

Abstract
Because of its impact on multiple biological pathways, heparanase has emerged as a major regulator of cancer, inflammation and other disease processes. Heparanase accomplishes this by degrading heparan sulfate which regulates the abundance and location of heparin-binding growth factors thereby influencing multiple signaling pathways that control gene expression, syndecan shedding and cell behavior. In addition, heparanase can act via non-enzymatic mechanisms that directly activate signaling at the cell surface. Clinical trials testing heparanase inhibitors as anti-cancer therapeutics are showing early signs of efficacy in patients further emphasizing the biological importance of this enzyme. This review focuses on recent developments in the field of heparanase regulation of cancer and inflammation, including the impact of heparanase on exosomes and autophagy, and novel mechanisms whereby heparanase regulates tumor metastasis, angiogenesis and chemoresistance. In addition, the ongoing development of heparanase inhibitors and their potential for treating cancer and inflammation are discussed.
Keywords: heparan sulfate, proteoglycan, heparanase, cancer, inflammation, autophagy, exosomes, heparanase inhibitors, metastasis, angiogenesis
Graphical Abstract
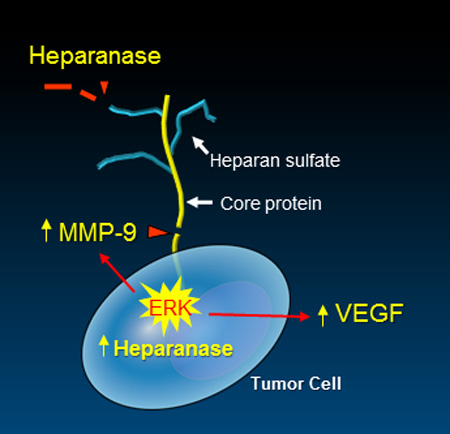
By degrading heparan sulfate, heparanase impacts multiple signaling pathways that control gene expression, syndecan shedding and cell behavior. Heparanase also activates signaling at the cell surface via non-enzymatic mechanisms. This review focuses on recent developments that provide new insight into mechanisms of heparanase-mediated regulation of cancer and inflammation, including its impact on exosomes, autophagy, angiogenesis, chemoresistance, cell migration and metastasis.
Heparanase in cancer
Heparanase is an endoglucuronidase that cleaves heparan sulfate, thereby regulating the structure and function of heparan sulfate proteoglycans and remodeling cell surfaces and the extracellular matrix. Much of our current knowledge regarding heparanase function is related to cancer, and numerous comprehensive reviews are available on that subject [1–3]. There is abundant evidence that heparanase plays a role in cancer. Analyses at the RNA or protein level demonstrate that heparanase expression is enhanced in almost all cancers examined to date including, for example, ovarian, pancreatic, myeloma, colon, bladder, brain, prostate, breast, liver and rhabdomyosarcoma [4–14]. Numerous clinical studies have consistently demonstrated that upregulated heparanase expression correlates with increased tumor size, tumor progression, enhanced metastasis and poor prognosis [1, 14–18]. Knockdown of heparanase expression or treatment of tumor bearing mice with compounds that inhibit heparanase enzyme activity markedly impair tumor progression further underscoring the potential of anti-heparanase therapy for multiple types of cancer [19–25]. Importantly, there is only a single, enzymatically active form of heparanase in humans, it is expressed in very low levels in normal tissues and heparanase knock-out animals exhibit no obvious deficits [17] implying that inhibition of heparanase will cause minimal side effects in cancer patients. Together these findings elevate heparanase as a highly desirable and druggable target for anti-cancer therapy, a view widely held by researchers in both academia and pharma and the topic of multiple reviews [1, 3, 15, 16, 21, 26–28].
Mechanistically, by cleaving heparan sulfate chains, the heparanase enzyme alters the structure and function of heparan sulfate proteoglycans and contributes to tumor-mediated remodeling of both cell surfaces and the extracellular matrix. These actions dynamically impact multiple regulatory pathways, most notably by augmenting the bioavailability of growth factors and cytokines bound to heparan sulfate [26]. In addition, heparanase expression by tumor cells initiates upregulation of expression of multiple genes that promote aggressive tumor behavior including vascular endothelial growth factor (VEGF), hepatocyte growth factor (HGF) and matrix metalloproteinase-9 (MMP-9) [29–33], among others. In addition, recent discoveries indicate that a major function of heparanase is to enhance the shedding of the cell surface proteoglycan syndecan-1 [18, 26, 34]. Shed syndecan-1 plays diverse roles in the tumor microenvironment including shuttling growth factors to both tumor and host cell surfaces and nucleating the formation of signaling complexes at the cell surface (discussed in detail below). For additional information on syndecans in cell signaling see ref. [35] in this minireview series.
Heparanase regulation of tumor progression via exosomes and autophagosomes
Heparanase regulates biogenesis, composition, and function of tumor cell-derived exosomes
Exosomes are powerful mediators of intercellular communication that drive tumor progression by regulating the behavior of tumor and host cells both locally within the tumor microenvironment and distally throughout the body [36]. Exosomes accomplish this regulatory function by docking with recipient cells and delivering their cargo of protein, DNA, mRNA and miRNA [36]. In cancer, secretion of exosomes often increases as tumors transit toward a more aggressive phenotype. The tumor-host crosstalk mediated by exosomes has multiple effects that can influence processes such as formation of the pre-metastatic niche, angiogenesis and host immune function [37–39]. Syndecan proteoglycans influence the biogenesis of exosomes through their interaction with the syntenin-ALG-2 interacting protein X (ALIX) complex [38, 40, 41]. Syntenin interacts with the syndecan core protein via two PDZ domains and to ALIX via three LYPXnL motifs [40]. ALIX then binds to ESCRT-III (endosomal-sorting complex required for transport), the machinery responsible for intraluminal vesicle formation at multivesicular endosomes. Importantly, heparanase activates the syndecan-syntenin-ALIX exosome pathway [40, 42]. Briefly, heparanase activity in endosomes trims long heparan sulfate chains into shorter ones, allowing clustering of syndecans through lateral interactions between their heparan sulfate chains [42, 43]. Heparanase-induced clustering is thought to stimulate the binding of syndecan cytoplasmic domains to the tandem PDZ domains of syntenin, driving ALIX-ESCRT-mediated sorting into exosomes [42–44]. Notably, heparanase activity also facilitates the recruitment of CD63 into exosomes, in a syntenin-dependent manner [42, 43]. Together, a complex picture is emerging in which syndecans, CD63 and possibly other membrane proteins that associate with endosomal syndecan and/or tetraspanin-enriched microdomains, are sorted into exosomes by a shared heparanase-syndecan-syntenin-ALIX pathway machinery [43, 44]. Heparanase-mediated effects on exosome biogenesis are best explained by heparanase acting on syndecan, promoting the assembly of syndecan in complexes that, by recruiting syntenin and ALIX–ESCRT, promote endosomal membranes to bud [44]. From that perspective, heparanase inhibitors as well as specific syntenin-PDZ inhibitors might be of particular interest for cancer, where both exosome release and heparanase are often elevated in the more aggressive forms of disease [26, 45]. In support of the above considerations, we have reported that in human cancer cells, when expression of heparanase is enhanced, or when tumor cells are exposed to exogenous heparanase, exosome secretion is dramatically increased [45]. This appears to rely on its enzymatic cleavage of heparan sulfate, because enzymatically inactive forms of the enzyme do not suffice [45]. Thus, heparanase released by tumor or host cells (e.g., macrophages) could diffuse within the microenvironment, impact neighboring tumor cells and enhance, among other effects, their secretion of exosomes. Heparanase also impacts exosome protein cargo as reflected by higher levels of syndecan-1, VEGF and HGF in exosomes secreted by heparanase-high expressing cells as compared to heparanase-low expressing cells [45]. In functional assays, exosomes from heparanase-high cells stimulated spreading of tumor cells on fibronectin and invasion of endothelial cells through extracellular matrix better than did exosomes secreted by heparanase-low cells, suggesting a role in promoting tumor cell spreading and angiogenesis [45].
Our finding that heparanase is present in exosomes raises the possibility that this is a means for its delivery to distal locations. Because of the known role of heparanase in promoting metastasis and angiogenesis, this may play a role in establishing niches to which tumor cells eventually home and grow. Our results indicate that heparanase promotes secretion of exosomes that interact with both tumor and host cells and drive them toward an aggressive tumor phenotype. Emerging data indicate that exosomes can act as barriers to anti-cancer therapy by interacting with tumor cells and enhancing their chemoresistance [46]. In fact, our ongoing studies reveal that heparanase enhances both exosome docking and exosome-mediated transfer of chemoresistance to tumor cells [47]. Collectively, it appears that heparanase helps drive exosome secretion, alters exosome composition and facilitates production and docking of exosomes that impact both tumor and host cell behavior thereby promoting tumor progression and chemoresistance [45].
Heparanase enhances tumor growth and chemoresistance by augmenting autophagy
In spite of its localization in a highly active protein degradation environment such as the lysosome, heparanase appears stable [48, 49] and exhibits a half-life of about 30 hours [50], relatively long compared with a t1/2 of 2–6 hours for transmembrane HSPGs and 25 minutes for GPI-anchored HSPGs [51]. Residence and accumulation of heparanase in lysosomes indicate that the enzyme may function in the normal physiology of this organelle. In a search for such function we revealed a role of lysosomal heparanase in modulating autophagy [52]. Autophagy is an evolutionarily conserved catabolic pathway through which cytoplasmic components, including macromolecules such as proteins and lipids as well as whole organelles, are sequestered into double-membrane vesicles called autophagosomes. Autophagosomes are subsequently fused with lysosomes, where the intracellular material is degraded and recycled. This process occurs at a basal level in every cell and is required to remove unfolded proteins and damaged organelles, thus maintaining cellular homeostasis. Autophagy is further induced by starvation and stress, promoting cancer cells survival by providing their metabolic needs [53, 54]. Our results indicate that heparanase promotes autophagy and that enhanced tumor growth and chemoresistance exerted by heparanase is mediated in part by augmenting autophagy [52]. This was concluded because reduced LC3-II (a protein that specifically associates with autophagosomes) levels are found in cells and tissues obtained from heparanase knockout mice as opposed to elevated LC3-II levels found in transgenic mice that over express heparanase [52]. Even higher induction of autophagy was evident in head and neck carcinoma and glioma cells over-expressing heparanase, in accordance with a strong pre-clinical and clinical correlation between heparanase expression and the progression of these malignancies [33, 55–60]. Notably, electron microscopy analyses of cells over-expressing heparanase revealed not only a higher number of autophagic vacuoles, but also abundant release of vesicles, likely exosomes, from the cell surface, further supporting the notion that heparanase enhances exosome secretion that contributes to tumor growth [45, 61] (Fig. 1).
Figure 1.
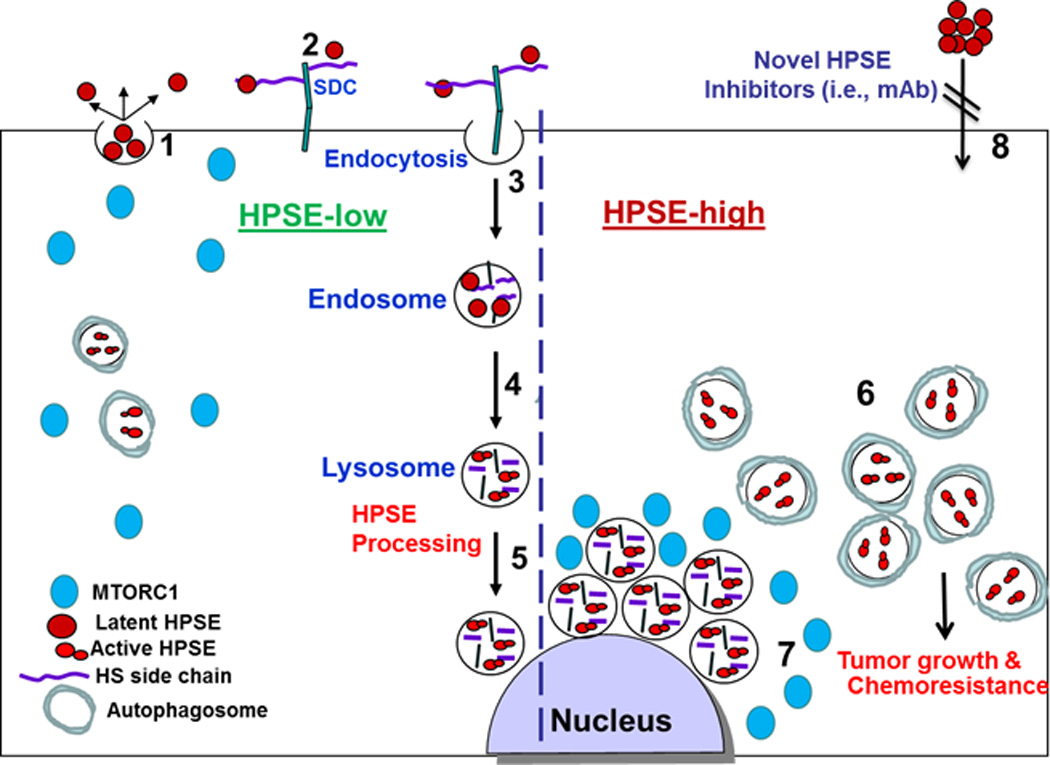
A schematic model of heparanase trafficking and function in autophagy. Once secreted (1), heparanase rapidly interacts with cell membrane HSPGs such as syndecans (SDC) (2), followed by a rapid endocytosis of the heparanase-HSPG complex (3). Conversion of endosomes to lysosomes (4) results in heparanase processing and activation (5). Typically, heparanase appears at perinuclear lysosomal vesicles (5). Lysosomal heparanase regulates the basal level of autophagy and resides within autophagosomes (HPSE-low). Cancer cells that exhibit high content of heparanase (HPSE-high) are endowed with increased autophagy (6) that promotes tumor growth and chemo resistance. Enhanced autophagy by heparanase is associated with reduced p70 S6-kinase phosphorylation levels and accumulation of mTOR1 at peri-nuclear areas (7) vs. more diffused distribution in control (HPSE-low) cells. Function of heparanase within the cell encourages the development of new class of inhibitors that will prevent heparanase uptake and lysosomal accumulation (8).
The mechanism underlying autophagy induction by heparanase is not entirely clear, but likely involves mTOR1 that plays a pivotal role in nutrient-sensing and autophagy regulation [62]. mTOR1 activity inhibits autophagy but under starvation its activity is repressed, leading to autophagy induction. We found that heparanase over-expression correlates with reduced mTOR1 activity, evident by decreased levels of p70 S6-kinase phosphorylation, an mTOR1 substrate. In contrast, heparanase-knockout cells exhibited increased mTOR1 activity and p70 S6-kinase phosphorylation [52]. Notably, mTOR1 appeared more diffusely scattered in control cells, whereas in cells with high content of heparanase, mTOR1 is found mostly in perinuclear regions, co-localizing with heparanase and LysoTracker, a dye that labels acidic lysosomal vesicles (Fig. 1). Our results imply that autophagy induction contributes to the pro-tumorigenic function of heparanase. This emerges from in vitro and in vivo experiments utilizing inhibitors of autophagy (chloroquine) and heparanase (PG545) alone or in combination [52]. Thus, combining chloroquine and PG545 in a tumor xenograft model resulted in significantly smaller and more differentiated tumors, suggesting that heparanase activity drives cancer cell de-differentiation as part of its pro-tumorigenic properties. Equally important is the ability of heparanase over-expression to confer resistance to stress, chemotherapy and targeted drugs [63], mediated, at least in part, by enhancing autophagy [52]. Indeed, diverse classes of anticancer drugs induce autophagy [64], thus attenuating tumor cell elimination, while autophagy inhibitors overcome chemoresistance [65, 66]. Based on this concept, chloroquine is currently being evaluated in clinical trials in combination with different classes of chemotherapeutic agents [65].
While traditional thinking envisions heparanase as an enzyme that functions extracellularly to cleave heparan sulfate and facilitate remodeling and ‘priming’ of the extracellular matrix (ECM), our results indicate that heparanase may also function inside cells [67]. From a translational point of view, targeting heparanase in the lysosome may be as important as its inhibition extracellularly, but the ability of currently available heparanase inhibitors to cross the plasma membrane and enter the cell is unclear. Alternatively, the pro-autophagy function of heparanase can be inhibited by inhibiting its cellular uptake and hence decreasing its lysosomal content [67]. This opens the way for the development of a new class of highly specific inhibitors (i.e., monoclonal antibodies) that prevent heparanase uptake by targeting its heparin-binding domain. Involvement of heparanase in exosome formation, autophagy and activation of innate immune cells (discussed below) indicate that it fulfills normal functions associated, for example, with vesicular traffic, lysosomal secretion, stress response, heparan sulfate turnover and immune surveillances. Unraveling these aspects of heparanase biology is ongoing and critical to our understanding of its multiple roles in health and disease. Interestingly, in addition to heparanase, proteoglycans have also been implicated in regulation of autophagy and inflammation and are the subject of a minireview within this series [68].
A novel heparanase-driven mechanism promoting both metastasis and angiogenesis
Metastasis is a multi-step process regulated by enzymes, growth factors and signaling from adhesion receptors [69, 70]. Historically, heparanase is thought to stimulate metastasis and angiogenesis by degrading extracellular matrix, thereby liberating heparan sulfate-bound growth factors and chemokines from the extracellular matrix or cell surfaces. These growth factors are then free to interact with high affinity signaling receptors on the surface of tumor or host cells. Using human myeloma cells as a model, we recently discovered a mechanism that shines new light on how heparanase promotes both metastasis and angiogenesis. Key to this mechanism is the ability of heparanase to promote shedding of syndecan-1. The heparan sulfate degrading activity of heparanase shortens the length of heparan sulfate chains on syndecan-1 leaving the core protein vulnerable to attack by proteases [71]. Heparanase also mediates upregulation of MMP-9 expression by tumor cells. MMP-9 cleaves the juxtamembrane region of syndecan-1 thereby releasing an intact ectodomain from the cell surface [29] [23]. (Fig. 2).
Figure 2.
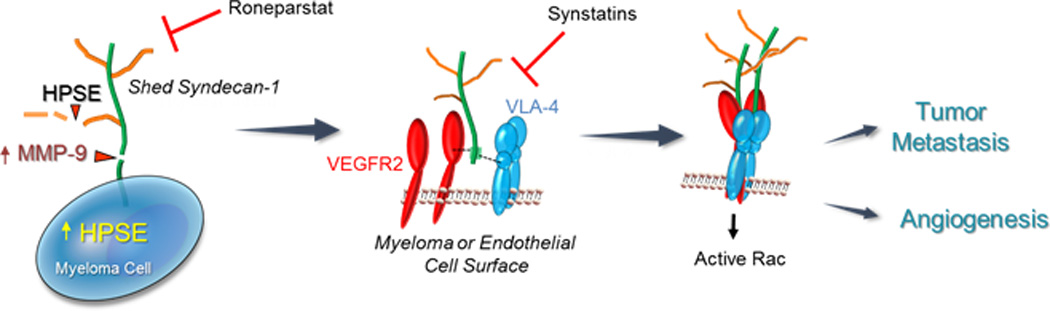
Heparanase activates a signaling mechanism that drives both tumor cell invasion and angiogenesis. (Left Panel) Myeloma cells express syndecan-1 on their cell surface composed of a core protein (green) and heparan sulfate chains (brown). Upregulation of heparanase (HPSE) expression by myeloma cells leads to trimming of syndecan-1 heparan sulfate chains, shortening their length and allowing increased access of proteases to the exposed syndecan-1 core protein. One such protease is MMP-9, a syndecan-1 sheddase whose expression is upregulated when heparanase is expressed by myeloma cells. MMP-9 cleaves the syndecan-1 core protein and the proteoglycan is shed from the cell surface. (Center Panel) Shedding of syndecan-1 exposes a cryptic domain within the juxtamembrane region of the core protein (green). Within this cryptic domain are amino acid sequences that bind to clustered VLA-4 (blue) and VEGFR2 (red) on the surface of myeloma cells or endothelial cells. (Right Panel) The coupling of VLA-4 and VEGFR2 receptors by shed syndecans activates VEGFR2 signaling that stimulates both cell invasion and endothelial tube formation. This signaling mechanism is inhibited by Roneparstat, a heparanase inhibitor that diminishes syndecan-1 shedding, or by synstatin peptides, peptide mimics of the syndecan-1 core protein that competitively inhibit binding of either VLA-4 or VEGFR-2 to shed syndecan-1.
Shedding exposes a cryptic domain on the syndecan-1 core protein that contains binding sites for very late antigen 4 (VLA-4) and vascular endothelial growth factor receptor 2 (VEGFR2). Coupling of these receptors by shed syndecan-1 activates VEGFR2 and localizes the receptor complex to the leading edge of the tumor cell where it stimulates invasion [72] (Fig. 2). Remarkably, this same heparanase-dependent mechanism is in play on endothelial cells where it potentiates endothelial tube formation. This mechanism adds to a growing list of receptor tyrosine kinases (IGF-1R, HER2, and EGFR) that rely on syndecan-mediated docking to an integrin in order to carry out critical roles in tumorigenesis and angiogenesis. As is the case here, the interactions are extracellular and accessible to synthesized peptides that mimic the capture site in the syndecan. These peptides are called “synstatins” or “SSTNs” and act to competitively disrupt the signaling mechanism [73–77]. In the case of VEGFR2 coupling to VLA-4, peptides based on either the VEGFR2 docking site (SSTNVEGFR2) or the VLA-4 docking site (SSTNVLA-4) in syndecan-1 serve to prevent tumor cell invasion or endothelial tube formation [72] (Fig. 2). This signaling mechanism is also highly dependent on heparanase as the initiating step, identifying a key and unanticipated modulating role for this enzyme. This role was confirmed by use of the heparanase inhibitor Roneparstat, a chemically modified non-anticoagulant heparin that diminishes syndecan-1 shedding and subsequent tumor invasion and angiogenesis [72].
Framing this information into the known steps of tumor cell metastasis, we envision the following. Syndecan-1 shed by tumor cells nucleates the coupling of VEGFR2 to VLA-4, initiating tumor cell migration out of an established lesion and into the circulation. To extravasate to a new site, the circulating tumor cells must bind and migrate through the endothelial cell layer of bone marrow capillaries, enter the bone marrow stroma, stimulate angiogenesis and grow. Binding of tumor cells to the vascular endothelium occurs via interaction of VLA-4 with vascular cell adhesion molecule 1 (VCAM-1), an adhesion receptor abundant on bone marrow endothelia [78, 79]. Although binding of tumor cells to VCAM-1 can occur in the absence of heparanase, the invasion through the endothelial layer and throughout the bone marrow stroma is likely to depend on heparanase, as it facilitates the invasive phenotype arising from the coupling of VEGFR2 to VLA-4 [72]. In addition, as cells invade, degradation of the heparan sulfate-rich subendothelial basement membrane is also facilitated by heparanase. The invading cells not only rely on VCAM-1 as they transit the endothelium, but also rely on it within the bone marrow as VCAM-1 is highly expressed on bone marrow stromal cells, along with fibronectin, another VLA-4 ligand, that is enriched in the bone marrow matrix. This same mechanism is likely to potentiate local angiogenesis, induced in part by heparanase expression and stimulation of syndecan-1 shedding in bone marrow endothelial cells, and potentially supplemented in part by syndecan-1 shed into the local environment from the tumor cells. In addition, heparanase can also potentiate VEGF dependent angiogenesis by enhancing VEGF expression in the tumor cells [30]. Finally, tumor cells that acquire this invasive phenotype due to heparanase-mediated shedding of syndecan-1 within the bone marrow may display a heightened ability to rely on it to re-enter the circulation, and engage and invade through the vascular endothelium at a distant site in a cyclic process that drives the spread of blood-borne tumor cells throughout the body.
Regarding the use of heparanase inhibitors to block tumor progression, at present there are three heparan sulfate mimics in early stage clinical trials in cancer patients, Roneparstat, Necuparanib and PG545 [22, 23, 25], for reviews see [1, 16, 26] [2]. Recently, using animal models, Roneparstat was found to be highly effective when used in combination with front line chemotherapeutic agents like bortezomib or melphalan against established and aggressive myeloma tumors growing within bone, or when used in combination with lapatinib to treat brain metastatic breast cancer [80, 81]. Additional opportunities for development of anti-heparanase therapeutics include monoclonal antibodies and use of the recently published crystal structure of heparanase for identification of small molecule inhibitors [82].
Heparanase in inflammation
The involvement of heparanase in immune reactions was first suggested by studies demonstrating heparan sulfate-degrading activity in immunocytes (neutrophils, macrophages, activated T-lymphocytes) that contribute to immune cell diapedesis and accumulation in target organs [83–89]. A role for heparanase in inflammatory responses was further supported by the finding that inhibitors of heparanase enzyme activity (i.e., heparin, synthetic heparin-mimicking compounds) had anti-inflammatory effects both in experimental and clinical settings [90–95].
The majority of early studies on the role of heparanase in inflammation focused on its ability to promote extravasation of immune cells. However, because heparan sulfate controls inflammatory responses at multiple levels, including sequestration of cytokines/chemokines in extracellular space, modulation of leukocyte interactions with endothelium and ECM, and initiation of innate immune responses through interactions with toll-like receptors (TLR) [96–104], enzymatic remodeling of heparan sulfate by heparanase appears to affect several aspects of inflammatory reactions. These include leukocyte recruitment, migration toward sites of inflammation, release of cytokines and chemokines anchored within the ECM or cell surfaces, as well as activation of innate immune cells.
Expanding a previous notion that immunocytes represent the principal source of the enzyme, more recent reports reveal a variety of cellular sources of heparanase in inflammation. Induction of heparanase was found to occur largely in epithelial and/or endothelial compartments in numerous inflammatory settings, including in vivo models of delayed type hypersensitivity [94], vascular injury [105], inflammatory bowel disease [106, 107], sepsis-associated lung injury [108] and autoimmune diabetes [109]. In addition, heparanase induction was found in auto-immune and auto-inflammatory human disorders, including rheumatoid arthritis [110], chronic obstructive pulmonary disease [111], Dengue disease [112], pleural empyema [113], inflammatory lung disease [108], ulcerative colitis and Crohn’s disease [107, 114]. Heparanase expression is induced in the presence of inflammatory cytokines [94, 107, 108, 115] or bacterial/viral infection [112, 113, 116].
In parallel with elucidation of cellular sources of the enzyme, accumulating experimental data reveal a complex picture of mechanisms employed by heparanase to modulate inflammatory responses. While many of these mechanisms (discussed below) are mediated by its well-characterized enzymatic function performed at the cell surface and within the extracellular compartment, other actions involve transcriptional regulation of the inflammatory phenotype in endothelial and immune cells by intracellular heparanase localized to the nucleus [117, 118]. Additionally, by virtue of heparanase ability to influence cell signaling independently of its enzymatic function, it was suggested that heparanase affects inflammatory cell responses via an unidentified cell surface receptor [119].
Mounting evidence suggests that heparanase profoundly influences the molecular physiology of innate immunocytes, including phagocytes (i.e., neutrophils, macrophages, dendritic cells), mast cells, and eosinophils [107, 108, 111, 120–125]. Inflammatory conditions in lungs seem to be one of the most extensively investigated anatomic sites in this respect [108, 111, 113, 126]. A recent report focused on heparanase-mediated degradation of the endothelial glycocalyx [108]. The glycocalyx, a thin gel-like layer that covers the luminal surface of endothelial cells lining blood vessels, is composed of heparan sulfate proteoglycans and glycoproteins [127, 128]. It acts as a barrier to circulating cells by limiting the availability of endothelial surface adhesion molecules to leukocytes [127, 128]. In a mouse model of sepsis-associated inflammatory lung disease, rapid induction of heparanase activity (via a tumor necrosis factor alpha-dependent mechanism) was demonstrated in pulmonary microvascular endothelial cells and correlated with neutrophil recruitment [108]. Heparanase induction was also found in biopsies of human inflammatory lung disease [108]. According to this report, sepsis associated loss of the pulmonary glycocalyx and endothelial hyperpermeability were attenuated in heparanase-null mice and in mice treated with inhibitors of heparanase enzymatic activity [108]. Another study, utilizing a dorsal air pouch inflammation model demonstrated that heparastatin (an iminosugar-based inhibitor of heparanase) potentially suppresses extravasation of neutrophils and monocytes by impairing the degradation of basement membrane heparan sulfate [129]. On the other hand, several recent reports combining heparanase knock out approach and/or inhibitors with in vivo models of airway inflammation found no significant effect of the enzyme in neutrophil recruitment/entrapment in the lung vasculature [111, 126]. Nevertheless, heparanase was critical for neutrophil accumulation in smoke-exposed lungs [126]. Even more perplexing, constitutive overexpression of heparanase in heparanase transgenic (Hpa-tg) mice was shown to attenuate intraluminal crawling of neutrophils in microvessels toward an extravascular chemokine source, reportedly due to reduction in endothelial surface heparan sulfate chain length and altered ability of truncated heparan sulfate to serve as a ligand for chemokines [120]. Additionally, studies exploring acute inflammatory phenotypes [130, 131] in Hpa-tg mice demonstrated that neutrophil recruitment and activation were attenuated in the presence of constitutively increased levels of heparanase. In light of the reported anti-inflammatory effects of heparin [90], increased levels of highly sulphated, "heparin-resembling" heparan sulfate fragments, which are constantly present in Hpa-tg as compared to wild type mice [132], may offer an explanation for the inhibitory effects of continuous heparanase overexpression on neutrophils in these settings [130, 131].
Unlike heparanase influence on neutrophils, its ability to modulate pro-inflammatory macrophage action remains less disputable and was highlighted in the setting of inflammatory bowel disease [107, 114], diabetic complications [133], pancreatic carcinoma associated inflammation [134], neointimal lesions following vascular injury [105] and atherosclerotic plaque progression toward vulnerability [121]. Modulation of toll-like receptor (TLR) signaling provides an attractive explanation for heparanase-mediated change in macrophage phenotype. Intact extracellular heparan sulfate inhibits TLR4 responses and macrophage activation, while its removal relieves this inhibition [102]. Indeed, incubation with active heparanase enzyme reduces the amount of intact heparan sulfate on the macrophage cell surface by 50% and significantly increases binding of fluorescent-labeled LPS (TLR4 ligand) by macrophages in vitro [107, 134], suggesting that degradation of cell-surface heparan sulfate by heparanase increases accessibility of the TLR. On the other hand, soluble heparan sulfate fragments released by heparanase degradation [135, 136], were found to stimulate TLR (in particular TLR4) signaling in vitro [102, 103, 137, 138] and in vivo [104].
The complexity of heparanase action in inflammatory processes is best exemplified by multiple levels of the enzyme involvement in type 1 diabetes and diabetic complications. In autoimmune diabetes, multiple roles were identified for heparanase produced by islet autoreactive T cells and inflammatory leukocytes. These roles include promotion of leukocyte migration from pancreatic blood vessels (i.e., across the sub-endothelial basement membrane and through the pancreatic ECM), aiding the passage of leukocytes across the islet basement membrane and depleting islet beta cells of the intracellular heparan sulfate needed for their survival [109, 139, 140]. In addition, heparanase was implicated in several inflammation-related complications of diabetes, notably – diabetic retinopathy [141] and diabetic nephropathy [133]. The latter condition is characterized by activation of immune cells and there is clear evidence for a significant role of chronic inflammation in its pathogenesis, highlighting the role of kidney-infiltrating macrophages [142–145]. In the diabetic kidney, macrophages activated by various elements of the diabetic milieu (e.g., high glucose [146], AGE [147, 148], albumin [149], free fatty acids [150]), release reactive oxygen species and proinflammatory cytokines such as tumor necrosis factor alpha or IL- 6 causing injury to podocytes and tubular cells [143, 144, 151]. Under these conditions heparanase that is overexpressed and post-translationally activated by Cathepsin L sustains continuous activation of kidney-damaging inflammatory macrophages eventually fostering chronic inflammation and renal injury [133].
Conclusions and perspectives
As investigation of heparanase continues, new and important roles for the enzyme are emerging. Recent studies demonstrating a role for heparanase in exosome formation, autophagy and activation of innate immune cells have further widened the scope of its influence. In addition, even though heparanase has long been associated with enhanced tumor metastasis and angiogenesis, the surprising discovery of a novel mechanism whereby heparanase induces shedding of syndecan-1 that then couples VEGFR2 and VLA-4 at the cell surface to promote metastasis and angiogenesis reminds us that there is still much to learn about mechanisms of heparanase action. Also, central to many of the downstream impacts of heparanase is its ability to regulate expression of genes for effectors such as HGF, MMP-9 and VEGF, yet our understanding of how heparanase regulates gene expression is not complete. Lastly, a remaining challenge in the field rests in the development of clinically effective inhibitors of heparanase that can be used to treat cancer and inflammatory diseases. The inhibitors currently in clinical trials are all modified heparins or heparin mimics and although they may prove effective, other more highly specific inhibitors such as monoclonal antibodies and small molecule chemical inhibitors are yet to be exploited. Further unraveling the mechanisms of action of heparanase and developing effective inhibitors of this enzyme are critical to our understanding of its multiple roles in health and disease.
Acknowledgments
This study was supported by research grants awarded to R.S. (CA138340) and to A.R. (CA139872) by the National Institutes of Health; to M.E. by the Israel Science Foundation (grant 806/14), the Mizutani Foundation for Glycoscience and the Legacy Heritage Biomedical Program of the Israel Science Foundation (grant No. 666/16); to I.V. by the Israel Science Foundation (grant 601/14), the ISF-UGB joint research program (grant No. 2277/15), and the Israel Cancer Research Fund (ICRF); and jointly to I.V. and R.S. by the United States-Israel Binational Science Foundation (BSF). I. V. is a Research Professor of the ICRF. We apologize for not citing all relevant articles due to space limitation.
Abbreviations
- ALIX
ALG-2 interacting protein X
- ECM
extracellular matrix
- ESCRT
endosomal-sorting complex required for transport
- HGF
hepatocyte growth factor
- HSPG
heparan sulfate proteoglycan
- MMP-9
matrix metalloproteinase 9
- SSTNs
synstatins
- TLR
toll-like receptor
- VCAM-1
vascular cell adhesion molecule 1
- VEGF
vascular endothelial growth factor
- VEGFR2
vascular endothelial growth factor receptor 2
- VLA-4
very late antigen 4
Footnotes
Author contributions
R.S., M.E., A.R., N.I. and I.V. wrote, reviewed and edited the manuscript. R.S. organized and submitted the manuscript.
References
- 1.Hammond E, Khurana A, Shridhar V, Dredge K. The role of heparanase and sulfatases in the modification of heparan sulfate proteoglycans within the tumor microenvironment and opportunities for novel cancer therapeutics. Front Oncol. 2014;4:195. doi: 10.3389/fonc.2014.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rivara S, Milazzo FM, Giannini G. Heparanase: a rainbow pharmacological target associated to multiple pathologies including rare diseases. Future Med Chem. 2016;8:647–680. doi: 10.4155/fmc-2016-0012. [DOI] [PubMed] [Google Scholar]
- 3.Barash U, Cohen-Kaplan V, Dowek I, Sanderson RD, Ilan N, Vlodavsky I. Proteoglycans in health and disease: new concepts for heparanase function in tumor progression and metastasis. FEBS J. 2010;277:3890–3903. doi: 10.1111/j.1742-4658.2010.07799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedmann Y, Vlodavsky I, Aingorn H, Aviv A, Peretz T, Pecker I, Pappo O. Expression of heparanase in normal, dysplastic, and neoplastic human colonic mucosa and stroma. Evidence for its role in colonic tumorigenesis. Am J Pathol. 2000;157:1167–1175. doi: 10.1016/S0002-9440(10)64632-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gohji K, Okamoto M, Kitazawa S, Toyoshima M, Dong J, Katsuoka Y, Nakajima M. Heparanase protein and gene expression in bladder cancer. J Urol. 2001;166:1286–1290. [PubMed] [Google Scholar]
- 6.Koliopanos A, Friess H, Kleeff J, Shi X, Liao Q, Pecker I, Vlodavsky I, Zimmermann A, Buchler MW. Heparanase expression in primary and metastatic pancreatic cancer. Cancer Res. 2001;61:4655–4659. [PubMed] [Google Scholar]
- 7.Marchetti D, Nicolson GL. Human heparanase: a molecular determinant of brain metastasis. Adv Enzyme Regul. 2001;41:343–359. doi: 10.1016/s0065-2571(00)00016-9. [DOI] [PubMed] [Google Scholar]
- 8.Hong X, Nelson KK, deCarvalho AC, Kalkanis SN. Heparanase expression of glioma in human and animal models. J Neurosurg. 2010;113:261–269. doi: 10.3171/2009.9.JNS09682. [DOI] [PubMed] [Google Scholar]
- 9.Ogishima T, Shiina H, Breault JE, Tabatabai L, Bassett WW, Enokida H, Li LC, Kawakami T, Urakami S, Ribeiro-Filho LA, Terashima M, Fujime M, Igawa M, Dahiya R. Increased heparanase expression is caused by promoter hypomethylation and up-regulation of transcriptional factor early growth response-1 in human prostate cancer. Clin Cancer Res. 2005;11:1028–1036. [PubMed] [Google Scholar]
- 10.Davidson B, Shafat I, Risberg B, Ilan N, Trope CG, Vlodavsky I, Reich R. Heparanase expression correlates with poor survival in metastatic ovarian carcinoma. Gynecol Oncol. 2007;104:311–319. doi: 10.1016/j.ygyno.2006.08.045. [DOI] [PubMed] [Google Scholar]
- 11.El-Assal ON, Yamanoi A, Ono T, Kohno H, Nagasue N. The clinicopathological significance of heparanase and basic fibroblast growth factor expressions in hepatocellular carcinoma. Clin Cancer Res. 2001;7:1299–1305. [PubMed] [Google Scholar]
- 12.Masola V, Maran C, Tassone E, Zin A, Rosolen A, Onisto M. Heparanase activity in alveolar and embryonal rhabdomyosarcoma: implications for tumor invasion. BMC Cancer. 2009;9:304. doi: 10.1186/1471-2407-9-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohen I, Pappo O, Elkin M, San T, Bar-Shavit R, Hazan R, Peretz T, Vlodavsky I, Abramovitch R. Heparanase promotes growth, angiogenesis and survival of primary breast tumors. Int J Cancer. 2006;118:1609–1617. doi: 10.1002/ijc.21552. [DOI] [PubMed] [Google Scholar]
- 14.Kelly T, Miao HQ, Yang Y, Navarro E, Kussie P, Huang Y, MacLeod V, Casciano J, Joseph L, Zhan F, Zangari M, Barlogie B, Shaughnessy J, Sanderson RD. High heparanase activity in multiple myeloma is associated with elevated microvessel density. Cancer Res. 2003;63:8749–8756. [PubMed] [Google Scholar]
- 15.Masola V, Secchi MF, Gambaro G, Onisto M. Heparanase as a target in cancer therapy. Curr Cancer Drug Targets. 2014;14:286–293. doi: 10.2174/1568009614666140224155124. [DOI] [PubMed] [Google Scholar]
- 16.Pisano C, Vlodavsky I, Ilan N, Zunino F. The potential of heparanase as a therapeutic target in cancer. Biochem Pharmacol. 2014;89:12–19. doi: 10.1016/j.bcp.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vlodavsky I, Beckhove P, Lerner I, Pisano C, Meirovitz A, Ilan N, Elkin M. Significance of heparanase in cancer and inflammation. Cancer Microenviron. 2012;5:115–132. doi: 10.1007/s12307-011-0082-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahtouk K, Hose D, Raynaud P, Hundemer M, Jourdan M, Jourdan E, Pantesco V, Baudard M, De Vos J, Larroque M, Moehler T, Rossi JF, Reme T, Goldschmidt H, Klein B. Heparanase influences expression and shedding of syndecan-1, and its expression by the bone marrow environment is a bad prognostic factor in multiple myeloma. Blood. 2007;109:4914–4923. doi: 10.1182/blood-2006-08-043232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edovitsky E, Elkin M, Zcharia E, Peretz T, Vlodavsky I. Heparanase gene silencing, tumor invasiveness, angiogenesis, and metastasis. J Natl Cancer Inst. 2004;96:1219–1230. doi: 10.1093/jnci/djh230. [DOI] [PubMed] [Google Scholar]
- 20.Cassinelli G, Lanzi C, Tortoreto M, Cominetti D, Petrangolini G, Favini E, Zaffaroni N, Pisano C, Penco S, Vlodavsky I, Zunino F. Antitumor efficacy of the heparanase inhibitor SST0001 alone and in combination with antiangiogenic agents in the treatment of human pediatric sarcoma models. Biochem Pharmacol. 2013;85:1424–1432. doi: 10.1016/j.bcp.2013.02.023. [DOI] [PubMed] [Google Scholar]
- 21.Casu B, Vlodavsky I, Sanderson RD. Non-anticoagulant heparins and inhibition of cancer. Pathophysiol Haemost Thromb. 2008;36:195–203. doi: 10.1159/000175157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dredge K, Hammond E, Handley P, Gonda TJ, Smith MT, Vincent C, Brandt R, Ferro V, Bytheway I. PG545, a dual heparanase and angiogenesis inhibitor, induces potent anti-tumour and anti-metastatic efficacy in preclinical models. Br J Cancer. 2011;104:635–642. doi: 10.1038/bjc.2011.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ritchie JP, Ramani VC, Ren Y, Naggi A, Torri G, Casu B, Penco S, Pisano C, Carminati P, Tortoreto M, Zunino F, Vlodavsky I, Sanderson RD, Yang Y. SST0001, a chemically modified heparin, inhibits myeloma growth and angiogenesis via disruption of the heparanase/syndecan-1 axis. Clin Cancer Res. 2011;17:1382–1393. doi: 10.1158/1078-0432.CCR-10-2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shafat I, Ben-Arush MW, Issakov J, Meller I, Naroditsky I, Tortoteto M, Cassinelli G, Lanzi C, Pisano C, Ilan N, Vlodavsky I, Zunino F. Preclinical and clinical significance of heparanase in Ewing's sarcoma. J Cell Mol Med. 2010 doi: 10.1111/j.1582-4934.2010.01190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou H, Roy S, Cochran E, Zouaoui R, Chu CL, Duffner J, Zhao G, Smith S, Galcheva-Gargova Z, Karlgren J, Dussault N, Kwan RY, Moy E, Barnes M, Long A, Honan C, Qi YW, Shriver Z, Ganguly T, Schultes B, Venkataraman G, Kishimoto TK. M402, a novel heparan sulfate mimetic, targets multiple pathways implicated in tumor progression and metastasis. PLoS One. 2011;6:e21106. doi: 10.1371/journal.pone.0021106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramani VC, Purushothaman A, Stewart MD, Thompson CA, Vlodavsky I, Au JL, Sanderson RD. The heparanase/syndecan-1 axis in cancer: mechanisms and therapies. FEBS J. 2013;280:2294–2306. doi: 10.1111/febs.12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McKenzie EA. Heparanase: a target for drug discovery in cancer and inflammation. Br J Pharmacol. 2007;151:1–14. doi: 10.1038/sj.bjp.0707182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang YF, Tang XD, Gao JH, Fang DC, Yang SM. Heparanase: a universal immunotherapeutic target in human cancers. Drug Discov Today. 2011;16:412–417. doi: 10.1016/j.drudis.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 29.Purushothaman A, Chen L, Yang Y, Sanderson RD. Heparanase stimulation of protease expression implicates it as a master regulator of the aggressive tumor phenotype in myeloma. J Biol Chem. 2008;283:32628–32636. doi: 10.1074/jbc.M806266200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Purushothaman A, Uyama T, Kobayashi F, Yamada S, Sugahara K, Rapraeger AC, Sanderson RD. Heparanase-enhanced shedding of syndecan-1 by myeloma cells promotes endothelial invasion and angiogenesis. Blood. 2010;115:2449–2457. doi: 10.1182/blood-2009-07-234757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramani VC, Yang Y, Ren Y, Nan L, Sanderson RD. Heparanase plays a dual role in driving hepatocyte growth factor (HGF) signaling by enhancing HGF expression and activity. J Biol Chem. 2011;286:6490–6499. doi: 10.1074/jbc.M110.183277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang Y, Ren Y, Ramani VC, Nan L, Suva LJ, Sanderson RD. Heparanase enhances local and systemic osteolysis in multiple myeloma by upregulating the expression and secretion of RANKL. Cancer Res. 2010;70:8329–8338. doi: 10.1158/0008-5472.CAN-10-2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cohen-Kaplan V, Naroditsky I, Zetser A, Ilan N, Vlodavsky I, Doweck I. Heparanase induces VEGF C and facilitates tumor lymphangiogenesis. Int J Cancer. 2008;123:2566–2573. doi: 10.1002/ijc.23898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang Y, Macleod V, Miao HQ, Theus A, Zhan F, Shaughnessy JD, Jr, Sawyer J, Li JP, Zcharia E, Vlodavsky I, Sanderson RD. Heparanase enhances syndecan-1 shedding: A novel mechanism for stimulation of tumor growth and metastasis. J Biol Chem. 2007;282:13326–13333. doi: 10.1074/jbc.M611259200. [DOI] [PubMed] [Google Scholar]
- 35.Afratis N. e a Syndecans: key regulators of cell signaling and biological functions. doi: 10.1111/febs.13940. [DOI] [PubMed] [Google Scholar]
- 36.Simons M, Raposo G. Exosomes--vesicular carriers for intercellular communication. Curr Opin Cell Biol. 2009;21:575–581. doi: 10.1016/j.ceb.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 37.Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, Garcia-Santos G, Ghajar C, Nitadori-Hoshino A, Hoffman C, Badal K, Garcia BA, Callahan MK, Yuan J, Martins VR, Skog J, Kaplan RN, Brady MS, Wolchok JD, Chapman PB, Kang Y, Bromberg J, Lyden D. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18:883–891. doi: 10.1038/nm.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peinado H, Lavotshkin S, Lyden D. The secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughts. Semin Cancer Biol. 2011;21:139–146. doi: 10.1016/j.semcancer.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 39.Zhang HG, Grizzle WE. Exosomes and cancer: a newly described pathway of immune suppression. Clin Can Res. 2011;17:959–964. doi: 10.1158/1078-0432.CCR-10-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baietti MF, Zhang Z, Mortier E, Melchior A, Degeest G, Geeraerts A, Ivarsson Y, Depoortere F, Coomans C, Vermeiren E, Zimmermann P, David G. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat Cell Biol. 2012;14:677–685. doi: 10.1038/ncb2502. [DOI] [PubMed] [Google Scholar]
- 41.Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, Moita CF, Schauer K, Hume AN, Freitas RP, Goud B, Benaroch P, Hacohen N, Fukuda M, Desnos C, Seabra MC, Darchen F, Amigorena S, Moita LF, Thery C. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol. 2010;12:19–30. doi: 10.1038/ncb2000. sup pp 1–13. [DOI] [PubMed] [Google Scholar]
- 42.Roucourt B, Meeussen S, Bao J, Zimmermann P, David G. Heparanase activates the syndecan-syntenin-ALIX exosome pathway. Cell Res. 2015;25:412–428. doi: 10.1038/cr.2015.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.David G, Zimmermann P. Heparanase tailors syndecan for exosome production. Mol Cell Oncol. 2016;3:e1047556. doi: 10.1080/23723556.2015.1047556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Friand V, David G, Zimmermann P. Syntenin and syndecan in the biogenesis of exosomes. Biol Cell. 2015;107:331–341. doi: 10.1111/boc.201500010. [DOI] [PubMed] [Google Scholar]
- 45.Thompson CA, Purushothaman A, Ramani VC, Vlodavsky I, Sanderson RD. Heparanase regulates secretion, composition, and function of tumor cell-derived exosomes. J Biol Chem. 2013;288:10093–10099. doi: 10.1074/jbc.C112.444562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Au Yeung CL, Co NN, Tsuruga T, Yeung TL, Kwan SY, Leung CS, Li Y, Lu ES, Kwan K, Wong KK, Schmandt R, Lu KH, Mok SC. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat Commun. 2016;7:11150. doi: 10.1038/ncomms11150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Purushothaman A, Bandari SK, Liu J, Mobley JA, Brown EE, Sanderson RD. Fibronectin on the surface of myeloma cell-derived exosomes mediates exosome-cell Interactions. J Biol Chem. 2016;291:1652–1663. doi: 10.1074/jbc.M115.686295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goldshmidt O, Nadav L, Aingorn H, Irit C, Feinstein N, Ilan N, Zamir E, Geiger B, Vlodavsky I, Katz BZ. Human heparanase is localized within lysosomes in a stable form. Exp Cell Res. 2002;281:50–62. doi: 10.1006/excr.2002.5651. [DOI] [PubMed] [Google Scholar]
- 49.Zetser A, Levy-Adam F, Kaplan V, Gingis-Velitski S, Bashenko Y, Schubert S, Flugelman MY, Vlodavsky I, Ilan N. Processing and activation of latent heparanase occurs in lysosomes. J Cell Sci. 2004;117:2249–2258. doi: 10.1242/jcs.01068. [DOI] [PubMed] [Google Scholar]
- 50.Gingis-Velitski S, Zetser A, Kaplan V, Ben-Zaken O, Cohen E, Levy-Adam F, Bashenko Y, Flugelman MY, Vlodavsky I, Ilan N. Heparanase uptake is mediated by cell membrane heparan sulfate proteoglycans. J Biol Chem. 2004;279:44084–44092. doi: 10.1074/jbc.M402131200. [DOI] [PubMed] [Google Scholar]
- 51.Egeberg M, Kjeken R, Kolset SO, Berg T, Prydz K. Internalization and stepwise degradation of heparan sulfate proteoglycans in rat hepatocytes. Biochim Biophys Acta. 2001;1541:135–149. doi: 10.1016/s0167-4889(01)00132-x. [DOI] [PubMed] [Google Scholar]
- 52.Shteingauz A, Boyango I, Naroditsky I, Hammond E, Gruber M, Doweck I, Ilan N, Vlodavsky I. Heparanase enhances tumor growth and chemoresistance by promoting autophagy. Cancer Res. 2015;75:3946–3957. doi: 10.1158/0008-5472.CAN-15-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rosenfeldt MT, Ryan KM. The multiple roles of autophagy in cancer. Carcinogenesis. 2011;32:955–963. doi: 10.1093/carcin/bgr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401–410. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Arvatz G, Barash U, Nativ O, Ilan N, Vlodavsky I. Post-transcriptional regulation of heparanase gene expression by a 3' AU-rich element. FASEB J. 2011;24:4969–4976. doi: 10.1096/fj.10-156372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cohen-Kaplan V, Doweck I, Naroditsky I, Vlodavsky I, Ilan N. Heparanase augments epidermal growth factor receptor phosphorylation: correlation with head and neck tumor progression. Cancer Res. 2008;68:10077–10085. doi: 10.1158/0008-5472.CAN-08-2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cohen-Kaplan V, Jrbashyan J, Yanir Y, Naroditsky I, Ben-Izhak O, Ilan N, Doweck I, Vlodavsky I. Heparanase induces signal transducer and activator of transcription (STAT) protein phosphorylation: preclinical and clinical significance in head and neck cancer. J Biol Chem. 2012;287:6668–6678. doi: 10.1074/jbc.M111.271346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Doweck I, Kaplan-Cohen V, Naroditsky I, Sabo E, Ilan N, Vlodavsky I. Heparanase localization and expression by head and neck cancer: correlation with tumor progression and patient survival. Neoplasia. 2006;8:1055–1061. doi: 10.1593/neo.06577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fux L, Feibish N, Cohen-Kaplan V, Gingis-Velitski S, Feld S, Geffen C, Vlodavsky I, Ilan N. Structure-function approach identifies a COOH-terminal domain that mediates heparanase signaling. Cancer Res. 2009;69:1758–1767. doi: 10.1158/0008-5472.CAN-08-1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zetser A, Bashenko Y, Edovitsky E, Levy-Adam F, Vlodavsky I, Ilan N. Heparanase induces vascular endothelial growth factor expression: correlation with p38 phosphorylation levels and Src activation. Cancer Res. 2006;66:1455–1463. doi: 10.1158/0008-5472.CAN-05-1811. [DOI] [PubMed] [Google Scholar]
- 61.Roucourt B, Meeussen S, Bao J, Zimmermann P, David G. Heparanase activates the syndecan-syntenin-ALIX exosome pathway. Cell Res. 2015;25:412–428. doi: 10.1038/cr.2015.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dunlop EA, Tee AR. mTOR and autophagy: A dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol. 2014 doi: 10.1016/j.semcdb.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 63.Ramani VC, Vlodavsky I, Ng M, Zhang Y, Barbieri P, Noseda A, Sanderson RD. Chemotherapy induces expression and release of heparanase leading to changes associated with an aggressive tumor phenotype. Matrix Biol. 2016 doi: 10.1016/j.matbio.2016.03.006. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Larrue C, Saland E, Boutzen H, Vergez F, David M, Joffre C, Hospital MA, Tamburini J, Delabesse E, Manenti S, Sarry JE, Recher C. Proteasome inhibitors induce FLT3-ITD degradation through autophagy in AML cells. Blood. 2016;127:882–892. doi: 10.1182/blood-2015-05-646497. [DOI] [PubMed] [Google Scholar]
- 65.Levy JM, Thorburn A. Targeting autophagy during cancer therapy to improve clinical outcomes. Pharmacol Ther. 2011;131:130–141. doi: 10.1016/j.pharmthera.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu WK, Coffelt SB, Cho CH, Wang XJ, Lee CW, Chan FK, Yu J, Sung JJ. The autophagic paradox in cancer therapy. Oncogene. 2012;31:939–953. doi: 10.1038/onc.2011.295. [DOI] [PubMed] [Google Scholar]
- 67.Ilan N, Shteingauz A, Vlodavsky I. Function from within: Autophagy induction by HPSE/heparanase-new possibilities for intervention. Autophagy. 2015;11:2387–2389. doi: 10.1080/15548627.2015.1115174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schaefer L. e a Proteoglycan neofunctions: regulation of inflammation and autophagy in tumor biology. doi: 10.1111/febs.13963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 70.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ramani VC, Pruett PS, Thompson CA, Delucas LD, Sanderson RD. Heparan sulfate chains of syndecan-1 regulate ectodomain shedding. J Biol Chem. 2012;287:9952–9961. doi: 10.1074/jbc.M111.330803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jung O, Trapp-Stamborski V, Purushothaman A, Jin H, Wang H, Sanderson RD, Rapraeger AC. Heparanase-induced shedding of syndecan-1/CD138 in myeloma and endothelial cells activates VEGFR2 and an invasive phenotype: prevention by novel synstatins. Oncogenesis. 2016;5:e202. doi: 10.1038/oncsis.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Beauvais DM, Ell BJ, McWhorter AR, Rapraeger AC. Syndecan-1 regulates alphavbeta3 and alphavbeta5 integrin activation during angiogenesis and is blocked by synstatin, a novel peptide inhibitor. J Exp Med. 2009;206:691–705. doi: 10.1084/jem.20081278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Beauvais DM, Jung O, Yang Y, Sanderson RD, Rapraeger AC. Syndecan-1 (CD138) suppresses apoptosis in multiple myeloma by activating IGF1 receptor: prevention by synstatinIGF1R inhibits tumor growth. Cancer research. 2016 doi: 10.1158/0008-5472.CAN-16-0232. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Beauvais DM, Rapraeger AC. Syndecan-1 couples the insulin-like growth factor-1 receptor to inside-out integrin activation. J Cell Sci. 2010;123:3796–3807. doi: 10.1242/jcs.067645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rapraeger AC. Synstatin: a selective inhibitor of the syndecan-1-coupled IGF1R-alphavbeta3 integrin complex in tumorigenesis and angiogenesis. The FEBS journal. 2013;280:2207–2215. doi: 10.1111/febs.12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang H, Jin H, Rapraeger AC. Syndecan-1 and syndecan-4 capture epidermal growth factor receptor family members and the alpha3 beta1 integrin via binding sites in their ectodomains: Novel synstatins prevent kinase capture and inhibit alpha6 beta4-integrin-dependent epithelial cell motility. J Biol Chem. 2015;290:26103–26113. doi: 10.1074/jbc.M115.679084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Van Reit I, Van Camp B. The involvement of adhesion molecules in the biology of multiple myeloma. Leuk Lymphoma. 1993;9:441–452. doi: 10.3109/10428199309145751. [DOI] [PubMed] [Google Scholar]
- 79.Vande Broek I, Vanderkerken K, Van Camp B, Van Riet I. Extravasation and homing mechanisms in multiple myeloma. Clin Exp Metastasis. 2008;25:325–334. doi: 10.1007/s10585-007-9108-4. [DOI] [PubMed] [Google Scholar]
- 80.Ramani VC, Zhan F, He J, Barbieri P, Noseda A, Tricot G, Sanderson RD. Targeting heparanase overcomes chemoresistance and diminishes relapse in myeloma. Oncotarget. 2016;7:1598–1607. doi: 10.18632/oncotarget.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang L, Ngo JA, Wetzel MD, Marchetti D. Heparanase mediates a novel mechanism in lapatinib-resistant brain metastatic breast cancer. Neoplasia. 2015;17:101–113. doi: 10.1016/j.neo.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wu L, Viola CM, Brzozowski AM, Davies GJ. Structural characterization of human heparanase reveals insights into substrate recognition. Nat Struct Mol Biol. 2015;22:1016–1022. doi: 10.1038/nsmb.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Naparstek Y, Cohen IR, Fuks Z, Vlodavsky I. Activated T lymphocytes produce a matrix-degrading heparan sulphate endoglycosidase. Nature. 1984;310:241–244. doi: 10.1038/310241a0. [DOI] [PubMed] [Google Scholar]
- 84.Matzner Y, Bar-Ner M, Yahalom J, Ishai-Michaeli R, Fuks Z, Vlodavsky I. Degradation of heparan sulfate in the subendothelial extracellular matrix by a readily released heparanase from human neutrophils. Possible role in invasion through basement membranes. J Clin Invest. 1985;76:1306–1313. doi: 10.1172/JCI112104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fridman R, Lider O, Naparstek Y, Fuks Z, Vlodavsky I, Cohen IR. Soluble antigen induces T lymphocytes to secrete an endoglycosidase that degrades the heparan sulfate moiety of subendothelial extracellular matrix. J Cell Physiol. 1987;130:85–92. doi: 10.1002/jcp.1041300113. [DOI] [PubMed] [Google Scholar]
- 86.Vlodavsky I, Eldor A, Haimovitz-Friedman A, Matzner Y, Ishai-Michaeli R, Lider O, Naparstek Y, Cohen IR, Fuks Z. Expression of heparanase by platelets and circulating cells of the immune system: possible involvement in diapedesis and extravasation. Invasion Metastasis. 1992;12:112–127. [PubMed] [Google Scholar]
- 87.Lider O, Baharav E, Mekori YA, Miller T, Naparstek Y, Vlodavsky I, Cohen IR. Suppression of experimental autoimmune diseases and prolongation of allograft survival by treatment of animals with low doses of heparins. J Clin Invest. 1989;83:752–756. doi: 10.1172/JCI113953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lider O, Mekori YA, Miller T, Bar-Tana R, Vlodavsky I, Baharav E, Cohen IR, Naparstek Y. Inhibition of T lymphocyte heparanase by heparin prevents T cell migration and T cell-mediated immunity. Eur J Immunol. 1990;20:493–499. doi: 10.1002/eji.1830200306. [DOI] [PubMed] [Google Scholar]
- 89.Yahalom J, Fibach E, Bar-Tana R, Fuks Z, Vlodavsky I. Differentiating human leukemia cells express heparanase that degrades heparan sulfate in subendothelial extracellular matrix. Leuk Res. 1988;12:711–717. doi: 10.1016/0145-2126(88)90003-3. [DOI] [PubMed] [Google Scholar]
- 90.Young E. The anti-inflammatory effects of heparin and related compounds. Thromb Res. 2008;122:743–752. doi: 10.1016/j.thromres.2006.10.026. [DOI] [PubMed] [Google Scholar]
- 91.Hershkoviz R, Mor F, Miao HQ, Vlodavsky I, Lider O. Differential effects of polysulfated polysaccharide on experimental encephalomyelitis, proliferation of autoimmune T cells, and inhibition of heparanase activity. J Autoimmun. 1995;8:741–750. doi: 10.1006/jaut.1995.0055. [DOI] [PubMed] [Google Scholar]
- 92.Parish CR, Hindmarsh EJ, Bartlett MR, Staykova MA, Cowden WB, Willenborg DO. Treatment of central nervous system inflammation with inhibitors of basement membrane degradation. Immunol Cell Biol. 1998;76:104–113. doi: 10.1046/j.1440-1711.1998.00722.x. [DOI] [PubMed] [Google Scholar]
- 93.Irony-Tur-Sinai M, Vlodavsky I, Ben-Sasson SA, Pinto F, Sicsic C, Brenner T. A synthetic heparin-mimicking polyanionic compound inhibits central nervous system inflammation. J Neurol Sci. 2003;206:49–57. doi: 10.1016/s0022-510x(02)00318-0. [DOI] [PubMed] [Google Scholar]
- 94.Edovitsky E, Lerner I, Zcharia E, Peretz T, Vlodavsky I, Elkin M. Role of endothelial heparanase in delayed-type hypersensitivity. Blood. 2006;107:3609–3616. doi: 10.1182/blood-2005-08-3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Floer M, Gotte M, Wild MK, Heidemann J, Gassar ES, Domschke W, Kiesel L, Luegering A, Kucharzik T. Enoxaparin improves the course of dextran sodium sulfate-induced colitis in syndecan-1-deficient mice. Am J Pathol. 2010;176:146–157. doi: 10.2353/ajpath.2010.080639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gotte M. Syndecans in inflammation. FASEB J. 2003;17:575–591. doi: 10.1096/fj.02-0739rev. [DOI] [PubMed] [Google Scholar]
- 97.Wang L, Fuster M, Sriramarao P, Esko JD. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat Immunol. 2005;6:902–910. doi: 10.1038/ni1233. [DOI] [PubMed] [Google Scholar]
- 98.Parish CR. The role of heparan sulphate in inflammation. Nat Rev Immunol. 2006;6:633–643. doi: 10.1038/nri1918. [DOI] [PubMed] [Google Scholar]
- 99.Taylor KR, Gallo RL. Glycosaminoglycans and their proteoglycans: host-associated molecular patterns for initiation and modulation of inflammation. FASEB J. 2006;20:9–22. doi: 10.1096/fj.05-4682rev. [DOI] [PubMed] [Google Scholar]
- 100.Bode L, Salvestrini C, Park PW, Li JP, Esko JD, Yamaguchi Y, Murch S, Freeze HH. Heparan sulfate and syndecan-1 are essential in maintaining murine and human intestinal epithelial barrier function. J Clin Invest. 2008;118:229–238. doi: 10.1172/JCI32335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Axelsson J, Xu D, Kang BN, Nussbacher JK, Handel TM, Ley K, Sriramarao P, Esko JD. Inactivation of heparan sulfate 2-O-sulfotransferase accentuates neutrophil infiltration during acute inflammation in mice. Blood. 2012;120:1742–1751. doi: 10.1182/blood-2012-03-417139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Brunn GJ, Bungum MK, Johnson GB, Platt JL. Conditional signaling by Toll-like receptor 4. FASEB J. 2005;19:872–874. doi: 10.1096/fj.04-3211fje. [DOI] [PubMed] [Google Scholar]
- 103.Johnson GB, Brunn GJ, Kodaira Y, Platt JL. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J Immunol. 2002;168:5233–5239. doi: 10.4049/jimmunol.168.10.5233. [DOI] [PubMed] [Google Scholar]
- 104.Akbarshahi H, Axelsson JB, Said K, Malmstrom A, Fischer H, Andersson R. TLR4 dependent heparan sulphate-induced pancreatic inflammatory response is IRF3-mediated. J Transl Med. 2011;9:219. doi: 10.1186/1479-5876-9-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Baker AB, Groothuis A, Jonas M, Ettenson DS, Shazly T, Zcharia E, Vlodavsky I, Seifert P, Edelman ER. Heparanase alters arterial structure, mechanics, and repair following endovascular stenting in mice. Circ Res. 2009;104:380–387. doi: 10.1161/CIRCRESAHA.108.180695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Quaglio AE, Castilho AC, Di Stasi LC. Experimental evidence of heparanase, Hsp70 and NF-kappaB gene expression on the response of anti-inflammatory drugs in TNBS-induced colonic inflammation. Life Sci. 2015;141:179–187. doi: 10.1016/j.lfs.2015.09.023. [DOI] [PubMed] [Google Scholar]
- 107.Lerner I, Hermano E, Zcharia E, Rodkin D, Bulvik R, Doviner V, Rubinstein AM, Ishai-Michaeli R, Atzmon R, Sherman Y, Meirovitz A, Peretz T, Vlodavsky I, Elkin M. Heparanase powers a chronic inflammatory circuit that promotes colitis-associated tumorigenesis in mice. J Clin Invest. 2011;121:1709–1721. doi: 10.1172/JCI43792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Schmidt EP, Yang Y, Janssen WJ, Gandjeva A, Perez MJ, Barthel L, Zemans RL, Bowman JC, Koyanagi DE, Yunt ZX, Smith LP, Cheng SS, Overdier KH, Thompson KR, Geraci MW, Douglas IS, Pearse DB, Tuder RM. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med. 2012;18:1217–1223. doi: 10.1038/nm.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ziolkowski AF, Popp SK, Freeman C, Parish CR, Simeonovic CJ. Heparan sulfate and heparanase play key roles in mouse beta cell survival and autoimmune diabetes. J Clin Invest. 2012;122:132–141. doi: 10.1172/JCI46177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li RW, Freeman C, Yu D, Hindmarsh EJ, Tymms KE, Parish CR, Smith PN. Dramatic regulation of heparanase activity and angiogenesis gene expression in synovium from patients with rheumatoid arthritis. Arthritis Rheum. 2008;58:1590–1600. doi: 10.1002/art.23489. [DOI] [PubMed] [Google Scholar]
- 111.Morris A, Wang B, Waern I, Venkatasamy R, Page C, Schmidt EP, Wernersson S, Li JP, Spina D. The role of heparanase in pulmonary cell recruitment in response to an allergic but not non-allergic stimulus. PLoS One. 2015;10:e0127032. doi: 10.1371/journal.pone.0127032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Puerta-Guardo H, Glasner DR, Harris E. Dengue Virus NS1 Disrupts the Endothelial Glycocalyx, Leading to Hyperpermeability. PLoS Pathog. 2016;12:e1005738. doi: 10.1371/journal.ppat.1005738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lapidot M, Barash U, Zohar Y, Geffen Y, Naroditsky I, Ilan N, Best LA, Vlodavsky I. Involvement of heparanase in empyema: implication for novel therapeutic approaches. J Clin Cell Immunol. 2015;6:pii:290. doi: 10.4172/2155-9899.1000290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Waterman M, Ben-Izhak O, Eliakim R, Groisman G, Vlodavsky I, Ilan N. Heparanase upregulation by colonic epithelium in inflammatory bowel disease. Mod Pathol. 2007;20:8–14. doi: 10.1038/modpathol.3800710. [DOI] [PubMed] [Google Scholar]
- 115.Chen G, Wang D, Vikramadithyan R, Yagyu H, Saxena U, Pillarisetti S, Goldberg IJ. Inflammatory cytokines and fatty acids regulate endothelial cell heparanase expression. Biochemistry. 2004;43:4971–4977. doi: 10.1021/bi0356552. [DOI] [PubMed] [Google Scholar]
- 116.Binder Gallimidi A, Fischman S, Revach B, Bulvik R, Maliutina A, Rubinstein AM, Nussbaum G, Elkin M. Periodontal pathogens Porphyromonas gingivalis and Fusobacterium nucleatum promote tumor progression in an oral-specific chemical carcinogenesis model. Oncotarget. 2015;6:22613–22623. doi: 10.18632/oncotarget.4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang F, Wang Y, Zhang D, Puthanveetil P, Johnson JD, Rodrigues B. Fatty acid-induced nuclear translocation of heparanase uncouples glucose metabolism in endothelial cells. Arterioscler Thromb Vasc Biol. 2012;32:406–414. doi: 10.1161/ATVBAHA.111.240770. [DOI] [PubMed] [Google Scholar]
- 118.He YQ, Sutcliffe EL, Bunting KL, Li J, Goodall KJ, Poon IK, Hulett MD, Freeman C, Zafar A, McInnes RL, Taya T, Parish CR, Rao S. The endoglycosidase heparanase enters the nucleus of T lymphocytes and modulates H3 methylation at actively transcribed genes via the interplay with key chromatin modifying enzymes. Transcription. 2012;3:130–145. doi: 10.4161/trns.19998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Blich M, Golan A, Arvatz G, Sebbag A, Shafat I, Sabo E, Cohen-Kaplan V, Petcherski S, Avniel-Polak S, Eitan A, Hammerman H, Aronson D, Axelman E, Ilan N, Nussbaum G, Vlodavsky I. Macrophage Activation by Heparanase Is Mediated by TLR-2 and TLR-4 and Associates With Plaque Progression. Arterioscler Thromb Vasc Biol. 2013;33:56–65. doi: 10.1161/ATVBAHA.112.254961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Massena S, Christoffersson G, Hjertstrom E, Zcharia E, Vlodavsky I, Ausmees N, Rolny C, Li JP, Phillipson M. A chemotactic gradient sequestered on endothelial heparan sulfate induces directional intraluminal crawling of neutrophils. Blood. 2010;116:1924–1931. doi: 10.1182/blood-2010-01-266072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Blich M, Golan A, Arvatz G, Sebbag A, Shafat I, Sabo E, Cohen-Kaplan V, Petcherski S, Avniel-Polak S, Eitan A, Hammerman H, Aronson D, Axelman E, Ilan N, Nussbaum G, Vlodavsky I. Macrophage Activation by Heparanase Is Mediated by TLR-2 and TLR-4 and Associates With Plaque Progression. Arterioscler Thromb Vasc Biol. 2012;33:56–65. doi: 10.1161/ATVBAHA.112.254961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Benhamron S, Nechushtan H, Verbovetski I, Krispin A, Abboud-Jarrous G, Zcharia E, Edovitsky E, Nahari E, Peretz T, Vlodavsky I, Mevorach D. Translocation of active heparanase to cell surface regulates degradation of extracellular matrix heparan sulfate upon transmigration of mature monocyte-derived dendritic cells. J Immunol. 2006;176:6417–6424. doi: 10.4049/jimmunol.176.11.6417. [DOI] [PubMed] [Google Scholar]
- 123.Benhamron S, Reiner I, Zcharia E, Atallah M, Grau A, Vlodavsky I, Mevorach D. Dissociation between mature phenotype and impaired transmigration in dendritic cells from heparanase-deficient mice. PLoS One. 2012;7:e35602. doi: 10.1371/journal.pone.0035602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang B, Jia J, Zhang X, Zcharia E, Vlodavsky I, Pejler G, Li JP. Heparanase affects secretory granule homeostasis of murine mast cells through degrading heparin. J Allergy Clin Immunol. 2011;128:1310–1317. e8. doi: 10.1016/j.jaci.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Poon IK, Goodall KJ, Phipps S, Chow JD, Pagler EB, Andrews DM, Conlan CL, Ryan GF, White JA, Wong MK, Horan C, Matthaei KI, Smyth MJ, Hulett MD. Mice deficient in heparanase exhibit impaired dendritic cell migration and reduced airway inflammation. Eur J Immunol. 2014;44:1016–1030. doi: 10.1002/eji.201343645. [DOI] [PubMed] [Google Scholar]
- 126.Petrovich E, Feigelson SW, Stoler-Barak L, Hatzav M, Solomon A, Bar-Shai A, Ilan N, Li JP, Engelhardt B, Vlodavsky I, Alon R. Lung ICAM-1 and ICAM-2 support spontaneous intravascular effector lymphocyte entrapment but are not required for neutrophil entrapment or emigration inside endotoxin-inflamed lungs. FASEB J. 2016;30:1767–1778. doi: 10.1096/fj.201500046. [DOI] [PubMed] [Google Scholar]
- 127.Weinbaum S, Tarbell JM, Damiano ER. The structure and function of the endothelial glycocalyx layer. Annu Rev Biomed Eng. 2007;9:121–167. doi: 10.1146/annurev.bioeng.9.060906.151959. [DOI] [PubMed] [Google Scholar]
- 128.Constantinescu AA, Vink H, Spaan JA. Endothelial cell glycocalyx modulates immobilization of leukocytes at the endothelial surface. Arterioscler Thromb Vasc Biol. 2003;23:1541–1547. doi: 10.1161/01.ATV.0000085630.24353.3D. [DOI] [PubMed] [Google Scholar]
- 129.Sue M, Higashi N, Shida H, Kogane Y, Nishimura Y, Adachi H, Kolaczkowska E, Kepka M, Nakajima M, Irimura T. An iminosugar-based heparanase inhibitor heparastatin (SF4) suppresses infiltration of neutrophils and monocytes into inflamed dorsal air pouches. Int Immunopharmacol. 2016;35:15–21. doi: 10.1016/j.intimp.2016.03.017. [DOI] [PubMed] [Google Scholar]
- 130.Li L, Wang B, Gao T, Zhang X, Hao JX, Vlodavsky I, Wiesenfeld-Hallin Z, Xu XJ, Li JP. Heparanase overexpression reduces carrageenan-induced mechanical and cold hypersensitivity in mice. Neurosci Lett. 2012;511:4–7. doi: 10.1016/j.neulet.2011.12.038. [DOI] [PubMed] [Google Scholar]
- 131.Zhang X, Wang B, O'Callaghan P, Hjertstrom E, Jia J, Gong F, Zcharia E, Nilsson LN, Lannfelt L, Vlodavsky I, Lindahl U, Li JP. Heparanase overexpression impairs inflammatory response and macrophage-mediated clearance of amyloid-beta in murine brain. Acta Neuropathol. 2012;124:465–478. doi: 10.1007/s00401-012-0997-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Escobar Galvis ML, Jia J, Zhang X, Jastrebova N, Spillmann D, Gottfridsson E, van Kuppevelt TH, Zcharia E, Vlodavsky I, Lindahl U, Li JP. Transgenic or tumor-induced expression of heparanase upregulates sulfation of heparan sulfate. Nat Chem Biol. 2007;3:773–778. doi: 10.1038/nchembio.2007.41. [DOI] [PubMed] [Google Scholar]
- 133.Goldberg R, Rubinstein AM, Gil N, Hermano E, Li JP, van der Vlag J, Atzmon R, Meirovitz A, Elkin M. Role of heparanase-driven inflammatory cascade in pathogenesis of diabetic nephropathy. Diabetes. 2014;63:4302–4313. doi: 10.2337/db14-0001. [DOI] [PubMed] [Google Scholar]
- 134.Hermano E, Meirovitz A, Meir K, Nussbaum G, Appelbaum L, Peretz T, Elkin M. Macrophage polarization in pancreatic carcinoma: role of heparanase enzyme. J Natl Cancer Inst. 2014;106 doi: 10.1093/jnci/dju332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Elkin M, Ilan N, Ishai-Michaeli R, Friedmann Y, Papo O, Pecker I, Vlodavsky I. Heparanase as mediator of angiogenesis: mode of action. FASEB J. 2001;15:1661–1663. doi: 10.1096/fj.00-0895fje. [DOI] [PubMed] [Google Scholar]
- 136.Kato M, Wang H, Kainulainen V, Fitzgerald ML, Ledbetter S, Ornitz DM, Bernfield M. Physiological degradation converts the soluble syndecan-1 ectodomain from an inhibitor to a potent activator of FGF-2. Nat Med. 1998;4:691–697. doi: 10.1038/nm0698-691. [DOI] [PubMed] [Google Scholar]
- 137.Goodall KJ, Poon IK, Phipps S, Hulett MD. Soluble heparan sulfate fragments generated by heparanase trigger the release of pro-inflammatory cytokines through TLR-4. PLoS One. 2014;9:e109596. doi: 10.1371/journal.pone.0109596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yu L, Wang L, Chen S. Endogenous toll-like receptor ligands and their biological significance. J Cell Mol Med. 2010;14:2592–2603. doi: 10.1111/j.1582-4934.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Simeonovic CJ, Ziolkowski AF, Wu Z, Choong FJ, Freeman C, Parish CR. Heparanase and autoimmune diabetes. Front Immunol. 2013;4:471. doi: 10.3389/fimmu.2013.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wang F, Wan A, Rodrigues B. The function of heparanase in diabetes and its complications. Can J Diabetes. 2013;37:332–338. doi: 10.1016/j.jcjd.2013.05.008. [DOI] [PubMed] [Google Scholar]
- 141.Abu El-Asrar AM, Alam K, Nawaz MI, Mohammad G, Van den Eynde K, Siddiquei MM, Mousa A, De Hertogh G, Geboes K, Opdenakker G. Upregulated Expression of Heparanase in the Vitreous of Patients With Proliferative Diabetic Retinopathy Originates From Activated Endothelial Cells and Leukocytes. Invest Ophthalmol Vis Sci. 2015;56:8239–8247. doi: 10.1167/iovs.15-18025. [DOI] [PubMed] [Google Scholar]
- 142.Lim AK, Tesch GH. Inflammation in diabetic nephropathy. Mediators Inflamm. 2012;2012:146154. doi: 10.1155/2012/146154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Navarro-Gonzalez JF, Mora-Fernandez C, Muros de Fuentes M, Garcia-Perez J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol. 2011;7:327–340. doi: 10.1038/nrneph.2011.51. [DOI] [PubMed] [Google Scholar]
- 144.Tesch GH. Macrophages and diabetic nephropathy. Semin Nephrol. 2010;30:290–301. doi: 10.1016/j.semnephrol.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 145.You H, Gao T, Cooper TK, Reeves WB, Awad AS. Macrophages Directly Mediate Diabetic Renal Injury. Am J Physiol Renal Physiol. 2013 doi: 10.1152/ajprenal.00141.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Shanmugam N, Reddy MA, Guha M, Natarajan R. High glucose-induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes. 2003;52:1256–1264. doi: 10.2337/diabetes.52.5.1256. [DOI] [PubMed] [Google Scholar]
- 147.Vlassara H, Brownlee M, Manogue KR, Dinarello CA, Pasagian A. Cachectin/TNF and IL-1 induced by glucose-modified proteins: role in normal tissue remodeling. Science. 1988;240:1546–1548. doi: 10.1126/science.3259727. [DOI] [PubMed] [Google Scholar]
- 148.Chow FY, Nikolic-Paterson DJ, Atkins RC, Tesch GH. Macrophages in streptozotocin-induced diabetic nephropathy: potential role in renal fibrosis. Nephrol Dial Transplant. 2004;19:2987–2996. doi: 10.1093/ndt/gfh441. [DOI] [PubMed] [Google Scholar]
- 149.Poteser M, Wakabayashi I. Serum albumin induces iNOS expression and NO production in RAW 267.4 macrophages. Br J Pharmacol. 2004;143:143–151. doi: 10.1038/sj.bjp.0705897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Cha JJ, Hyun YY, Lee MH, Kim JE, Nam DH, Song HK, Kang YS, Lee JE, Kim HW, Han JY, Cha DR. Renal protective effects of toll-like receptor 4 signaling blockade in type 2 diabetic mice. Endocrinology. 2013;154:2144–2155. doi: 10.1210/en.2012-2080. [DOI] [PubMed] [Google Scholar]
- 151.Soldatos G, Cooper ME. Diabetic nephropathy: important pathophysiologic mechanisms. Diabetes Res Clin Pract. 2008;82(Suppl 1):S75–S79. doi: 10.1016/j.diabres.2008.09.042. [DOI] [PubMed] [Google Scholar]