

Abstract
There is currently no effective treatment for acute exacerbation of idiopathic pulmonary fibrosis (IPF). We herein report the case of a patient with acute exacerbation of IPF which was treated with nintedanib, an intracellular inhibitor of tyrosine kinases, and showed improvement of the condition. An 84‐year‐old man with IPF was admitted to our hospital because of dry cough and worsening of dyspnoea within last 1 month. He presented with hypoxemia, and chest high‐resolution computed tomography (HRCT) revealed new, bilateral multifocal ground‐glass opacities superimposed on the background of lung fibrosis. After exclusion of alternative causes, acute exacerbation of IPF was diagnosed and we started treatment with nintedanib of 300 mg/day. This resulted in the gradual improvement of his condition and HRCT findings without administering antibiotics or corticosteroids. Serum Krebs von den Lungen‐6 and surfactant protein D levels increased at acute exacerbation and subsequently decreased. This case suggests that nintedanib therapy may have possible benefits in acute exacerbation of IPF.
Keywords: Acute exacerbation, idiopathic pulmonary fibrosis, KL‐6, nintedanib
Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive, debilitating condition of the ageing population with a devastating prognosis. Some patients with IPF experience acute respiratory worsening of unknown aetiology during their clinical course, known as acute exacerbation of IPF 1. There is currently no effective treatment for this condition, including steroid therapy, and its prognosis is extremely poor. We herein report a case of acute exacerbation of IPF which was treated with nintedanib, an intracellular inhibitor of tyrosine kinases including the receptors for the fibroblast growth factor (FGF), platelet‐derived growth factor (PDGF), and vascular endothelial growth factor (VEGF).
Case Report
An 84‐year‐old man was admitted to our hospital in February 2016 because of dry cough and worsening of dyspnoea within last 1 month. He had been diagnosed as having IPF based on the clinical and high‐resolution computed tomography (HRCT) findings in June 2014. Since then he was followed up without specific therapy for IPF and had a stable condition until December 2015. At regular follow‐up visit 3 months previously, chest X‐ray showed slight infiltrative shadows in the lower lung fields (Fig. 1A) and HRCT of the chest showed basal and peripheral predominant reticular abnormality with honeycombing combined with emphysema (Fig. 2A). Pulmonary function tests showed forced expiratory volume in one second of 89.7% predicted, forced vital capacity (FVC) of 92.8% predicted, and diffusing capacity of lung for carbon monoxide (DLco) of 54.4% predicted. He was an ex‐smoker with a history of 52 pack‐years. His medical history included atrial fibrillation, gastro‐oesophageal reflux, hyperlipidaemia, emphysema, and chronic sinusitis and he was taking bisoprolol, ethyl icosapentate, rabeprazole sodium, inhalant of tiotropium bromide hydrate, and olodaterol hydrochloride. On admission his temperature was 37°C and fine crackles were noted at both lung bases. His blood tests showed a white blood cell count of 7020/μL, Hb of 14.1 g/dL, platelet count of 16.3 × 104, and C‐reactive protein (CRP) of 0.5 mg/dL. Serum Krebs von den Lungen‐6 (KL‐6) and surfactant protein D (SP‐D) levels were 1034 U/mL (normal: <500 U/mL) and 354.0 ng/mL (normal: <110 ng/mL), respectively. Both values were clearly increased compared with those examined in November 2015 (556 U/mL and 268.9 ng/mL, respectively). Serological tests for atypical pathogens including Mycoplasma pneumoniae and Chlamydophila (Chlamydia) pneumoniae were negative as were β‐D glucan (<5.0 pg/mL). B‐type natriuretic peptide was 56.2 pg/mL (normal range: <18.4 pg/mL) without clinical evidence of congestive heart failure. Echocardiography demonstrated ejection fraction of 69% and estimated right ventricular systolic pressure of 40 mmHg. Arterial blood gas measurement while breathing room air revealed hypoxia (PaO2 54.7 Torr) with PaCO2 as 32.2 Torr and pH as 7.490. FVC decreased to 77.8% predicted and DLco was not performed. Smear and cultures of sputum showed no evidence of respiratory pathogens. Chest X‐ray (Fig. 1B) showed bilateral infiltrative shadows in the lower lung fields, which were worsened compared with those obtained 3 months previously (Fig. 1A). HRCT scan revealed new, bilateral multifocal ground‐glass opacities superimposed on the background of lung fibrosis (Fig. 2B). Acute exacerbation of IPF was diagnosed and we started treatment with nintedanib of 300 mg/day. This resulted in the gradual improvement of his condition without administering antibiotics or corticosteroids. He was discharged on the 11th day without oxygen inhalation. Follow‐up chest X‐ray (Fig. 1C) and HRCT scan (Fig. 2C) after the 1 month of nintedanib administration revealed improvement of ground‐glass opacities. The patient continued to take nintedanib without significant side effects and the KL‐6 and SP‐D values gradually decreased as follows: 916 U/mL and 455.2 ng/mL in March, 775 U/mL and 357.7 ng/mL in May, 488 U/mL and 269.6 ng/mL in August 2016, respectively. FVC was restored to 89.8% in May 2016.
Figure 1.
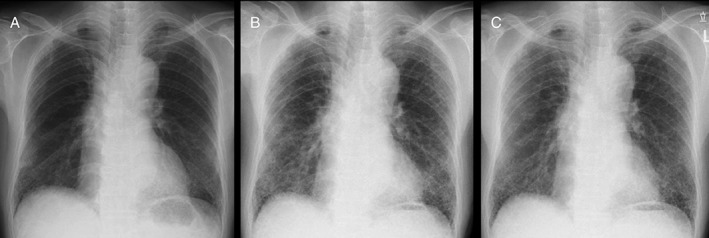
Chest X‐ray (A) October 2015, (B) January 2016, (C) March 2016, showing bilateral infiltrative shadows in the lower lung fields in (B), which were worsened compared with (A), and improvement after nintedanib therapy in (C).
Figure 2.
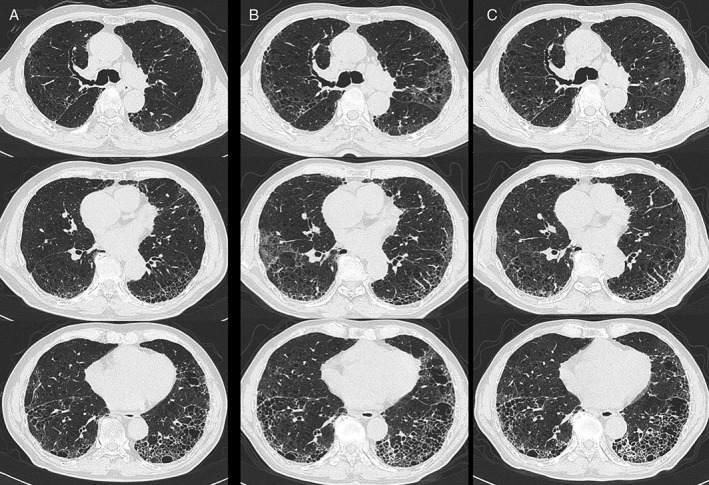
Chest high‐resolution computed tomography scan (A) November 2015, (B) January 2016, (C) March 2016, showing new, bilateral multifocal ground‐glass opacities in (B) superimposed on the background of lung fibrosis and emphysema in (A), and improvement after nintedanib therapy in (C).
Discussion
Nintedanib has been recently approved for the treatment of IPF based on a large‐scale randomized controlled trial demonstrating a decreased rate of disease progression as measured by FVC in well‐defined patients with IPF 2. In addition, a significant benefit in favour of nintedanib on time to first investigator‐reported acute exacerbation was observed in the pooled analysis 3. So preventative administration of nintedanib may reduce acute exacerbation.
On the other hand, to our knowledge, no cases of acute exacerbation treated with nintedanib were found in the literature. Although the present case showed multifocal HRCT changes, and it might have a better prognosis according to a HRCT study in acute exacerbation 4, it nevertheless deserves attention since our patient survived without administration of antibiotics or high‐dose corticosteroids. Published reports from both human and experimental lung fibrosis indicate that fibrotic areas have fewer blood vessels, whereas adjacent non‐fibrotic tissue is highly vascularized 5. In case of acute exacerbation, non‐fibrotic tissue are targeted and resulted in the histopathologic finding of diffuse alveolar damage. The experimental evidence supports nintedanib having anti‐angiogenic efficacy and significant inhibitory effects of nintedanib on inflammation and fibrosis were also demonstrated in bleomycin‐induced lung fibrosis 6. Furthermore, although direct hemoperfusion with a polymyxin B‐immobilized fibre column (PMX‐DHP) has attracted attention as a promising therapy for acute exacerbation of IPF, it has been reported that PMX‐fibre removed profibrotic cytokines such as FGF, PDGF, and transforming growth factor‐β in patients with acute exacerbation of IPF 7. From these observations, nintedanib may attenuate acute lung injury in acute exacerbation of IPF.
In summary, this case report suggests that nintedanib therapy may have possible benefits in acute exacerbation of IPF. Further experience and clinical studies are needed to verify this promising approach.
Disclosure Statement
H. Tomioka reported receiving lecture fees from Boehringer Ingelheim, Dai‐ichi Sankyo, and Novartis. H. Takada disclosed no potential conflicts of interest.
Appropriate written informed consent was obtained for publication of this case report and accompanying images.
Tomioka H. and Takada H. (2017) Treatment with nintedanib for acute exacerbation of idiopathic pulmonary fibrosis. Respirology Case Reports, 5 (2), e00215. doi: 10.1002/rcr2.215.
Associate Editor: Dr Conroy Wong
References
- 1. Raghu G, Collard HR, Egan JJ, et al. 2011. An official ATS/ERS/JRS/ALAT statement‐idiopathic pulmonary fibrosis: evidence‐based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 183:788–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Richeldi L, du Bois RM, Raghu G, et al. 2014. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 370:2071–2082. [DOI] [PubMed] [Google Scholar]
- 3. Richeldi L, Cottin V, du Bois RM, et al. 2016. Nintedanib in patients with idiopathic pulmonary fibrosis: combined evidence from the TOMORROW and INPULSIS® trials. Respir. Med. 113:74–79. [DOI] [PubMed] [Google Scholar]
- 4. Akira M, Hamada H, Sakatani M, et al. 1997. CT findings during phase of accelerated deterioration in patients with idiopathic pulmonary fibrosis. AJR Am. J. Roentgenol. 168:79–83. [DOI] [PubMed] [Google Scholar]
- 5. Ebina M, Shimizukawa M, Shibata N, et al. 2004. Heterogeneous increase in CD34‐positive alveolar capillaries in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 169:1203–1208. [DOI] [PubMed] [Google Scholar]
- 6. Wollin L, Maillet I, Quesniaux V, et al. 2014. Antifibrotic and anti‐inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J. Pharmacol. Exp. Ther. 349:209–220. [DOI] [PubMed] [Google Scholar]
- 7. Oishi K, Mimura‐Kimura Y, Miyasho T, et al. 2013. Association between cytokine removal by polymyxin B hemoperfusion and improved pulmonary oxygenation in patients with acute exacerbation of idiopathic pulmonary fibrosis. Cytokine 61:84–89. [DOI] [PubMed] [Google Scholar]