

Abstract
Pantothenamides are N-substituted pantothenate derivatives which are known to exert antimicrobial activity through interference with coenzyme A (CoA) biosynthesis or downstream CoA-utilizing proteins. A previous report has shown that replacement of the ProR methyl group of the benchmark N-pentylpantothenamide with an allyl group (R-anti configuration) yielded one of the most potent antibacterial pantothenamides reported so far (MIC of 3.2 μM for both sensitive and resistant Staphylococcus aureus). We describe herein a synthetic route for accessing the corresponding R-syn diastereomer using a key diastereoselective reduction with Baker’s yeast, and report on the scope of this reaction for modified systems. Interestingly, whilst the R-anti diastereomer is the only one to show antibacterial activity, the R-syn isomer proved to be significantly more potent against the malaria parasite (IC50 of 2.4 ± 0.2 μM). Our research underlines the striking influence that stereochemistry has on the biological activity of pantothenamides, and may find utility in the study of various CoA-utilizing systems.
Keywords: pantothenamides, antibacterial, antiplasmodial, baker’s yeast, coenzyme A
Infectious diseases remain a major contributing factor to worldwide mortality. Moreover, the development of antimicrobial resistance is raising significant concerns about the increasingly limited efficacy of currently available treatments.1 There have been considerable efforts towards discovering and characterising novel therapeutic targets for antimicrobial drugs. One such target which has emerged as a promising point-of-attack is coenzyme A (CoA) biosynthesis and its associated cellular processes.2 This ubiquitous cofactor is required for a diverse set of biological functions and is essential to all organisms. Most rely on the exogenous uptake of its natural precursor pantothenate (vitamin B5), and extend it into CoA through a 5-step biotransformation.3–4 The inherent differences in the CoA biosynthetic machinery between humans and microbial pathogens suggest that this pathway can be exploited for therapeutic applications.1 In fact, a variety of pantothenate analogues have been evaluated for antibacterial, antiplasmodial and antifungal properties.2
An important class of such compounds are the N-substituted pantothenamides, including the benchmark molecule in the field, N-pentylpantothenamide (Figure 1).5 This compound was shown to have potent activity against Escherichia coli (in low pantothenate media)5 and Staphylococcus aureus.6 It was later found that pantothenamides were, in fact, being extended by the CoA biosynthetic enzymes into CoA analogues which affected downstream targets such as the acyl carrier protein necessary for fatty acid synthesis.7–8 It has been suggested that pantothenamides may also act by inhibiting the CoA biosynthetic pathway.9–10 While much of the research has focused on antibacterials, targeting the CoA pathway is also a promising strategy for antiplasmodials. For example, the provitamin pantothenol as well as a range of other pantothenate analogues have been shown to repress the proliferation of the malarial parasite Plasmodium falciparum.11–15
Fig. 1.

Structure of pantothenate and N-pentylpantothenamide analogues.
A synthetic route for accessing geminal dialkyl-substituted pantothenamide derivatives was recently reported by some of us.16 In this study, several compounds were synthesized and evaluated for antibacterial properties which revealed that larger substituents were not well tolerated at the gem-dimethyl position. This led to the identification of a methyl-allyl derivative (1) with potent antibacterial activity against both sensitive and resistant S. aureus.16 It was envisaged that these structure-activity relationships (SARs) could be extended through modification of the stereochemistry of the alkyl-substituted pantoyl fragment. In designing a target, we opted to focus on the 2-methyl-allyl derivative because of its clear superiority in antibacterial activity assays,16 and to maintain the R-configuration at C-3 based on previous studies suggesting that this is the preferred stereochemistry.2 We report here on a methodology for accessing the 2S,3R-syn allyl-substituted isomer (2), and on the contrasting profiles of diastereomers 1 and 2 with regards to antibacterial and antiplasmodial activities.
Synthetically, the pantothenamide structure has been obtained through a sequence of amide couplings on a modified pantoyl fragment.16 In the synthesis of geminal dialkyl-substituted pantothenamide derivatives, the stereochemistry at the quaternary carbon is determined by the initial configuration of the alcohol in the starting material, and can be controlled via two successive alkylations anti to the alcohol.16 This synthetic methodology, however, only provides access to 2R,3R-anti analogues diastereoselectively. Reversing the configuration at the quaternary center by reversing the order of the two alkylation reactions, e.g. adding the larger alkyl group before methylation, proceeds with poor selectivity and produces inseparable mixtures of 2R,3R-anti and 2R,3S-syn analogues.16–17 The difficulty in obtaining the syn product by reversing the alkylation sequence warrants an alternate synthetic route. In order to access the novel 2S,3R diastereomer, we envisaged inverting the stereochemistry of the fragment through oxidation, followed by diastereoselective reduction. We expected this last step to pose the greatest challenge, due to the unfavourable energetic barrier associated with forming the syn product. Thus, L-(−)-malic acid (3S-alcohol) is used here to access the syn isomer (2) through inversion of stereochemistry, while D-(−)-malic acid (3R-alcohol) was previously used directly to synthesize the anti isomer (1).16
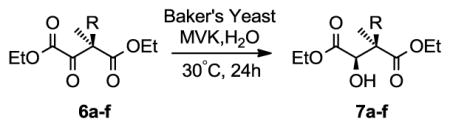
In order to generate the alkyl-substituted pantoyl fragment with the desired stereochemistry, L-(−)-malic acid was first esterified under mild acidic conditions (Scheme 1). The Frater-Seebach method of alkylating chiral β-hydroxy esters was used to install the methyl and various alkyl groups onto 3 with excellent diastereoselectivity.17 The addition of two equivalents of strong base generates a di-anion which forms a six-membered ring chelate.17 The stereoconfiguration of the secondary alcohol directs the electrophilic addition from the less hindered face, thereby yielding the anti product with a consistently high diastereomeric ratio (dr; as measured by NMR of the crude sample). Swern oxidation was used to afford the prochiral ketone 6a in good yield, which was subsequently reduced to the R-alcohol 7a.
Scheme 1.

Synthesis of compounds 7a–f.
Several commercially available chemical reducing agents were tested to evaluate their ability to yield the syn alcohol product from 6a. As shown in Table S1, typical agents such as DIBAL-H, NaBH4 and Zn2(BH4)2, as well as chiral reducing agents such as (R)-CBS and (S)-CBS showed poor diastereoselectivity and yielded predominantly the anti-product. The potential of biocatalysts was thus explored next. To this end, we envisaged utilizing a whole cell mixture of common Baker’s yeast (Saccharomyces cerevisiae) to reduce 6a to the syn product 7a diastereoselectively. There is precedence for Baker’s yeast to reduce α- and β-ketoesters to enantiopure alcohols.18 Baker’s yeast expresses several reducing enzymes which can be selectively inhibited or favoured by varying the reaction conditions.19 Pre-treatment of the yeast with cross-linking agents such as methyl-vinyl ketone (MVK), was found to favor syn-selectivity and prevent over-reduction to the diol.20 Heat-denaturation (50°C, 30 min) also achieved the same goal and even worked synergistically with MVK.21–22
Indeed, reduction of 6a with Baker’s yeast yielded the desired syn product (dr of >99:1) in 68% yield after purification. This high selectivity was only possible when the yeast was pre-incubated with MVK at 50°C for 30 min before addition of the ketone. The dr ratio was determined by integrating the characteristic NMR peaks for the methyl group at the quaternary carbon, specifically the signal at 1.16 ppm from the anti product,16 and the signal at 1.05 ppm from the syn product. The absolute stereochemistry of the product was confirmed by derivatization using enantiomeric auxiliary reagents and subsequent 1H NMR analysis as described by Seco et al.23 (R)- and (S)-methoxyphenylacetic acid (MPA) were thus coupled to 7a to generate the diastereoisomeric MPA derivatives for NMR analysis. To evaluate the scope of the Baker’s yeast reaction, a series of α-ketosuccinates (6a–f) was synthesized and reduced. The dr was measured by NMR on the crude product, and the yield was calculated after purification (Table 1). Interestingly, all compounds showed excellent diastereoselectivity (dr ≥ 98:1, with one exception at 80:20), although the yield of the reactions generally decreased with increasing bulk of the alkyl substituents, consistent with larger groups not being well tolerated in the binding pocket of the reductase of interest. The low yields observed in some of the examples are largely attributed to product recovery issues resulting from inefficient extraction from the complex matrix. Overall, Baker’s yeast shows excellent stereoselectivity for the reduction of these systems.
Table 1.
Baker’s yeast reduction of di-alkyl substituted α-ketomalonates.
| |||
|---|---|---|---|
| Compound | R | dra | Yieldb (%) |
| 7a | allyl | >99:1 | 68 |
| 7b | propargyl | 80:20 | 65 |
| 7c | ethyl | 98:1 | 37 |
| 7d | propyl | >99:1 | 31 |
| 7e | hexyl | >99:1 | 21 |
| 7f | isobutyl | 98:1 | 26 |
Diastereomeric ratio determined by NMR;
isolated yield.
With the stereochemistry established, we were able to extend the intermediates into full pantothenamides as shown in Scheme 2. As mentioned above, the methyl-allyl derivative 7a was selected for extension based on the reported antibacterial activity of 1.16 Thus, 7a was reduced to the corresponding triol using LiAlH4 and selectively protected as the six-membered ring anisaldehyde acetal 8. A Dess-Martin oxidation afforded the aldehyde 9, which was further oxidized to the acid 10 via Pinnick oxidation. Due to its instability, the crude acid (10) was used directly in the amide coupling to deprotected amine 13. Finally, the acetal protecting group of 11 was cleaved using 90% aqueous acetic acid to produce the desired pantothenamide 2.
Scheme 2.

Synthesis of compound 2.16
The 2S,3R-syn pantothenamide 2 was tested for antibacterial activity against S. aureus (sensitive and MRSA) but no appreciable effect could be detected at the highest concentration tested (500 μM; data not shown). This is in sharp contrast to the 2R,3R-anti pantothenamide 1, which previously showed potent antibacterial activity (MIC of 3.2 μM for both strains).9 Interestingly, this trend was reversed when 1 and 2 were evaluated for their antiplasmodial activity. As shown in Figure 2, although both diastereomers inhibited in vitro parasite growth, the syn derivative 2 (IC50 of 2.4 ± 0.2 μM, n = 6) appeared to be considerably more potent (p ≪ 0.001) when compared to its anti-isomer 1 (31 ± 4 μM, n = 3). Moreover, increasing the extracellular pantothenate concentration (from 1 to 50 μM) resulted in a higher IC50 for 2 (13 ± 1 μM, n = 3, p ≪ 0.001), consistent with 2 inhibiting parasite growth by targeting CoA biosynthesis or utilization (Figure 2). Compounds 1 and 2 were also tested in human cells and did not show significant toxicity (see Figure S1).
Fig. 2.

Antiplasmodial activity of the syn-isomer 2 in medium containing 1 μM pantothenate (black circles) or 50 μM pantothenate (white circles), in comparison with the anti-isomer 1 in medium containing 1 μM pantothenate (black triangles). Error bars represent SEM from 3–6 independent experiments.
A successful method was established for the synthesis of 2S,3R-syn 2-methyl-alkyl pantothenamide derivatives. A key step of this process involved the use of Baker’s yeast for the stereoselective reduction of α-ketosuccinates. Stereochemical modification of the pantoyl fragment significantly influenced the biological activity and antimicrobial selectivity of these compounds. This suggests that such alterations may be applied to study other CoA-utilizing systems. For example, N-substituted pantothenamides have found use as chemical biology tools and as mechanistic probes of CoA-dependent enzymes. Specific applications include both in vitro and in-cell labelling of carrier proteins,24–26 as well as prodrug activation by the CoA biosynthetic pathway.27 Thus, we expect the methodology developed herein to find utility in various areas.
Supplementary Material
Acknowledgments
This work was supported by the Center in Green Chemistry and Catalysis, as well as by research grants from the Canadian Institute of Health Research (CIHR) and the Natural Sciences and Engineering Research Council of Canada (NSERC) to K.A. A.H. thanks CIHR and the CIHR-McGill Drug Development Training Program (DDTP) for financial support. We are grateful to the Canberra branch of the Australian Red Cross Blood Service for the provision of erythrocytes and to Dr. Giel van Dooren (ANU) for assistance with the HFF assays.
Footnotes
Supporting information available, including experimental procedures, characterization of all compounds, as well as Figure S1, NMR spectra and selected HPLC traces. This material is available via the Internet.
References
- 1.Vicente M, Hodgson J, Massidda O, Tonjum T, Henriques-Normark B, Ron EZ. FEMS Microbiol Rev. 2006;30:841. doi: 10.1111/j.1574-6976.2006.00038.x. [DOI] [PubMed] [Google Scholar]
- 2.Spry C, Kirk K, Saliba KJ. FEMS Microbiol Rev. 2008;32:56. doi: 10.1111/j.1574-6976.2007.00093.x. [DOI] [PubMed] [Google Scholar]
- 3.Genschel U. Mol Biol Evol. 2004;21:1242. doi: 10.1093/molbev/msh119. [DOI] [PubMed] [Google Scholar]
- 4.Gerdes SY, Scholle MD, D’Souza M, Bernal A, Baev MV, Farrell M, Kurnasov OV, Daugherty MD, Mseeh F, Polanuyer BM, Campbell JW, Anantha S, Shatalin KY, Chowdhury SA, Fonstein MY, Osterman AL. J Bacteriol. 2002;184:4555. doi: 10.1128/JB.184.16.4555-4572.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clifton G, Bryant SR, Skinner CG. Arch Biochem Biophys. 1970;137:523. doi: 10.1016/0003-9861(70)90470-4. [DOI] [PubMed] [Google Scholar]
- 6.Choudhry AE, Mandichak TL, Broskey JP, Egolf RW, Kinsland C, Begley TP, Seefeld MA, Ku TW, Brown JR, Zalacain M, Ratnam K. Antimicrob Agents Chemother. 2003;47:2051. doi: 10.1128/AAC.47.6.2051-2055.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strauss E, Begley TP. J Biol Chem. 2002;277:48205. doi: 10.1074/jbc.M204560200. [DOI] [PubMed] [Google Scholar]
- 8.Zhang YM, Frank MW, Virga KG, Lee RE, Rock CO, Jackowski S. J Biol Chem. 2004;279:50969. doi: 10.1074/jbc.M409607200. [DOI] [PubMed] [Google Scholar]
- 9.Thomas J, Cronan JE. Antimicrob Agents Chemother. 2010;54:1374. doi: 10.1128/AAC.01473-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Virga KG, Zhang YM, Leonardi R, Ivey RA, Hevener K, Park HW, Jackowski S, Rock CO, Lee RE. Bioorg Med Chem. 2006;14:1007. doi: 10.1016/j.bmc.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 11.de Villiers M, Macuamule C, Spry C, Hyun YM, Strauss E, Saliba KJ. ACS Med Chem Lett. 2013;4:784. doi: 10.1021/ml400180d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saliba KJ, Kirk K. Mol Biochem Parasitol. 2005;141:129. doi: 10.1016/j.molbiopara.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 13.Saliba KJ, Ferru I, Kirk K. Antimicrob Agents Chemother. 2005;49:632. doi: 10.1128/AAC.49.2.632-637.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spry C, Chai CL, Kirk K, Saliba KJ. Antimicrob Agents Chemother. 2005;49:4649. doi: 10.1128/AAC.49.11.4649-4657.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spry C, Macuamule C, Lin Z, Virga KG, Lee RE, Strauss E, Saliba KJ. PLoS One. 2013;8:e54974. doi: 10.1371/journal.pone.0054974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akinnusi TO, Vong K, Auclair K. Bioorg Med Chem. 2011;19:2696. doi: 10.1016/j.bmc.2011.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frater G, Mueller U, Guenther W. Tetrahedron. 1984;40:1269. [Google Scholar]
- 18.Nakamura K, Yamanaka R, Matsuda T, Harada T. Tetrahedron: Asymmetry. 2003;14:2659. [Google Scholar]
- 19.Shieh WR, Gopalan AS, Sih CJ. J Am Chem Soc. 1985;107:2993. [Google Scholar]
- 20.Nakamura K, Kawai Y, Miyai T, Ohno A. Tetrahedron Lett. 1990;31:3631. [Google Scholar]
- 21.Nakamura K, Kawai Y, Ohno A. Tetrahedron Lett. 1991;32:2927. [Google Scholar]
- 22.Nakamura K, Kondo S, Kawai Y, Hida K, Kitano K, Ohno A. Tetrahedron: Asymmetry. 1996;7:409. [Google Scholar]
- 23.Seco JM, Quinoa E, Riguera R. Tetrahedron: Asymmetry. 2001;12:2915. [Google Scholar]
- 24.Clarke KM, Mercer AC, La Clair JJ, Burkart MD. J Am Chem Soc. 2005;127:11234. doi: 10.1021/ja052911k. [DOI] [PubMed] [Google Scholar]
- 25.Meier JL, Mercer AC, Rivera H, Jr, Burkart MD. J Am Chem Soc. 2006;128:12174. doi: 10.1021/ja063217n. [DOI] [PubMed] [Google Scholar]
- 26.Worthington AS, Burkart MD. Org Biomol Chem. 2006;4:44. doi: 10.1039/b512735a. [DOI] [PubMed] [Google Scholar]
- 27.Vong K, Tam IS, Yan X, Auclair K. ACS Chem Biol. 2012;7:470. doi: 10.1021/cb200366u. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.