli E. 2011. Oxidative stress and air pollution exposure. J Toxicol 2011 487074, doi: 10.1155/2011/487074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TC, Yet I, Tsai PC, Bell JT. 2015. coMET: visualisation of regional epigenome-wide association scan results and DNA co-methylation patterns. BMC Bioinformatics 16 131, doi: 10.1186/s12859-015-0568-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melén E, Granell R, Kogevinas M, Strachan D, Gonzalez JR, Wjst M, et al. Genome-wide association study of body mass index in 23 000 individuals with and without asthma. Clin Exp Allergy. 2013;43:463–474. doi: 10.1111/cea.12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min JY, Lim SO, Jung G. Downregulation of catalase by reactive oxygen species via hypermethylation of CpG island II on the catalase promoter. FEBS Lett. 2010;584:2427–2432. doi: 10.1016/j.febslet.2010.04.048. [DOI] [PubMed] [Google Scholar]
- Minelli C, Wei I, Sagoo G, Jarvis D, Shaheen S, Burney P. Interactive effects of antioxidant genes and air pollution on respiratory function and airway disease: a HuGE review. Am J Epidemiol. 2011;173:603–620. doi: 10.1093/aje/kwq403. [DOI] [PubMed] [Google Scholar]
- Nadeau K, McDonald-Hyman C, Noth EM, Pratt B, Hammond SK, Balmes J, et al. Ambient air pollution impairs regulatory T-cell function in asthma. J Allergy Clin Immunol. 2010;126:845–852.e10. doi: 10.1016/j.jaci.2010.08.008. [DOI] [PubMed] [Google Scholar]
- Nagiah S, Phulukdaree A, Naidoo D, Ramcharan K, Naidoo RN, Moodley D, et al. Oxidative stress and air pollution exposure during pregnancy: a molecular assessment. Hum Exp Toxicol. 2015;34:838–847. doi: 10.1177/0960327114559992. [DOI] [PubMed] [Google Scholar]
- NIST (National Institute of Standards and Technology) What are outliers in the data? In: NIST/SEMATECH e-Handbook of Statistical Methods. 2013 http://www.itl.nist.gov/div898/handbook/prc/section1/prc16.htm [accessed 18 April 2016]
- Pedersen M, Giorgis-Allemand L, Bernard C, Aguilera I, Andersen AM, Ballester F, et al. Ambient air pollution and low birthweight: a European cohort study (ESCAPE). Lancet Respir Med. 2013;1:695–704. doi: 10.1016/S2213-2600(13)70192-9. [DOI] [PubMed] [Google Scholar]
- Perera F, Tang WY, Herbstman J, Tang D, Levin L, Miller R, et al. 2009. Relation of DNA methylation of 5’-CpG island of ACSL3 to transplacental exposure to airborne polycyclic aromatic hydrocarbons and childhood asthma. PLoS One 4 e4488, doi: 10.1371/journal.pone.0004488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM, Avol E, Gauderman WJ, Linn WS, Navidi W, London SJ, et al. A study of twelve Southern California communities with differing levels and types of air pollution. II. Effects on pulmonary function. Am J Respir Crit Care Med. 1999;159:768–775. doi: 10.1164/ajrccm.159.3.9804144. [DOI] [PubMed] [Google Scholar]
- Pidsley R, Wong CCY, Volta M, Lunnon K, Mill J, Schalkwyk LC. 2013. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics 14 293, doi: 10.1186/1471-2164-14-293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinti M, Gibellini L, Liu Y, Xu S, Lu B, Cossarizza A. Mitochondrial lon protease at the crossroads of oxidative stress, ageing and cancer. Cell Mol Life Sci. 2015;72:4807–4824. doi: 10.1007/s00018-015-2039-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlén SE, Greco D, et al. 2012. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One 7 e41361, doi: 10.1371/journal.pone.0041361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond RC, Simpkin AJ, Woodward G, Gaunt TR, Lyttleton O, McArdle WL, et al. Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: findings from the Avon Longitudinal Study of Parents and Children (ALSPAC). Hum Mol Genet. 2015;24:2201–2217. doi: 10.1093/hmg/ddu739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossner P, Jr, Tulupova E, Rossnerova A, Libalova H, Honkova K, Gmuender H, et al. Reduced gene expression levels after chronic exposure to high concentrations of air pollutants. Mutat Res. 2015;780:60–70. doi: 10.1016/j.mrfmmm.2015.08.001. [DOI] [PubMed] [Google Scholar]
- Rossnerova A, Tulupova E, Tabashidze N, Schmuczerova J, Dostal M, Rossner P, Jr, et al. Factors affecting the 27K DNA methylation pattern in asthmatic and healthy children from locations with various environments. Mutat Res. 2013;741:18–26. doi: 10.1016/j.mrfmmm.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Ryberg E, Larsson N, Sjögren S, Hjorth S, Hermansson NO, Leonova J, et al. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. 2007;152:1092–1101. doi: 10.1038/sj.bjp.0707460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salam MT, Byun HM, Lurmann F, Breton CV, Wang X, Eckel SP, et al. Genetic and epigenetic variations in inducible nitric oxide synthase promoter, particulate pollution, and exhaled nitric oxide levels in children. J Allergy Clin Immunol. 2012;129:232–239.e7. doi: 10.1016/j.jaci.2011.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaughnessy DT, McAllister K, Worth L, Haugen AC, Meyer JN, Domann FE, et al. 2014. Mitochondria, energetics, epigenetics, and cellular responses to stress. Environ Health Perspect 122 1271 1278, doi: 10.1289/ehp.1408418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strimmer K. fdrtool: a versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics. 2008;24:1461–1462. doi: 10.1093/bioinformatics/btn209. [DOI] [PubMed] [Google Scholar]
- Tang WY, Levin L, Talaska G, Cheung YY, Herbstman J, Tang D, et al. 2012. Maternal exposure to polycyclic aromatic hydrocarbons and 5’-CpG methylation of interferon-γ in cord white blood cells. Environ Health Perspect 120 1195 1200, doi: 10.1289/ehp.1103744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29:189–196. doi: 10.1093/bioinformatics/bts680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touleimat N, Tost J. Complete pipeline for Infinium® Human Methylation 450K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics. 2012;4:325–341. doi: 10.2217/epi.12.21. [DOI] [PubMed] [Google Scholar]
- Verriele M, Schoemaecker C, Hanoune B, Leclerc N, Germain S, Gaudion V, et al. The MERMAID study: indoor and outdoor average pollutant concentrations in 10 low energy school buildings in France. Indoor Air. 2015;26:702–713. doi: 10.1111/ina.12258. [DOI] [PubMed] [Google Scholar]
- Voraphani N, Gladwin MT, Contreras AU, Kaminski N, Tedrow JR, Milosevic J, et al. An airway epithelial iNOS-DUOX2-thyroid peroxidase metabolome drives Th1/Th2 nitrative stress in human severe asthma. Mucosal Immunol. 2014;7:1175–1185. doi: 10.1038/mi.2014.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (World Health Organization) WHO Guidelines for Indoor Air Quality—Selected Pollutants. 2010 http://www.euro.who.int/__data/assets/pdf_file/0009/128169/e94535.pdf [accessed 18 April 2016] [PubMed]
- Wichmann J, Lind T, Nilsson MAM, Bellander T. PM2.5, soot and NO2 indoor–outdoor relationships at homes, pre-schools and schools in Stockholm, Sweden. Atmos Environ. 2010;44:4536–4544. [Google Scholar]
- Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright RJ, Brunst KJ. Programming of respiratory health in childhood: influence of outdoor air pollution. Curr Opin Pediatr. 2013;25:232–239. doi: 10.1097/MOP.0b013e32835e78cc. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Liu Y, Duan F, Qin M, Wu F, Sheng W, et al. 2015. Transcriptomic analyses of the biological effects of airborne PM2.5 exposure on human bronchial epithelial cells. PLoS One 10 e0138267, doi: 10.1371/journal.pone.0138267 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.