

Abstract
Albumin is the most abundant circulating protein in plasma and has recently emerged as a versatile protein carrier for drug targeting and for improving the pharmacokinetic profile of peptide or protein based drugs. Three drug delivery technologies related to albumin have been developed, which include the coupling of low-molecular weight drugs to exogenous or endogenous albumin, conjugating bioactive proteins by albumin fusion technology (AFT), and encapsulation of drugs into albumin nanoparticles. This review article starts with a brief introduction of human serum albumin (HSA), and then summarizes the mainstream chemical strategies of developing HSA binding molecules for coupling with drug molecules. Moreover, we also concisely condense the recent progress of the most important clinical applications of HSA-binding platforms, and specify the current challenges that need to be met for a bright future of HSA-binding.
1. Introduction
Human serum albumin (HSA) is the most abundant protein in plasma, and it can serve as a versatile carrier for drug delivery as well as for prolonging the active profile of fast-clearance drugs.1–3 Besides being a key drug-delivery protein in blood, it also undertakes the transportation of many essential biomolecules, such as fatty acids, hormones and amino acids.4,5 HSA has a notably long half-life (19 days) in blood circulation.6,7 HSA is produced in the liver cells as preproalbumin, and then modified by Golgi vesicles to give secreted albumin. Approximately 13–14 g of albumin is secreted into the intravascular system each day, and the extravascular HSA will return to intravascular circulation through the lymphatic system.8,9 The degradation of HSA is highly dependent on the interaction with albumin receptors, including gp18, gp30, megalin and the neonatal Fc receptor (FcRn).
HSA is widely used as a carrier for small molecule drugs and imaging probes.10–19 It is biodegradable and non-toxic and lacks immunogenicity, making it an excellent candidate as an excipient for vaccines and many other pharmaceuticals.20,21 In addition, HSA is robust against chemical modifications and can be stable in the pH range of 4–9 at 60 °C for as long as 10 h. Therefore, the amino acid residues on albumin can be readily linked with therapeutic drugs, imaging reporters and targeting molecules through chemical conjugation. Albumin is also found to specifically target tumor regions because of its enhanced permeability and retention (EPR) effect as well as albumin receptor binding, which is a unique advantage as the carrier for tumor-targeted drug delivery.22,23
There has been long-standing interest in developing a general strategy that can effectively prolong the active profile of pharmaceuticals. Among the currently developed methods, conjugating pharmaceuticals to albumin-binding molecules is one of the most commonly used approaches due to its high efficiency and minimum side effect.24
In the past two decades, a number of advances in HSA-binding therapeutics have been approved by the Food and Drug Administration (FDA), and many more are under active clinical investigation (Table 1). These successful discoveries are of significance to a broad spectrum of healthcare, especially for cancer therapy and diabetes treatment. Overall, the development is often derived from a new understanding of HSA chemistry, followed by a smart application designed to solve an emerging clinical challenge. For instance, the development and market approval of Abraxane, a paclitaxel albumin nanoparticle, became a landmark for both nanomedicine and albumin-based drug delivery technology with annual sales of $850 million in 2014. Indeed, the thoughts and rationales of these successes are greatly inspiring, not only for the development of future HSA-binding therapeutics, but also to the most general audiences including chemists, biologists and clinical doctors who have interest in new drug development. For a better understanding of these exciting progresses, we would like to share our review to guide the biological design, chemical screening and clinical application of HSA-based drugs, with focus on the strategy of in vivo binding that are most practical for clinical use.
Table 1.
| Company sponsoring clinical study | Brand name or drag code | Molecular type | Status | Indication of clinical studies | Clinical trial identifier number | Ref. |
|---|---|---|---|---|---|---|
| Novo Nordisk | Levemir | Fatty acid insulin conjugate | Approved | Diabetes |
NCT00655044 NCTOO806897 |
4 |
| Novo Nordisk | Liraglutide | Fatty acid peptide conjugate | Approved | Diabetes | NCT01795248 | 4 |
| GlaxoSmithKline | Albiglutide | Peptide HSA conjugate | Approved | Diabetes |
NCT01357889 NCTOO849017 |
160 and 161 |
| Abraxis BioScience | Abraxane | Nanoparticle albumin bound small molecule | Approved | Breast cancer, lung cancer and prostate cancer |
NCT01307891 NCT02027428 NCT00732836 |
4 and 167 |
| Nycomed Amersham | Nanocoll | 99mTc macroaggregated HSA | Approved | SPECT scan for breast cancer and rheumatoid arthritis | NCT00929032 | 20 |
| Human Genome Sciences | Albinterferon | Interferon alpha (IFN-α) HSA conjugate | Phase III | Hepatitis C | NCT00724776 | 20 |
| Innovive Pharmaceuticals | Aldoxorubicin | Doxorubicin maleimide conjugate | Phase III | Soft tissue sarcomas, small cell lung cancer |
NCT01673438 NCT02235688 |
4 |
| Conjuchem Inc. | CJC-1134 | Peptide maleimide conjugate | Phase II | Diabetes |
NCT01514149 NCT00638716 |
152 |
| Fujisawa Deutschland GmbH | MTX–HSA | Methotrexate HSA conjugates | Phase II | Metastatic translational cell cancer | EORTC30951 EORTC20947 |
25 |
2. General strategy to develop HSA-conjugated drugs
2.1. In vitro covalent conjugation
To start with, amide coupling based on lysine residue is the most classical method for in vitro covalent HSA conjugation (Fig. 1A–D).28–34 At present, the functional moieties often contain p-isothiocyanate (p-SCN) or NHS ester (N-hydroxysuccinimide) that can be obtained by in situ activation. However, these methods are not site-specific and always lead to a mixture of mono and multiple modified HSA.18,19,35–39
Fig. 1.

General methods of covalently conjugating small molecules onto albumin. (A) The coupling molecule is activated in situ by using classical coupling reagents such as N,N′-dicyclohexylcarbodiimide (DCC), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), hydroxybenzotriazole (HOBT) and 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), and then attached onto lysine residue of HSA under weakly basic conditions. (B) The coupling molecule is activated as an N-hydroxysuccinimide (NHS) ester prior to be conjugated onto lysine residue of HSA under weakly basic conditions. (C) The coupling molecule is activated as a tetrafluorophenyl (TFP) ester prior to being conjugated onto lysine residue of HSA under weakly basic conditions. (D) The coupling molecule is modified to contain p-isothiocyanate (p-SCN), and then attached onto lysine residue of HSA under weakly basic conditions. (E) The coupling molecule is modified to contain maleimide moieties, and then attached onto cysteine residue of HSA under weakly basic conditions.
To meet the challenge, an optimized coupling method was developed to perform the conjugation on cysteine-34 instead of lysines on HSA, and has provided better-defined HSA–drug conjugates that have high purity with a constant drug-loading ratio, a minimal alteration of the three-dimensional protein structure and a preset breaking point. However, as the cysteine-34 position on commercially available HSA is largely blocked by cysteine, homocysteine as well as other sulfhydryl containing compounds, the HSA is a mixture of mercaptalbumin and nonmercaptalbumin and only approximately 20–60% of them contain free sulfhydryl groups. To solve this problem, Mansour et al. developed a one-step procedure of selectively reducing HSA with dithiothreitol (Cleland’s reagent), giving approximately one sulfhydryl group for each HSA molecule (Fig. 1E). In the next step, the reduced HSA is directly coupled with the maleimide modified drugs such as doxorubicin maleimide. Compared with the free doxorubicin, the HSA-conjugated version was significantly better on curing murine renal carcinoma (RENCA) at equitoxic dose.40
In vitro covalent conjugation has been widely used in preparing HSA-based drugs (Fig. 2). For instance, to radiolabel HSA with radiometals for diagnostic imaging, radiometal chelators will be linked on HSA by incubating NHS-activated ester of DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) together with HSA under weakly basic conditions (pH = 8–9).41,42 18F-HSA, 68Ga-DOTA–HSA, 111In-DTPA–HSA (DTPA is the abbreviation for diethylene triamine pentaacetic acid) and Gd-DTPA–HSA have been considered as blood-pool imaging reagents by using positron emission tomography (PET), single-photon emission computed tomography (SPECT) and magnetic resonance image (MRI), respectively.28,43–45 In convention, radioactive HSA is prepared by multiple-step radiosynthesis. Nevertheless, by taking the advantage of the development of milder and more efficient radiolabeling strategies,46–50 one-step HSA labeling is likely to be practical in the near future. In addition, peptide and small molecular drugs have also been conjugated on the lysine residues of HSA to develop advanced HSA-binding imaging probe and therapeutics.28,43–45
Fig. 2.

Representative HSA drugs based on in vitro conjugation: (a) 111In-labeled HSA for single-photon emission computed tomography (SPECT);51 (b) Gd-labeled HSA for magnetic resonance imaging (MRI);52 (c) 68Ga-labeled HSA for positron emission tomography (PET);53 (d) doxorubicin HSA conjugates for cancer chemotherapy with less side effect;54 (e) 18F-labeled HSA conjugates for PET;44 (f) CysCOOH HSA conjugates for photothermal therapy;55 (g) 3,5-diiodo-thyronine HSA conjugates for antibody production in animals;56 (h) exendin-4 peptide HSA conjugates for the treatment of type 2 diabetes.57
Fusion protein technology (FPT) is a special way of in vitro conjugating HSA with functional moieties yet has been broadly used in preparing recombinant HSA/protein. By doing so, albumin protein conjugates are genetically engineered by putting together the genes of the two molecules and expressing the albumin fusion proteins in yeast strains (Fig. 3).
Fig. 3.

(A) The construction of the recombinant protein that fuses HSA and a peptide of interest. (B) Schematic structure of a representative HSA fusion protein Albuferon. Reprinted with permission from ref. 58 and 4. Copyright 2007 (ref. 58) European Peptide Society and John Wiley & Sons, Ltd. and copyright 2008 (ref. 4) Elsevier B.V.
Clinically, one such albumin fusion protein is Albuferon, a fusion protein of albumin and interferon α-2b for the treatment of hepatitis C.59 A number of other albumin fusion proteins have entered early clinical trials. These include fusion proteins with low-molecular weight peptides such as β-natriuretic peptide and glucagon-like peptide 1, as well as fusion proteins with cytokines. Albuleukin, an albumin fusion protein with recombinant interleukin-2 that has shown promising antitumor efficacy against murine renal cell carcinoma and melanoma.55,59
2.2. In vivo covalent conjugation
Kratz et al. established a strategy that exploits endogenous HSA as a drug carrier.60 In this therapeutic strategy, the prodrug binds rapidly and selectively to the cysteine-34 position of circulating serum albumin after intravenous administration thereby generating a macromolecular transport form of the drug in situ in the blood. Indeed, the strategy of in vivo HSA conjugation would have several advantages over in vitro synthesized drug albumin conjugates: (a) the use of commercial and possibly pathogenic albumin is avoided; (b) easy to use and inexpensive to manufacture; and (c) the related quality control is simple, which is comparable to any other low-molecular weight drug candidates.
The macromolecular prodrug approach targets the cysteine-34 position of albumin. A HPLC analysis demonstrates that approximately 70% of circulating albumin in the blood stream is mercaptalbumin (HMA) that contains an accessible cysteine-34.4,61,62 Moreover, the free thiol group of cysteine-34 of HSA is an unusual feature of an extracellular protein. As known, only three other major proteins that contain free cysteine residues in human plasma: (1) apolipoprotein B-100 of low-density lipoprotein (LDL) which has two cysteine residues (Cys-3734 and Cys-4190) located at the C-terminal end of the protein,63–65 (2) fibronectin which has two cryptic, free sulfhydryl groups,66 and (3) R1-antitrypsin which has a single cysteine residue (Cys-232).66–68 However, the sulfhydryl groups in these proteins do not react readily with sulfhydryl reagents under physiological conditions and are normally linked to either cysteine or glutathione in blood circulation. Therefore, the free thiol group on HSA, cysteine-34 of endogenous albumin, is a unique amino acid on the surface of a circulating protein, which is capable of further conjugation.69
Proof of concept was obtained with the (6-maleimidocaproyl) hydrazone derivative of doxorubicin (DOXO-EMCH) that rapidly and selectively binds to circulating albumin within a few minutes (Fig. 4). Inspired by translational research with DOXO-EMCH, many albumin-binding prodrugs have been developed (Fig. 5). These prodrugs often consist of an anticancer drug, the maleimide group as the thiol-binding moiety and an enzymatically cleavable peptide linker. Examples include doxorubicin prodrugs that are cleaved by matrix metalloproteases 2 and 9,40 cathepsin B,70 urokinase plasminogen (uPA) or prostate-specific antigen (PSA),71,72 methotrexate prodrugs that are cleaved by cathepsin B or plasmin,73 and camptothecin prodrugs that are cleaved by cathepsin B or unidentified proteases.74 In addition, maleimide derivatives with 5-fluorouracil analogues and platinum(II) complexes have been developed.75
Fig. 4.

(A) X-ray structure of human serum albumin in which the cysteine-34 position is marked as shown; (B) chemical structure of the (6-maleimidocaproyl) hydrazone derivative of doxorubicin (DOXO-EMCH); (C) a schematic description of in vivo thiol–maleimide conjugation.
Fig. 5.

Structures of selected albumin-binding maleimide modified prodrugs. (a) Doxorubicin prodrug that is cleaved by cathepsin B;76 (b) and (c) albumin-binding prodrugs with Pt(II) complexes;75 (d) camptothecin prodrug that is cleaved by cathepsin B;73 and (e) doxorubicin prodrug that is cleaved by prostate-specific antigen (PSA).40,60,77,78
2.3. In vitro non-covalent HSA binding
Besides covalently connecting HSA with small functional molecules, non-covalent van der Waals force or electronic interaction is another approach that can be used for HSA binding.79,80 For instance, certain radiometals can form robust conjugates with macro-aggregated albumin (MAA) without using any chelators, the resulting complexes (111In-MAA and 99mTc-MAA, Fig. 6A and B) have been widely used in clinical diagnosis, especially for lung perfusion and for detecting gastrointestinal bleeding by SPECT.20,51,81–83 To form this self-assembled capsule, firstly the intramolecular disulfide bonds of HSA are partially reduced by using glutathione (GSH) to give free sulfhydryl groups. Then, the pretreated HSA/ water solution is mixed with small drugs in triaryl butyl alcohol (TBA). Here, TBA is used as the anti-solvent for albumin and water is used as the anti-solvent for the small molecular drugs. In the mixed solution, HSA and small molecular drugs would precipitate out because of the decreased solubility of both HSA and small molecular drugs. At last, this suspension is further incubated at 37 °C to form interamolecular disulfide to give small molecular drug loaded HSA nanoparticles (Fig. 6C).20,84
Fig. 6.

Representative strategies of in vitro non-covalent HSA binding. (A) 111In-labeled aggregated HSA for SPECT. (B) 99mTc-labeledaggregated HSA for SPECT. (C) Schematic description of the preparation of self-cross linked HSA nanoparticles. Reprinted with the kind permission from ref. 84. Copyright 2007 John Wiley & Sons, Ltd. (D) HSA coated iron oxide nanoparticles as multiple functional theranostic platform. Reprinted with permission from ref. 91. Copyright 2010 Elsevier B.V.
In addition, in vitro non-covalent HSA binding is also commonly used in preparing HSA–nanoparticle complexes, especially for the purpose of imaging and therapy.2,26–28,85–90 For instance, IONPs (iron oxide nanoparticles) were incubated with dopamine to become moderately hydrophilic before being doped into HSA matrices via non-covalent binding. In this case, a physical capsule is formed between HSA and IONP that can load small molecular drugs with high efficiency. Additionally, as shown in Fig. 6D, the HSA matrix is capable of carrying fluorophores and radioactive reporters, therefore this type of HSA-binding nanoparticle can serve as a multiple functional platform for the purpose of both in vivo imaging and drug delivery.
2.4. In vivo non-covalent HSA targeting
The three-dimensional crystal structure of HSA was solved in early 1990s (Fig. 7).92 It is a heart-shaped protein with three homogeneous domains, and each domain is composed of two subdomains that own the same structural motifs. Notably, HSA is one of the smallest proteins in human plasma. Both size and abundance explain the fact that the transportation of many metabolic compounds and therapeutic drugs is related to HSA by non-covalent binding. These HSA ligand-binding pockets are a series of hydrophobic cavities in subdomains II and III. Indeed, the design of HSA-binding molecules is mainly based on the structures of binding pockets, which is also the key to determine the physical performance of HSA.
Fig. 7.

Crystal structure of albumin illustrating (A) small molecule binding site 1 and site 2 and (B) fatty acid (FA) binding site.93 Reprinted with the kind permission from ref. 93. Copyright 2005 Elsevier Ltd.
Little was known about the variety of binding sites of HSA until an interesting study was reported in 1975,94 which was about the surprisingly different binding affinities of a number of fluorescent molecules for HSA. Changing the side chain on the amino acid moiety of the dansylamino acids was found to substantially affect the binding of these compounds to HSA. In fact, the binding of the dansylamino acids to HSA varied both in the number of binding sites and in the binding tightness to these sites, suggesting that electrostatic and dipolar forces as well as steric factors play a role in both strength and specificity of binding. This study corroborates with the results of Ghuman et al., who figured out based on circular dichroism measurements that the aromatic portion of flufenamic acid was inserted into a hydrophobic crevice on albumin while the carboxylate anion was associated with a cation that is around the gate of a binding pocket.93 Overall, there are two high affinity binding sites for small heterocyclic or aromatic compounds (located on subdomains IIA and IIIA),94 two to three dominant long-chain fatty acid binding sites (located on subdomains IB and IIIB), and two distinct metal-binding sites, making a total of six dominant areas of ligand association to albumin.95 In this part, we will elaborate on the chemistry of design, synthesis and screening of the small organic albumin-binding entities according to the specific binding sites.
2.4.1. HSA binding site 1
Binding site 1 is an essential pocket of HSA to carry and deliver small molecules in blood circulation. The interior environment of the pocket is predominantly apolar but is composed of two polar residues: an inner one towards the bottom and an outer polar residue near the entrance (Fig. 8). Therefore, the molecules binding to pocket 1 generally contain a lipophilic aromatic structure in the middle and spherically surrounded by negative charges. Many dye molecules bind to domain II with high binding affinities (Table 2).
Fig. 8.

(A) Drug binding to site 1 in HSA (defatted). The detailed binding conformation is shown for 3-carboxy-4-methyl-5-propyl-2-furanpropanoic acid (CMPF), in which the drug is shown in a stick representation with a semi-transparent van der Waals surface. Sticks color-coded by atom type indicate selected side-chains; hydrogen bonds are shown as yellow dashed lines. (B) Top view of the superposition of CMPF bound to site 1 in defatted HSA. Drugs are presented as a stick model with carbon atoms colored orange, nitrogen atoms in blue and oxygen atoms in red.85 Reprinted with permission from ref. 93. Copyright 2005 Elsevier Ltd.
Table 2.
Structure, binding affinity, number of binding for some classical binders to HSA binding site 193,96,97




Among them, Evans blue (EB) dye, as a good example, exhibits high affinity for binding site 1 on serum albumin. EB is an important tool in many physiologic and clinical investigations because of its high affinity for serum albumin, and has been used in clinical practice for almost 90 years as a way of determining patient plasma volume.98 By taking advantage of the high in vivo binding affinity of EB to albumin, Niu et al. developed a NOTA (1,4,7-triazacyclononane-N,N-triacetic acid) conjugate of a truncated form of Evans blue (NEB) for in vivo albumin labeling. 18F-labeling was achieved by complexing with 18F-aluminum fluoride (18F-AlF), and 68Ga and 64Cu labeling was accomplished through standard chelation chemistry (Fig. 9).99–101
Fig. 9.

(A) Schematic structure of supramolecular system of Evans blue that binds to the site 1 on HSA. As shown, Evans blue dye exhibits strong tendency towards self-assembly to form stable, continuous, ribbon-like supramolecules when it binds to HSA. This self-assembling capability is also found to essentially correlate with the capacity of protein binding;102 Reprinted with permission from ref. 102. Copyright 2000 John Wiley & Sons, Ltd. (B) Synthesis and 18F-AlF radiolabeling of NOTA(1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid)-trucated Evans blue conjugate (NEB).99
2.4.2. HSA binding site 2
Different from site 1, binding site 2 has a single main polar patch, located close to one side of the entrance of the binding pocket (Fig. 10). Based on the protein docking study, the hydrophobic binding cleft is about 16 Å deep and about 8 Å wide in the albumin molecule with a cationic group located near the surface. Therefore, as shown in Table 3, most of the binders to site 2 are lipophilic carboxylate derivatives. Nevertheless, a negative charge is not required for the molecule that binds to site 2. For example, diazepam, a basic drug molecule that exists mainly in the un-ionized form at neutral pH, also binds with high affinity for site 2. The presence of a positive charge often precludes binding to site 2. As shown in Table 3, aliphatic amines with chain lengths C-3 to C-12 do not have measurable binding to site 2 although fatty acids with the same side chains are micromolar binders to the same binding pocket.93
Fig. 10.

(A) Binding of indoxyl sulphate to site 2 in HSA. Indoxyl sulphate is shown in a stick representation with a semi-transparent van der Waals surface. Color-coding is the same as shown in Fig. 8. (B) Top view of the superposition of indoxyl sulphate bound to site 2 in HSA along with a semi-transparent surface.85 Reprinted with permission from ref. 93. Copyright 2005 Elsevier Ltd.
Table 3.
Structure, molecular length, binding affinity of some classical binders to HSA binding site 293,96,97





Recently, Neri et al. reported a class of 4-(p-iodophenyl)-butyric acid derivatives that display stable non-covalent interaction with binding site 2. These HSA-binding tags were selected based on the strategy of Systematic Evolution of Ligands by Exponential Enrichment (SELEX). The candidate pool is a DNA-encoded chemical library with more than six hundred oligonucleotide-compound conjugates. After selection, the DNA sequences of stronger albumin binders were amplified by PCR and decoded on oligonucleotide microarrays. The corresponding signal intensities were normalized after selection against the intensities of compounds selected on empty resin (Fig. 11A and B). The selected HSA-binding molecules are listed in Fig. 11C. Interestingly, some of the selected HSA-binding molecules are structurally similar and featured by the basic structure of a 4-phenylbutanoic acid moiety, with different hydrophobic substituents on the phenyl ring (Fig. 11C). Notably, one of these HSA-binding tags has been applied into several pharmaceutical systems to tune their clearance from blood circulation, such as elongation of the pharmaceutical profile of fast clearing drugs (Fig. 11D), improved performance of MRI contrast agents (Fig. 11E), and reduced kidney uptake of radiotherapeutic drugs (Fig. 21).103
Fig. 11.

(A) Microarray readout of the selections performed against inactivated resin and resin displaying HSA (right panel). The spots corresponding to the enriched compounds 428 and 539 are enlarged (center); (B) enrichment of compounds in selections for HSA binding (compound numbers are indicated). (C) Structures of the molecules identified as potential binders; (D) pharmacokinetic studies of fluorescein (black), 428-D-Lys-FAM (blue), 622-D-Lys-FAM (red), and phenethylamine-FAM (green) after injection in two mice each. As shown, the plasma concentration time course of 177Lu-labeled MSA is listed here for comparison; (E) fluorescein angiography images in mice were recorded over 1 h after injection of 50 nmol of fluorescein (top row) and 428-D-Lys-FAM (bottom row).104 Reprinted with permission from ref. 104. Copyright 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
Fig. 21.

(A) Chemical structure of cm09. (B and C) SPECT/CT images of KB tumor-bearing mice injected with 177Lu-cm09 (B) and 177Lu-EC0800 (C). Accumulation of radioactivity was found in FR-positive tumors (white arrows) and kidneys (yellow arrows). Images show a significantly improved tumor-to-kidney ratio (1.0 vs. 0.2) at 1, 4, 24, and 72 h after injection in mice that received 177Lu-cm09, compared with mice that received 177Lu-EC0800. (D) Internal radiation therapy protocol. (E) Average relative tumor size over time under different treatment regimens. (F) Relative body weight of mice under different therapies. (G) Survival curves of mice from groups A–E. (A, dark blue) control group. (B, green) unlabeled cm09. (C, red) 1 × 20 MBq of 177Lu-cm09. (D, violet) 2 × 10 MBq of 177Lu-cm09. (E, light blue) 3 × 7 MBq of 177Lu-cm09.182,183 Reprinted with permission from ref. 210. Copyright 2013 Society of Nuclear Medicine and Molecular Imaging.
2.4.3. Fatty acid modification for HSA binding
When Kendall accomplished HSA crystallization in 1941, he found that the product contained a small amount of free fatty acid (FA).105 In addition, other researchers noted that the lipids extracted from blood plasma contained small quantities of FA as well.105–107 In the following decades, multiple binding sites were found for FA, and the binding affinity of fatty acid for HSA is mildly strong with an association constant in the range of 10−4 to 10−6 M−1.108–112 As fatty acids are commercially inexpensive and can be readily attached to other pharmaceutical moieties, conjugating FA onto GLP (glucagon-like peptide) or insulin has been an effective way to develop long-acting antidiabetic therapeutics. For example, GLP-1 analog exendin-4 has been modified by two fatty acids: lauric acid (LUA, C12) and palmitic acid (PAA, C16) at its two lysine residues. The resulting FA-exendin-4 conjugates were tested as regulators of blood glucose to cure type 2 diabetes, and showed a notably longer blood circulation profile over exendin-4 (Table 4).113 Additionally, the FA acylated insulin has also been developed as a long-circulating anti-diabetic drug. It binds at the long-chain fatty acid binding sites, but the binding affinity is lower than that of the free fatty acids and depends to a relatively small degree on the number of carbon atoms in the fatty acid. This FA insulin conjugate showed a prolonged circulatory half-life,114 but FA modification is not applicable to a broad set of molecules because of its negative effect on solubility.
Table 4.
3. Medical applications based on HSA-conjugates or HSA-binding moieties
3.1. Blood pool imaging agents
3.1.1. The efficacy of MRI contrast agents is improved by HSA-binding
The interest in the investigation of the binding ability towards HSA of paramagnetic complexes based on Gd(III), the most used T1 contrast agents, is driven by two main reasons. First, the pharmacokinetic and pharmacodynamic properties of a HSA-binding contrast agent can be essentially effected by HSA, as the contrast agent is usually administered intravenously while HSA is the most predominant protein in the blood.115–118 After binding, the blood clearance of the contrast agent will be slowed down, and consequently the blood half-life and intravascular retention, will be increased.119–122 Thus, HSA-binding has been primarily considered for the visualization of vascular structures and for detecting regions with abnormal vascular permeability. Also, HSA binding can significantly improve the efficacy of these agents because the water proton relaxation time is strongly dependent on the tumbling motion of the metal complex.123–127
As described previously, the presence of hydrophobic moieties as well as hydrophilic negatively charged groups are the basic structural requirements for binding pocket 2 of HSA, most of the work in this field has been focused on the design of metal complexes matching such features.124,125
Gadofosveset or MS-325 (trade name: Ablavar, (trisodium 2-(R)-[(4,4-diphenylcyclohexyl) phosphonooxymethyl]diethylene-triaminepentaacetatoaquo gadolinium)) is a clinically approved gadolinium (Gd) based blood-pool MRI contrast agent (Fig. 12A) as an aid in diagnosing aortoiliac occlusive disease in patients with known or suspected peripheral vascular disease (PVD) or abdominal aortic aneurysm (AAA).117 As a result of transient binding to HSA, gadofosveset has ten times the signal-enhancing power of existing contrast agents as well as prolonged retention in the blood (Fig. 12B and C). This enables rapid acquisition of high-resolution magnetic resonance angiography (MRA) using standard MRI machines. Moreover, HSA binding offers an additional benefit beyond localization in the blood pool. The contrast agent begins to spin much more slowly, at the rate albumin spins, causing a relaxivity gain that produces a substantially brighter signal than would be possible with freely circulating gadolinium (Table 4).117,126–128
Fig. 12.

(A) Chemical structure of gadofosveset. (B) Observed longitudinal ( , circles) and transverse ( , squares) relaxivity for 0.1 mM gadofosveset in the presence (filled symbols) and in the absence (open symbols) of 22.5% (w/v) HSA at 37 °C, phosphate-buffered saline, pH 7.4. (C) Observed longitudinal ( , circles) and transverse ( , squares) relaxivity for 0.1 mM gadofosveset in the presence (filled symbols) and in the absence (open symbols) of 22.5% (w/v) HSA at 37 °C, phosphate-buffered saline, pH 7.4. Reprinted with permission from ref. 127. Copyright 2002 American Chemical Society (D–G) Comparable coronal projections of (D) conventional angiography, (E) gadofosveset enhanced MR angiography, (F) two-dimensional TOF MR angiography, and (G) a transverse reconstruction of a steady-state gadofosveset dataset showing stenoses (arrows) in both right and left common iliac arteries.132 Reprinted with permission from ref. 126. Copyright 2007 Radiological Society of North America.
The extended blood half-life of gadofosveset also results in a longer time period for imaging, which allows the radiologist to perform multiple imaging experiments and to image under steady-state conditions (Fig. 12G as an example).58,133–135 In addition to imaging peripheral vascular disease and coronary artery disease (Fig. 12D and F), current trials are being conducted to evaluate gadofosveset as an aid in diagnosing breast cancer and to identify myocardial perfusion defects with delayed high-resolution imaging.136–138
Another promising case of developing HSA-binding MRI contrast agent, 428-D-Lys-β-Ala-DTPA-Gd (Fig. 13A), was contributed by the Neri group, which also targets binding site 2.104 The dissociation constant of 428-D-Lys-β-Ala-DTPA-Gd to HSA was determined by ITC at 37 °C (Kd = 3.3 μM, Fig. 13B), while Gd-DTPA had negligible binding to HSA. Pharmacokinetic profiles were studied in mice by injecting DTPA and 428-D-Lys-β-Ala-DTPA complexed with 177Lu, thus allowing quantification by gamma-counting. Similar to the situation encountered with the fluorescein derivatives, the plasma concentration of DTPA-177Lu decreased rapidly and was no longer detectable at 60 min after injection, whereas 428-D-Lys-β-Ala-DTPA-177Lu displayed a substantially slower biphasic pharmacokinetic profile (Fig. 13C; DTPA-177Lu: t1/2 = 8.6 min vs. 428-D-Lys-bAla-DTPA-177Lu: t1/2 = 408 min). The rapid extravasation of DTPA-Gd in comparison to 428-D-Lys-β-Ala-DTPA-Gd was also observed by MRI procedures following intravenous injection of the contrast agents. MRI analysis of major blood vessels of the brain revealed a slower decrease of signal intensities in those injected with 428-D-Lys-β-Ala-DTPA-Gd (Fig. 13C–E).
Fig. 13.

(A) Chemical structure of 428-D-Lys-β-Ala-DTPA-Gd. (B) Kd value of 428-D-Lys-DTPA-Gd to HSA determined by isothermal titration calorimetry (ITC) at 37 °C. (C) Pharmacokinetic studies of DTPA-177Lu (filled symbols) and 428-D-Lys-β-Ala-DTPA-177Lu (empty symbols) after i.v. injection in mice. The plasma concentration time course of 177Lu-labeled mouse serum albumin is given for comparison. (D) Transverse MR images of the mouse head indicating the region of interest (ROI) used to select the blood vessel. (E) Time course of the MR signal intensity in the ROI after injection of Gd-DTPA (left panels) and 428-D-Lys-β-Ala-DTPA-Gd (right panels).104 Reprinted with permission from ref. 104. Copyright 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
3.1.2. Radiolabeled HSA as the blood pool imaging agents
Although many radiolabeled HSA derivatives have been developed as blood pool agents for radionuclide imaging,139–144 the true revolution came from the recent report of 18F-NEB (Fig. 14A, NOTA conjugated truncated Evans blue), of which the preparation has been described previously.99 Within a few minutes after tracer injection, 18F-NEB reached the highest SUV value in the blood. Afterwards, a slow but steady clearance of the radioactivity was observed from the blood, due to the turnover of albumin from blood circulation and slight dissociation of 18F-NEB from albumin. As shown in Fig. 14, this in vivo labeling strategy can be applied to blood-pool imaging to evaluate the cardiac function under both physiologic and pathologic conditions (Fig. 14B–D). This method can also be used to evaluate vascular permeability in tumors, inflammatory diseases, and ischemic or infarcted lesions.
Fig. 14.

(A) Chemical structure of 18F-NEB and 68Ga-NEB. (B) Series of maximum-intensity-projection PET images in normal mice after intravenous injection of either 18F-AlF-NEB or 18F-FB-MSA. Each mouse received around 3.7 MBq of radioactivity. Images were reconstructed from a 60 min dynamic scan. (C) Time-activity curves of ROIs outlined over muscle, heart, liver, and bladder regions on 18F-AlF-NEB PET images. (D) Time-activity curves of ROIs outlined over muscle, heart, liver, and bladder regions on 18F-FB-MSA PET images. Reprinted with permission from ref. 99. Copyright 2014 Society of Nuclear Medicine and Molecular Imaging. (E) Multiple time-point whole-body maximum intensity projection PET images of a female healthy volunteer at 5, 10, 15, 30, 45, 60, 75, and 90 min after intravenous administration of 68Ga-NEB. Reprinted with permission from ref. 101. Copyright 2014 Society of Nuclear Medicine and Molecular Imaging.
Soon after the establishment of 18F-NEB, a first-in-human study was successfully performed with 68Ga-labeled NEB (Fig. 14A). After intravenous injection, majority of the radioactivity was retained in the blood circulation due to the stable interaction of 68Ga-NEB with serum albumin (Fig. 14B). A dosimetry study confirmed the safety with acceptable absorbed doses by critical organs even with multiple injections for one patient.
Overall, as a blood pool imaging agent, the preliminary clinical studies of 68Ga-NEB demonstrate the value of differentiating hepatic hemangioma from other benign or malignant focal hepatic lesions. In addition, NEB can be easily labeled with different positron emitters of various half-lives and demonstrates promising pharmacokinetics in humans, warranting further clinical applications of NEB-based PET tracers.
3.1.3. Labeled HSA for lymph node mapping
Besides being a blood pool imaging agent, radiolabeled or fluorophore attached HSA is often used to noninvasively identify the lymph nodes for cancer diagnosis or guiding surgery.145–147 For instance, 99mTc-HSA has been successfully applied to map sentinel lymph nodes for identifying the patients with melanoma and regional nodal micrometastasis, and exhibits a statistically better concordance rate than the radiotracers without HSA-conjugation.148,149
As another good example, 18F-NEB (Fig. 14A) has also been applied to accurately locate sentinel lymph nodes.100 After local injection, both 18F-AlF-NEB and EB form complexes with endogenous albumin in the interstitial fluid and allow for visualizing the lymphatic system. Positron emission tomography (PET) and/or optical imaging of LNs was performed in three different animal models including a hind limb inflammation model, an orthotropic breast cancer model, and a metastatic breast cancer model (Fig. 15). In these three models, the LNs can be distinguished clearly by using the blue color and the fluorescence signal from EB as well as the PET signal from 18F-NEB, suggesting that this combination of 18F-NEB and EB is potentially useful for mapping sentinel LNs and provide intraoperative guidance for clinical diagnosis.
Fig. 15.
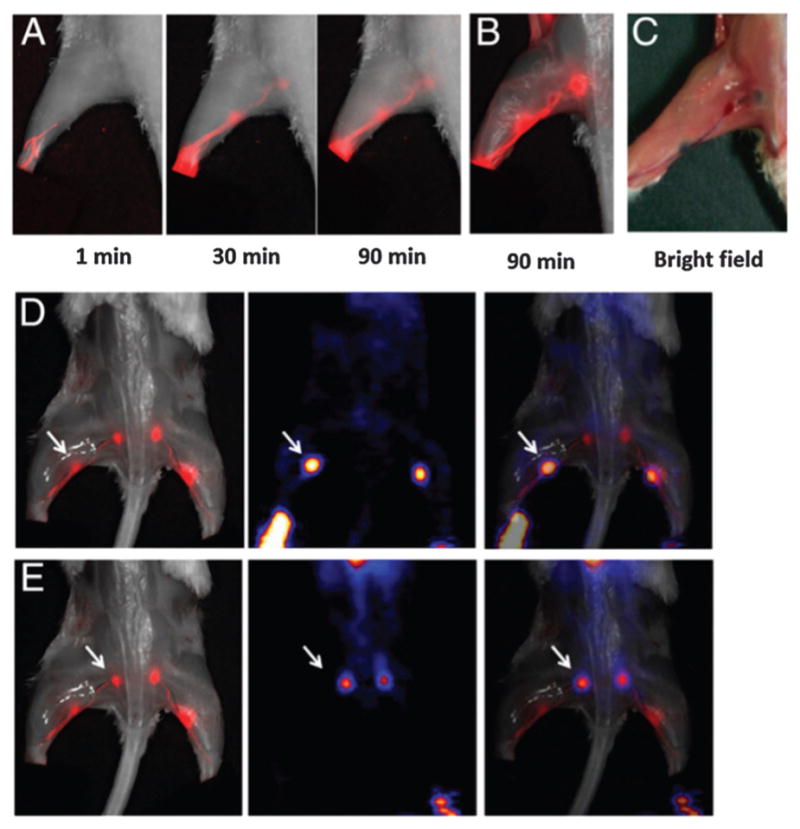
(A) Longitudinal fluorescence imaging of the lymphatic system after hock injection of 18F-AlF-NEB/EB. LNs and lymphatic vessels are clearly visible. (B) Ex vivo optical imaging of LNs without skin. (C) Photograph of the same mice to show the blue color within the LNs. (D) Co-registration of the optical image (left) and the PET image (Middle) to present the popliteal LNs, indicated by a white arrow. (E) Co-registration of the optical image (left) and the PET image (middle) to present the sciatic LNs, indicated by a white arrow. The mice were euthanized at 90 min after hock injection of 18F-AlF-NEB/EB and the skin was removed.100 Reprinted with permission from ref. 100. Copyright 2015 National Academy of Sciences.
3.2. HSA as a regulating platform for managing the blood sugar level
One of the most important clinical applications of the HSA-binding strategy is to elongate the blood circulation of anti-diabetic drugs. Up to now, three HSA-binding anti-diabetic drugs have been approved by U.S. FDA, and at least ten more candidates are under clinical tests.150–154
Glucagon-like peptide (GLP)-1 is a 30-amino acid peptide hormone secreted from gut endocrine cells in response to nutrient ingestion that promotes nutrient assimilation through regulation of gastrointestinal motility and islet hormone secretion.155 Infusion of GLP-1 into normal or diabetic human subjects stimulates insulin and inhibits glucagon secretion, thereby indirectly modulating peripheral glucose uptake and control of hepatic glucose production, therefore can enhance GLP-1 action for the treatment of type 2 diabetes.156
A major challenge for the therapeutic use of regulatory peptides, including native GLP-1, is a short circulating t1/2, due principally to rapid enzymatic inactivation and/or renal clearance. Although infusion of native GLP-1 is highly effective in lowering blood glucose in subjects with type 2 diabetes, a single subcutaneous injection of the native peptide is quickly degraded and disappears from the circulation within minutes.157 Hence, the majority of pharmaceutical approaches to the development of GLP-1 mimetic agents have focused on the development of long-acting degradation-resistant peptides, such as Albugon, monoExendin-4 HSA (E1HSA), bisExendin-4 HSA (E2HSA), and so on. The pharmaceutical characteristics and pharmacokinetic properties of these HSA-binding or HSA containing anti-diabetic drugs are summarized in Table 5.
Table 5.
Summary of HSA-based blood glucose regulatory drugs. AUCs were extrapolated by the formula of AUC = 1/2 × [(2 × biological half-life value + peak time value) × peak effect value]20,57,113,152,160–162,211
| Sample | Chemical construction | t1/2 (h) | CL/F (mL h−1 kg−1) | Vd (mL kg −1) | Cmax (ng mL−1) | AUC (ng h mL−1) | MRT (h) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Exenatide | Peptide | 0.58 ± 0.09 | 861.2 ± 164.0 | 1534.8 ± 415.9 | 0.20 ± 0.06 | 0.39 ± 0.12 | 1.39 ± 0.15 | 20 |
| Albugon | Peptide HSA perfusion | 108 ± 13 | 14.3 ± 2.8 | 91.7 ± 13.6 | 614 ± 136 | 175 000 ± 37 000 | 94.8 ± 24.0 | 152, 160 and 161 |
| E1HSA | Peptide HSA perfusion | 56.7 | 2.1 | 102 | N.A. | 206815.9 | 66.7 | 162 |
| E2HSA | Double peptide HSA perfusion | 53.4 ± 8.0 | 1.66 ± 0.27 | 125.5 ± 8.8 | 1810.1 ± 198.7 | 179182 ± 27 148 | 78.3 ± 6.2 | 57 |
| LUA-M1 | Peptide fatty acid conjugates | 4.0 ± 0.5 | 6.61 ± 2.06 | 38.87 ± 15.30 | 892.66 ± 249.31 | 6940.4 ± 2571.5 | 7.85 ± 0.32 | 113 |
| Ex-PEG (5 kDa) | Peptide PEG conjugates | 6.1 ± 0.8 | 37.5 ± 13.5 | 310 ± 79.1 | 76.1 ± 14.5 | 101.0 ± 25.8 | 7.9 ± 0.3 | 211 |
| Ex-PEG (20 kDa) | Peptide PEG conjugates | 49.4 ± 7.7 | 4.4 ± 0.4 | 272.9 ± 63.2 | 154.0 ± 5.3 | 893.5 ± 119.3 | 53.1 ± 0.6 | 211 |
| Ex-PEG (40 kDa) | Peptide PEG conjugates | 76.4 ± 7.4 | 2.3 ± 0.3 | 259.0 ± 61.6 | 148.1 ± 24.5 | 1780.7 ± 279.9 | 78.9 ± 0.4 | 211 |
Albugon, or E1HSA, is a recombinant exendin-4-human serum albumin (HSA) fusion protein which retains the GLP-1 receptor binding activity of exendin-4 and as such is expected to exert glucose lowering effects with a prolonged duration (Fig. 16A).158 In order to effectively bind to the GLP-1 receptor, HSA was fused at the C-terminus of Ex4 and a 5-aa linker (GGGGS) was inserted between them.159 To determine the in vivo bioactivity of E1HSA, an oral glucose tolerance test (OGTT) was performed in diabetic db/db mice by a single injection of E1HSA. As shown in Fig. 16B–D, glucose tolerance in diabetic db/db mice was effectively improved by E1HSA over the control group. In addition, an obvious dose-effect relationship was observed between the postdose serum glucose concentration and injection dose. Moreover, postdose time-course observation indicated that the glucose-lowering effect of E1HSA lasted for at least 24 hours.57
Fig. 16.

(A) Schematic structure of E1HSA; (B) E1HSA lowers the blood glucose in db/db mice by an oral glucose tolerance test (OGTT). Single dose of E1HSA (0.3, 1, and 3 mg kg−1) or HSA (3 mg kg−1) were injected intraperioneally in mice. OGTT was carried out at various times postdose to evaluate the duration of E1HSA action (B): 12 h; (C): 24 h; (D): 36 h. Values are expressed as means ± SE; n = 5 mice per group.158 Reprinted with permission from ref. 58. Copyright 2007 European Peptide Society and John Wiley & Sons, Ltd.
3.3. HSA as a carrier for precision cancer therapy
HSA has long been a versatile drug carrier for developing effective anti-cancer agents. Upon binding to HSA, both the pharmaco-kinetics and pharmaceutical profiles of chemotherapeutic drugs may be changed to give better drug delivery efficiency as well as a less side effect. In this section, we summarize some of the recent developments in the field of HSA–drug conjugates, with the focus on chemotherapy and radiotherapy for cancers.
3.3.1. Albumin-bound drug nanoparticle increases the therapeutic index of conventional chemotherapy drugs
In general, paclitaxel and other chemotherapeutic drugs are hydrophobic and thereby have poor solubility in blood circulation (Fig. 17A).2,164 To solve the problem, organic agents including polyethylated castor oil (Cremophor® EL) and ethanol are required in their clinical formulations as their vehicles.165,166 Nevertheless, these vehicles often cause severe toxicities, requiring prolonged infusion or premedication to reduce the risk of hypersensitivity reaction. Interestingly, albumin binds to many types of hydrophobic molecules in a reversible manner and consequently can help to transport the drugs in the body.10,167 Moreover, as an intrinsic protein carrier in the blood, utilizing albumin as the drug vehicle avoids the risk of hypersensitivity reaction caused by the artificial formulation, is thus capable of serving as a clinically safer platform to deliver hydrophobic drugs in the body.
Fig. 17.

(A) Chemical structure of paclitaxel, which is a hydrophobic small molecule with poor solubility in the blood. (B and C) Representative structures of small drug loaded albumin nanoparticles, and the diameters of this complex is between 80 to 150 nm with a mean value of 130 nm. Reprinted with permission from ref. 163. Copyright 2010 Elsevier B.V. (D) Process of gp60-mediated transcytosis of albumin across the vascular endothelium. The endothelial transcytosis of albumin is started by binding to the 60 kDa glycoprotein (gp60) receptor on the cell surface. This interaction induces caveolin and results in invagination and pinching off of the endothelial cell membrane, thereby concentrating and transporting the albumin complex into vesicular structures denoted as caveolae (“little caves”).
Besides the reduced toxicity and less immunogenicity, albumin also assists the transportation of plasma constituents through endothelial cells via albumin receptor binding. Traditionally, only the unbound drugs were thought to be able to penetrate the vascular wall via junctional gaps between endothelial cells.168 Nonetheless, a selective transportation mechanism was disclosed recently that albumin-bound molecules can cross vascular endothelium through albumin transcytosis.169–171 This process, illustrated in Fig. 17D, is thought to play a key role in delivering proteins across the vascular endothelium in order to meet the nutritional needs of cells. Because of its abnormal requirement of nutrition, tumors often take a higher level of albumin than healthy tissues, and thereby albumin-bound drugs can be delivered to tumor with better selectivity.
SPARC, which is short for the secreted protein that is acidic and rich in cysteine, is an extracellular matrix glycoprotein that is essentially related to tumor metastasis. It has been shown to be overexpressed on cancer cells and associated with poor prognosis in a number of tumors.172 Interestingly, recent evidence suggests that albumin exhibits high binding affinity to SPARC,173–176 and the tumor secretion of SPARC also plays a key role for the high tumor uptake of albumin.177,178 Therefore, the SPARC-inducing effect accumulates albumin to the areas of tumor that may further improve the delivery efficiency for albumin bound drugs (Fig. 17B and C).
Small molecular drug loaded albumin nanoparticles can be prepared in a number of ways, namely, desolvation,179,180 emulsification,181,182 thermal gelation,183 nanospray drying,184 nab-technology,163 and self-assembly.185 Here, we will mainly focus on nab™-Technology, which is a biologically interactive delivery system that uses the biochemical properties of albumin to increase drug delivery to tumors. The first commercial product using this technology, Abraxane (nab-paclitaxel), is a solvent-free, 130 nm albumin particle form of paclitaxel. An in vitro experiment conducted by human lung microvessel endothelial cells indicated that the transportation of fluorescently labelled paclitaxel is about 4.2-fold greater rate across an endothelial cell monolayer when formulated as nab-paclitaxel than CrEL-paclitaxel (CrEL: Cremophor®EL, polyethylated castor oil). Because of its comparatively better efficacy for cancer treatment, Abraxane was approved by FDA in 2005 for the treatment of breast cancer cases where cancer did not respond to other chemotherapy (Fig. 18). In 2012 and 2013, Abraxane received approval from FDA to be used for the treatment of non-small cell lung cancer (NSCLC) as well as advanced prostate cancer because of its less toxicity during the treatment.
Fig. 18.

(A) Time to disease progression in a phase III comparative trial of nab-paclitaxel versus CrEL-paclitaxel. Reprinted with permission from ref. 164. Copyright 2005 American Society of Clinical Oncology. (B) Better efficacy of albumin-bound paclitaxel, compared with polyethylated castor oil-based paclitaxel in women with metastatic breast cancer. Reprinted with permission from ref. 163. Copyright 2010 Elsevier B.V.
3.3.2. HSA–nanoparticle (NP) complex as a theranostic platform for diagnostic imaging and small molecular drug delivery
In the past decade, the HSA–NP complex has been developed as a common nanoplatform with both imaging and therapeutic functions, denoted as “nanotheranostics”.2,26–28,85–90 For instance, after coupling with targeting ligands and imaging moieties, iron oxide nano-particles (IONPs) can provide many potential applications including multimodality imaging and therapy. In addition, HSA coated nanoparticles generally give reduced accumulation in mononuclear phagocytic system-related organs over the naked nanoparticles.85,87–91,186 In a pilot study, doxorubicin (Dox) was encapsulated into the HINPs (HSA coated iron oxide nanoparticles). About 0.5 mg of Dox and 1 mg of IONPs (iron oxide nanoparticles) could be loaded based on 10 mg of HSA matrices. The resulting D-HINPs (Dox loaded HINPs) could release Dox in a sustained fashion and effectively suppressed tumor growth that was much better than free Dox on a 4T1 murine breast cancer xenograft model.186
This strategy was then extended to load other types of small molecules and to build a multimodal-imaging platform (Fig. 19). This combinational MRI/PET/NIRF theranostics nanosystem is capable of integrating the strengths of high anatomical resolution (MRI), in vitro validation (NIRF), quantitative evaluation (PET) and cancer treatment, and therefore can be a platform technology in theranostics.27,89–91,187
Fig. 19.

(A) A brief scheme to describe the preparation of albumin-coated IONP. Reprinted with permission from ref. 87. Copyright 2012 American Chemical Society (B) MR images taken before, and 1 and 4 h after the injection of NPs (6 mg of Fe per mL). As illustrated here, the contrast enhancement was decreased from 26.1% to 5.2% and then 4.3% at 0 h, 1 h and 4 h p.i., which was the result of tumor accumulation of HINPs. Reprinted with permission from ref. 87. Copyright 2011 American Chemical Society. (C) Schematic illustration of the multi-functional HSA–IONPs. (D) Representative in vivo NIRF images of mouse injected with HSA–IONPs. Images were acquired 1 h, 4 h and 18 h post injection. (E) In vivo PET imaging results of mouse injected with HSA–IONPs. Images were acquired by 1 h, 4 h and 18 hours of post injection. (F) MRI images acquired before and 18 h post injection.87,89,90,186 Reprinted with permission from ref. 91. Copyright 2010 Elsevier B.V.
3.3.3. Small molecule HSA conjugates for cancer chemotherapy
The first HSA–drug conjugate that was evaluated in clinical trials was a HSA-conjugated chemotherapeutic drug: methotrexate–HSA conjugate (MTX–HSA). A phase I study with 17 patients treated with weekly MTX–HSA77 found that two patients with renal cell carcinoma and one patient with mesothelioma responded to MTX–HSA therapy (one partial response, two minor responses). However, the clinical trial stopped at phase II as no objective response was seen with metastatic renal carcinoma.78 The failure was most likely attributed to the drawbacks of MTX–HSA, such as the unclear chemical structure and unclear metabolic pathway of MTX–HSA.
By taking advantage of the in vivo maleimide–HSA conjugation strategy which was detailed in Section 2.2, DOXO-EMCH was highly effective in preclinical tumor models (Fig. 20). As expected, there was a pronounced difference between the levels of DOXO-EMCH and doxorubicin in the serum of MDA-MB-435 tumor mice. A good antitumor effect was achieved at 3 × 16 mg kg−1 doxorubicin equivalents and complete remission was found at 3 × 24 mg kg−1. Notably, preliminary toxicity studies in nude mice showed that the maximum tolerated dose of DOXO-EMCH was approximately 4.5 times higher than that of free doxorubicin.188
Fig. 20.

(A) Biodistribution study in MDA-MB-435 xenografted mice with radiolabeled doxorubicin or DOXO-EMCH (organ values were corrected for blood volume); (B) curves depicting tumor growth inhibition of subcutaneously implanted MDA-MB-435 tumor under therapy with doxorubicin and DOXO-EMCH.188,189 Reprinted with permission from ref. 4. Copyright 2008 (ref. 4) Elsevier B.V.
DOXO-EMCH entered clinical trial in 2007, and was renamed INNO-206 or Aldoxorubicin in 2008. The on-going clinical studies suggest that INNO-206 can be administered safely at higher doses in patients than free doxorubicin, resulting in better efficacy compared with the currently available anthracyclines to treat several types of cancer.188,190,191
3.3.4. Radiolabeled HSA-conjugate for internal radiotherapy of cancer
In 2013, Shibili and his colleagues from ETH reported a strategy in which a DOTA–folate conjugate was coupled with a small molecule albumin binder, denoted as cm09. Radiolabeled folic acid derivatives have been used for folate receptor (FR) targeted imaging and therapy.192–194 However, using folate-based radiopharmaceuticals for therapy has long been regarded as an unattainable goal because of the poor tumor-to-kidney uptake ratio. As known, the rapid clearance of DOTA–folate conjugates from the blood circulation is generally considered as an advantage over the other targeting strategies.195–197 It is because rapid clearance usually gives high tumor-to-background contrast and therefore minimizes the exposure of major organs to the therapeutic probe.198–204 However, this pharmacokinetics is a double-bladed sword that is also responsible for the relatively low uptake of folate conjugates in tumor tissue and an extremely high accumulation of radioactivity in the kidneys.205–209 In addition, once the folate conjugate is cleared from blood into the renal system, most of them would be strongly trapped by the folate-binding protein in the kidneys, and therefore the kidney uptake of folate will not decrease over time.209 To solve this problem, this group reasoned that a HSA-binding radio-pharmaceutical could change this dissatisfying situation as prolonged blood circulation could improve the tumor uptake, and reduce the problematic renal accumulation of the DOTA–folate conjugates.182,183
As shown in Fig. 21, installation of an albumin-binding entity into the structure of a folate-based radioconjugate improved the overall tissue distribution significantly. Tumor uptake was doubled, and kidney retention was reduced to 30% of the value obtained with folate conjugates without an albumin-binding entity. In addition, tumor growth inhibition was observed without radiotoxic side effects.103
4. Conclusions and outlook
As the key circulating protein in the blood circulation, albumin has been an excellent delivery platform for a number of endogenous and exogenous compounds. It has also been used to extend blood half-life and reduce renal clearance of both imaging probes and therapeutic drugs.
An ideal albumin-binding imaging probe may not only have a slow clearance from the blood, but also truly reflect a clear biological pathway in the body, viz., the signal it provides needs to be correlated with circulation, metabolism and bioactivity of natural albumin. To accomplish this goal, the labeled albumin should be indistinguishable with natural albumin for in vivo bioactivity, and thus the imaging tag should be small in size, free of charge and stable in vivo. In addition, detachment of imaging reporters from the imaging probe should be avoided as it often gives misleading information for clinical diagnosis, and therefore the binding strategy to HSA has to be robust, covalent and irreversible, though some of the non-covalent binding strategies (e.g. Evans blue NOTA derivatives) also give promising results in the clinic. In addition, considering the clinical practice and operational simplicity, the HSA-binding imaging probe would better be a small molecule with unambiguous definition of chemistry; consequently the in vivo targeting strategy will be one of the choices for the future development of HSA-binding imaging probes.
In addition, in order to develop a more convenient and possibly less expensive treatment for diabetes, a HSA-binding blood glucose regulator would ideally have the longest if possible glucose-lowering effect without an apparent side effect to the patients. Meanwhile, to design a better HSA binding cancer therapeutic drug, the key here is to improve the tumor specificity, meaning increasing the tumor uptake while reducing the unnecessary cytotoxicity on healthy tissues. If possible, on-site drug release would be preferred, as it may essentially reduce the side effect since lower therapeutic dose would be applied to the patients.
In conclusion, as in vitro HSA conjugation chemistry has been well established, it is believed that the future of HSA-binding chemistry should focus on developing new in vivo HSA binders, either covalent or non-covalent. In addition, when a functional moiety is covalently coupled to HSA, the nonspecific adsorption of small molecules onto HSA is hard to be removed or purified, which is always a concern but can be avoided by in vivo targeting approaches. In the case of in vivo covalent binding, a faster and more bio-orthogonal conjugation method is in great need to improve the efficiency and selectivity of the binding reaction. For in vivo non-covalent binders, systematic chemical screening is necessary to develop a series of HSA binders toward different binding sites with various binding affinities. It is noteworthy that the strongest binder is not always in favor, as we may need a balance between blood retention and clearance in certain circumstances. Moreover, the space linker between the HSA-binding moiety and the functional molecule also needs more comprehensive investigation, and the ultimate goal would be a linker design that does not compromise the function of the albumin binder as well as the molecules of interest.
Biographies

Zhibo Liu
Zhibo (Zippo) Liu received his BS in Chemistry from Nanjing University in 2010, and PhD in Chemistry from the University of British Columbia in 2014. Under the supervision of Drs David M. Perrin, Kuo-Shyan Lin, and François Bénard, he developed a broadly applicable one-step 18F-labeling method based on a novel organo-trifluoroborate. Right after graduation, he joined the Laboratory of Molecular Imaging and Nanomedicine (LOMIN) at National Institutes of Health (NIH) as a postdoctoral research fellow under the supervision of Dr Xiaoyuan (Shawn) Chen. His research focuses on developing albumin-binding imaging probes as well as utilizing boramino acid to identify tumor from inflammation for early cancer diagnosis.

Xiaoyuan Chen
Xiaoyuan (Shawn) Chen received his PhD in chemistry from the University of Idaho in 1999. After being a faculty member at the University of Southern California and Stanford University, he joined the Intramural Research Program of the NIBIB in 2009 as a Senior Investigator and Chief of the Laboratory of Molecular Imaging and Nanomedicine (LOMIN). Dr Chen has published over 500 papers (H-index = 90) and numerous books and book chapters. He sits on the editorial board of over 10 peer-reviewed journals and is the founding editor of the journal Theranostics. His lab focuses on developing molecular imaging probes and nanotechnologies for early diagnosis of disease, monitoring therapy responses, and guiding drug discovery/development.
References
- 1.Kramer PA. J Pharm Sci. 1974;63:1646–1647. doi: 10.1002/jps.2600631044. [DOI] [PubMed] [Google Scholar]
- 2.Cho K, Wang X, Nie S, Chen Z, Shin DM. Clin Cancer Res. 2008;14:1310–1316. doi: 10.1158/1078-0432.CCR-07-1441. [DOI] [PubMed] [Google Scholar]
- 3.Wunder A, Muller-Ladner U, Stelzer E, Neumann E, Sinn H, Gay S, Fiehn C. Arthritis Res Ther. 2003;170:4793–4801. doi: 10.4049/jimmunol.170.9.4793. [DOI] [PubMed] [Google Scholar]
- 4.Kratz F. J Controlled Release. 2008;132:171–183. doi: 10.1016/j.jconrel.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 5.Koehler MFT, Zobel K, Beresini MH, Caris LD, Combs D, Paasch BD, Lazarus RA. Bioorg Med Chem Lett. 2002;12:2883–2886. doi: 10.1016/s0960-894x(02)00610-8. [DOI] [PubMed] [Google Scholar]
- 6.Putnam FW. In: All About Albumin. Peters T, editor. Academic Press; San Diego: 1995. pp. xi–xiii. [Google Scholar]
- 7.Sleep D, Cameron J, Evans LR. Biochim Biophys Acta. 2013;1830:5526–5534. doi: 10.1016/j.bbagen.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 8.Anderson CL, Chaudhury C, Kim J, Bronson CL, Wani MA, Mohanty S. Trends Immunol. 2006;27:343–348. doi: 10.1016/j.it.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 9.Kim J, Hayton WL, Robinson JM, Anderson CL. Clin Immunol. 2007;122:146–155. doi: 10.1016/j.clim.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elzoghby AO, Samy WM, Elgindy NA. J Controlled Release. 2012;157:168–182. doi: 10.1016/j.jconrel.2011.07.031. [DOI] [PubMed] [Google Scholar]
- 11.Neumann E, Frei E, Funk D, Becker MD, Schrenk H-H, Müller-Ladner U, Fiehn C. Expert Opin Drug Delivery. 2010;7:915–925. doi: 10.1517/17425247.2010.498474. [DOI] [PubMed] [Google Scholar]
- 12.Verrecchia T, Spenlehauer G, Bazile DV, Murry-Brelier A, Archimbaud Y, Veillard M. J Controlled Release. 1995;36:49–61. [Google Scholar]
- 13.Lu W, Zhang Y, Tan Y-Z, Hu K-L, Jiang X-G, Fu S-K. J Controlled Release. 2005;107:428–448. doi: 10.1016/j.jconrel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 14.Baron MH, Baltimore D. Cell. 1982;28:395–404. doi: 10.1016/0092-8674(82)90357-9. [DOI] [PubMed] [Google Scholar]
- 15.Bansal R, Prakash J, Ruijter Md, Beljaars L, Poelstra K. Mol Pharmaceutics. 2011;8:1899–1909. doi: 10.1021/mp200263q. [DOI] [PubMed] [Google Scholar]
- 16.Ambros V, Baltimore D. J Biol Chem. 1978;253:5263–5266. [PubMed] [Google Scholar]
- 17.Schilling U, Friedrich EA, Sinn H, Schrenk HH, Clorius JH, Maier-Borst W. Int J Radiat Appl Instrum, Part B. 1992;19:685–695. doi: 10.1016/0883-2897(92)90103-6. [DOI] [PubMed] [Google Scholar]
- 18.Tilton RD, Robertson CR, Gast AP. J Colloid Interface Sci. 1990;137:192–203. [Google Scholar]
- 19.Lenkei R, Onica D, Ghetie V. Experientia. 1977;33:1046–1047. doi: 10.1007/BF01945961. [DOI] [PubMed] [Google Scholar]
- 20.Elsadek B, Kratz F. J Controlled Release. 2012;157:4–28. doi: 10.1016/j.jconrel.2011.09.069. [DOI] [PubMed] [Google Scholar]
- 21.Sleep D. Expert Opin Drug Delivery. 2014;12:793–812. doi: 10.1517/17425247.2015.993313. [DOI] [PubMed] [Google Scholar]
- 22.Heneweer C, Holland JP, Divilov V, Carlin S, Lewis JS. J Nucl Med. 2011;52:625–633. doi: 10.2967/jnumed.110.083998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cavalu S, Damian G, Dânşoreanu M. Biophys Chem. 2002;99:181–188. doi: 10.1016/s0301-4622(02)00182-5. [DOI] [PubMed] [Google Scholar]
- 24.Wunder A, Müller-Ladner U, Stelzer EHK, Funk J, Neumann E, Stehle G, Pap T, Sinn H, Gay S, Fiehn C. J Immunol. 2003;170:4793–4801. doi: 10.4049/jimmunol.170.9.4793. [DOI] [PubMed] [Google Scholar]
- 25.Bolling C, Graefe T, Lübbing C, Jankevicius F, Uktveris S, Cesas A, Meyer-Moldenhauer WH, Starkmann H, Weigel M, Burk K, Hanauske AR. Invest New Drugs. 2006;24:521–527. doi: 10.1007/s10637-006-8221-6. [DOI] [PubMed] [Google Scholar]
- 26.Wang W, Huang Y, Zhao S, Shao T, Cheng Y. Chem Commun. 2013;49:2234–2236. doi: 10.1039/c3cc38397k. [DOI] [PubMed] [Google Scholar]
- 27.Zhao S, Wang W, Huang Y, Fu Y, Cheng Y. MedChemComm. 2014;5:1658–1663. [Google Scholar]
- 28.Mier W, Hoffend J, Krämer S, Schuhmacher J, Hull WE, Eisenhut M, Haberkorn U. Bioconjugate Chem. 2005;16:237–240. doi: 10.1021/bc034216c. [DOI] [PubMed] [Google Scholar]
- 29.Spanoghe M, Lanens D, Dommisse R, Van der Linden A, Alderweireldt F. Magn Reson Imaging. 1992;10:913–917. doi: 10.1016/0730-725x(92)90445-6. [DOI] [PubMed] [Google Scholar]
- 30.Aldini R, Roda A, Labate AM, Cappelleri G, Roda E, Barbara L. J Lipid Res. 1982;23:1167–1173. [PubMed] [Google Scholar]
- 31.Hettick JM, Siegel PD, Green BJ, Liu J, Wisnewski AV. Anal Biochem. 2012;421:706–711. doi: 10.1016/j.ab.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong K, Cleland LG, Poznansky MJ. Agents Actions. 1980;10:231–239. doi: 10.1007/BF02025941. [DOI] [PubMed] [Google Scholar]
- 33.Shen WC, Ryser HJ. Proc Natl Acad Sci U S A. 1978;75:1872–1876. doi: 10.1073/pnas.75.4.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lau S, Graham B, Cao N, Boyd BJ, Pouton CW, White PJ. Mol Pharmaceutics. 2012;9:71–80. doi: 10.1021/mp2002522. [DOI] [PubMed] [Google Scholar]
- 35.McMenamy RH, Madeja MI, Watson F. J Biol Chem. 1968;243:2328–2336. [PubMed] [Google Scholar]
- 36.Choi JY, Jeong JM, Yoo BC, Kim K, Kim Y, Yang BY, Lee Y-S, Lee DS, Chung J-K, Lee MC. Nucl Med Biol. 2011;38:371–379. doi: 10.1016/j.nucmedbio.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 37.Zhang YZ, Wang X, Feng Y, Li J, Lim CT, Ramakrishna S. Biomacromolecules. 2006;7:1049–1057. doi: 10.1021/bm050743i. [DOI] [PubMed] [Google Scholar]
- 38.Håkansson L, Venge PER. APMIS. 1994;102:308–316. doi: 10.1111/j.1699-0463.1994.tb04880.x. [DOI] [PubMed] [Google Scholar]
- 39.Hopf U, Büschenfelde K-HMz, Dierich MP. J Immunol. 1976;117:639–645. [PubMed] [Google Scholar]
- 40.Mansour AM, Drevs J, Esser N, Hamada FM, Badary OA, Unger C, Fichtner I, Kratz F. Cancer Res. 2003;63:4062–4066. [PubMed] [Google Scholar]
- 41.Becker JM, Wilchek M, Katchalski E. Proc Natl Acad Sci U S A. 1971;68:2604–2607. doi: 10.1073/pnas.68.10.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kasina S, Rao TN, Srinivasan A, Sanderson JA, Fitzner JN, Reno JM, Beaumier PL, Fritzberg AR. J Nucl Med. 1991;32:1445–1451. [PubMed] [Google Scholar]
- 43.Even GA, Green MA. Int J Radiat Appl Instrum, Part B. 1989;16:319–321. doi: 10.1016/0883-2897(89)90014-7. [DOI] [PubMed] [Google Scholar]
- 44.Chang YS, Jeong JM, Lee Y-S, Kim HW, Rai GB, Lee SJ, Lee DS, Chung J-K, Lee MC. Bioconjugate Chem. 2005;16:1329–1333. doi: 10.1021/bc050086r. [DOI] [PubMed] [Google Scholar]
- 45.Wu S-Y, Kuo J-W, Chang T-K, Liu R-S, Lee R-C, Wang S-J, Lin W-J, Wang H-E. Nucl Med Biol. 2012;39:1026–1033. doi: 10.1016/j.nucmedbio.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 46.Liu ZB, Li Y, Lozada J, Schaffer P, Adam MJ, Ruth TJ, Perrin DM. Angew Chem, Int Ed. 2013;52:2303–2307. doi: 10.1002/anie.201208551. [DOI] [PubMed] [Google Scholar]
- 47.Liu Z, Chen H, Chen K, Shao Y, Kiesewetter DO, Niu G, Chen X. Sci Adv. 2015;1:e1500694. doi: 10.1126/sciadv.1500694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Šimeček J, Notni J, Kapp TG, Kessler H, Wester H-J. Mol Pharmaceutics. 2014;11:1687–1695. doi: 10.1021/mp5000746. [DOI] [PubMed] [Google Scholar]
- 49.Bernard-Gauthier V, Bailey JJ, Liu Z, Wängler B, Wängler C, Jurkschat K, Perrin DM, Schirrmacher R. Bioconjugate Chem. 2015 doi: 10.1021/acs.bioconjchem.5b00560. [DOI] [PubMed] [Google Scholar]
- 50.Liu Z, Li Y, Lozada J, Wong MQ, Greene J, Lin K-S, Yapp D, Perrin DM. Nucl Med Biol. 2013;40:841–849. doi: 10.1016/j.nucmedbio.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 51.Palmowski M, Goedicke A, Vogg A, Christ G, Mühlenbruch G, Kaiser H, Günther R, Kuhl C, Mottaghy F, Behrendt F. Eur Radiol. 2013;23:3062–3070. doi: 10.1007/s00330-013-2916-9. [DOI] [PubMed] [Google Scholar]
- 52.Ogan MD, Schmiedl U, Moseley ME, Grodd W, Paajanen H, Brasch RC. Invest Radiol. 1987;22:665–671. [PubMed] [Google Scholar]
- 53.Todica A, Brunner S, Böning G, Lehner S, Nekolla S, Wildgruber M, Übleis C, Wängler C, Sauter M, Klingel K, Cumming P, Bartenstein P, Schirrmacher R, Franz W, Hacker M. Mol Imaging Biol. 2013;15:441–449. doi: 10.1007/s11307-013-0618-y. [DOI] [PubMed] [Google Scholar]
- 54.Kratz F, Drevs J, Bing G, Stockmar C, Scheuermann K, Lazar P, Unger C. Bioorg Med Chem Lett. 2001;11:2001–2006. doi: 10.1016/s0960-894x(01)00354-7. [DOI] [PubMed] [Google Scholar]
- 55.Rong P, Huang P, Liu Z, Lin J, Jin A, Ma Y, Niu G, Yu L, Zeng W, Wang W, Chen X. Nanoscale. 2015;7:16330–16336. doi: 10.1039/c5nr04428f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burke CW, Shakespear RA. J Endocrinol. 1975;65:133–138. doi: 10.1677/joe.0.0650133. [DOI] [PubMed] [Google Scholar]
- 57.Zhang L, Wang L, Meng Z, Gan H, Gu R, Wu Z, Gao L, Zhu X, Sun W, Li J, Zheng Y, Dou G. Biochem Biophys Res Commun. 2014;445:511–516. doi: 10.1016/j.bbrc.2014.02.045. [DOI] [PubMed] [Google Scholar]
- 58.Liu X, Bi X, Huang J, Jerecic R, Carr J, Li D. Invest Radiol. 2008;43:663–668. doi: 10.1097/RLI.0b013e31817ed1ff. [DOI] [PubMed] [Google Scholar]
- 59.Melder R, Osborn B, Riccobene T, Kanakaraj P, Wei P, Chen G, Stolow D, Halpern W, Migone T-S, Wang Q, Grzegorzewski K, Gallant G. Cancer Immunol Immunother. 2005;54:535–547. doi: 10.1007/s00262-004-0624-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kratz F, Müller-Driver R, Hofmann I, Drevs J, Unger C. J Med Chem. 2000;43:1253–1256. doi: 10.1021/jm9905864. [DOI] [PubMed] [Google Scholar]
- 61.Etoh T, Miyazaki M, Harada K, Nakayama M, Sugii A. J Chromatogr B: Biomed Sci Appl. 1992;578:292–296. doi: 10.1016/0378-4347(92)80428-s. [DOI] [PubMed] [Google Scholar]
- 62.Era S, Hamaguchi T, Sogami M, Kuwata K, Suzuki E, Miura K, Kawai K, Kitazawa Y, Okabe H, Noma A, Miyata S. Int J Pept Protein Res. 1988;31:435–442. doi: 10.1111/j.1399-3011.1988.tb00900.x. [DOI] [PubMed] [Google Scholar]
- 63.Coleman RD, Kim TW, Gotto AM, Jr, Yang C-y. Biochim Biophys Acta. 1990;1037:129–132. doi: 10.1016/0167-4838(90)90111-r. [DOI] [PubMed] [Google Scholar]
- 64.Ferguson E, Singh RJ, Hogg N, Kalyanaraman B. Arch Biochem Biophys. 1997;341:287–294. doi: 10.1006/abbi.1997.9975. [DOI] [PubMed] [Google Scholar]
- 65.Yang CY, Kim TW, Weng SA, Lee BR, Yang ML, Gotto AM. Proc Natl Acad Sci U S A. 1990;87:5523–5527. doi: 10.1073/pnas.87.14.5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Smith DE, Mosher DF, Johnson RB, Furcht LT. J Biol Chem. 1982;257:5831–5838. [PubMed] [Google Scholar]
- 67.Larsson LJ, Lindahl P, Hallén-Sandgren C, Björk I. Biochem J. 1987;243:47–54. doi: 10.1042/bj2430047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zissimopoulos S, Marsh J, Stannard L, Seidel M, Lai FA. Biochem J. 2014;459:265–273. doi: 10.1042/BJ20131061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kratz F, Warnecke A, Scheuermann K, Stockmar C, Schwab J, Lazar P, Drückes P, Esser N, Drevs J, Rognan D, Bissantz C, Hinderling C, Folkers G, Fichtner I, Unger C. J Med Chem. 2002;45:5523–5533. doi: 10.1021/jm020276c. [DOI] [PubMed] [Google Scholar]
- 70.Schmid B, Chung D-E, Warnecke A, Fichtner I, Kratz F. Bioconjugate Chem. 2007;18:702–716. doi: 10.1021/bc0602735. [DOI] [PubMed] [Google Scholar]
- 71.Chung D-E, Kratz F. Bioorg Med Chem Lett. 2006;16:5157–5163. doi: 10.1016/j.bmcl.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 72.Kratz F, Mansour A, Soltau J, Warnecke A, Fichtner I, Unger C, Drevs J. Arch Pharm. 2005;338:462–472. doi: 10.1002/ardp.200500130. [DOI] [PubMed] [Google Scholar]
- 73.Warnecke A, Fichtner I, Saß G, Kratz F. Arch Pharm. 2007;340:389–395. doi: 10.1002/ardp.200700025. [DOI] [PubMed] [Google Scholar]
- 74.Schmid B, Warnecke A, Fichtner I, Jung M, Kratz F. Bioconjugate Chem. 2007;18:1786–1799. doi: 10.1021/bc0700842. [DOI] [PubMed] [Google Scholar]
- 75.Warnecke A, Fichtner I, Garmann D, Jaehde U, Kratz F. Bioconjugate Chem. 2004;15:1349–1359. doi: 10.1021/bc049829j. [DOI] [PubMed] [Google Scholar]
- 76.Abu Ajaj K, Graeser R, Fichtner I, Kratz F. Cancer Chemother Pharmacol. 2009;64:413–418. doi: 10.1007/s00280-009-0942-8. [DOI] [PubMed] [Google Scholar]
- 77.Hartung G, Stehle G, Sinn H, Wunder A, Schrenk HH, Heeger S, Kränzle M, Edler L, Frei E, Fiebig HH, Heene DL, Maier-Borst W, Queisser W. Clin Cancer Res. 1999;5:753–759. [PubMed] [Google Scholar]
- 78.Vis A, van der Gaast A, van Rhijn B, Catsburg T, Schmidt C, Mickisch G. Cancer Chemother Pharmacol. 2002;49:342–345. doi: 10.1007/s00280-001-0417-z. [DOI] [PubMed] [Google Scholar]
- 79.Ross PD, Subramanian S. Biochemistry. 1981;20:3096–3102. doi: 10.1021/bi00514a017. [DOI] [PubMed] [Google Scholar]
- 80.Grymonpré KR, Staggemeier BA, Dubin PL, Mattison KW. Biomacromolecules. 2001;2:422–429. doi: 10.1021/bm005656z. [DOI] [PubMed] [Google Scholar]
- 81.Bledin AG, Kantarjian HM, Kim EE, Wallace S, Chuang VP, Patt YZ, Haynie TP. Am J Roentgenol. 1982;139:711–715. doi: 10.2214/ajr.139.4.711. [DOI] [PubMed] [Google Scholar]
- 82.Watanabe N, Shirakami Y, Tomiyoshi K, Oriuchi N, Hirano T, Higuchi T, Inoue T, Endo K. J Nucl Med. 1997;38:1590–1592. [PubMed] [Google Scholar]
- 83.Miskowiak J, Nielsen SL, Munck O. Radiology. 1981;141:499–504. doi: 10.1148/radiology.141.2.6457317. [DOI] [PubMed] [Google Scholar]
- 84.Zhao F, Shen G, Chen C, Xing R, Zou Q, Ma G, Yan X. Chem – Eur J. 2014;20:6880–6887. doi: 10.1002/chem.201400348. [DOI] [PubMed] [Google Scholar]
- 85.Lee H-Y, Li Z, Chen K, Hsu AR, Xu C, Xie J, Sun S, Chen X. J Nucl Med. 2008;49:1371–1379. doi: 10.2967/jnumed.108.051243. [DOI] [PubMed] [Google Scholar]
- 86.Swierczewska M, Lee S, Chen X. Mol Imaging. 2011;10:3–16. [PMC free article] [PubMed] [Google Scholar]
- 87.Quan Q, Xie J, Gao H, Yang M, Zhang F, Liu G, Lin X, Wang A, Eden HS, Lee S, Zhang G, Chen X. Mol Pharmaceutics. 2011;8:1669–1676. doi: 10.1021/mp200006f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gao J, Chen K, Xie R, Xie J, Lee S, Cheng Z, Peng X, Chen X. Small. 2010;6:256–261. doi: 10.1002/smll.200901672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Huang J, Bu L, Xie J, Chen K, Cheng Z, Li X, Chen X. ACS Nano. 2010;4:7151–7160. doi: 10.1021/nn101643u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xie J, Liu G, Eden HS, Ai H, Chen X. Acc Chem Res. 2011;44:883–892. doi: 10.1021/ar200044b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xie J, Chen K, Huang J, Lee S, Wang J, Gao J, Li X, Chen X. Biomaterials. 2010;31:3016–3022. doi: 10.1016/j.biomaterials.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.He XM, Carter DC. Nature. 1992;358:209–215. doi: 10.1038/358209a0. [DOI] [PubMed] [Google Scholar]
- 93.Ghuman J, Zunszain PA, Petitpas I, Bhattacharya AA, Otagiri M, Curry S. J Mol Biol. 2005;353:38–52. doi: 10.1016/j.jmb.2005.07.075. [DOI] [PubMed] [Google Scholar]
- 94.Sudlow G, Birkett DJ, Wade DN. Mol Pharmacol. 1975;11:824–832. [PubMed] [Google Scholar]
- 95.Dockal M, Carter DC, Rüker F. J Biol Chem. 1999;274:29303–29310. doi: 10.1074/jbc.274.41.29303. [DOI] [PubMed] [Google Scholar]
- 96.Peters T., Jr . In: All About Albumin. Peters T, editor. Academic Press; San Diego: 1995. pp. 76–132. [DOI] [Google Scholar]
- 97.Wanwimolruk S, Birkett DJ, Brooks PM. Mol Pharmacol. 1983;24:458–463. [PubMed] [Google Scholar]
- 98.Crooke AC, Morris CJO. J Physiol. 1942;101:217–223. doi: 10.1113/jphysiol.1942.sp003976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Niu G, Lang L, Kiesewetter DO, Ma Y, Sun Z, Guo N, Guo J, Wu C, Chen X. J Nucl Med. 2014;55:1150–1156. doi: 10.2967/jnumed.114.139642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang Y, Lang L, Huang P, Wang Z, Jacobson O, Kiesewetter DO, Ali IU, Teng G, Niu G, Chen X. Proc Natl Acad Sci U S A. 2015;112:208–213. doi: 10.1073/pnas.1414821112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang J, Lang L, Zhu Z, Li F, Niu G, Chen X. J Nucl Med. 2015;56:1609–1614. doi: 10.2967/jnumed.115.159640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Skowronek M, Roterman I, Konieczny L, Stopa B, Rybarska J, Piekarska B. J Comput Chem. 2000;21:656–667. doi: 10.1016/s0097-8485(99)00089-3. [DOI] [PubMed] [Google Scholar]
- 103.Müller C, Schibli R. Front Oncol. 2013;3:249. doi: 10.3389/fonc.2013.00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dumelin CE, Trüssel S, Buller F, Trachsel E, Bootz F, Zhang Y, Mannocci L, Beck SC, Drumea-Mirancea M, Seeliger MW, Baltes C, Müggler T, Kranz F, Rudin M, Melkko S, Scheuermann J, Neri D. Angew Chem, Int Ed. 2008;47:3196–3201. doi: 10.1002/anie.200704936. [DOI] [PubMed] [Google Scholar]
- 105.Goodman DS. J Am Chem Soc. 1958;80:3892–3898. [Google Scholar]
- 106.Chen RF. J Biol Chem. 1967;242:173–181. [PubMed] [Google Scholar]
- 107.Spector AA. J Lipid Res. 1975;16:165–179. [PubMed] [Google Scholar]
- 108.van der Vusse GJ. Drug Metab Pharmacokinet. 2009;24:300–307. doi: 10.2133/dmpk.24.300. [DOI] [PubMed] [Google Scholar]
- 109.Alvarez JG, Storey BT. Mol Reprod Dev. 1995;42:334–346. doi: 10.1002/mrd.1080420311. [DOI] [PubMed] [Google Scholar]
- 110.Richieri GV, Anel A, Kleinfeld AM. Biochemistry. 1993;32:7574–7580. doi: 10.1021/bi00080a032. [DOI] [PubMed] [Google Scholar]
- 111.Curry S, Brick P, Franks NP. Biochim Biophys Acta. 1999;1441:131–140. doi: 10.1016/s1388-1981(99)00148-1. [DOI] [PubMed] [Google Scholar]
- 112.Fletcher JE, Spector AA, Ashbrook JD. Biochemistry. 1971;10:3229–3232. doi: 10.1021/bi00793a011. [DOI] [PubMed] [Google Scholar]
- 113.Chae SY, Choi YG, Son S, Jung SY, Lee DS, Lee KC. J Controlled Release. 2010;144:10–16. doi: 10.1016/j.jconrel.2010.01.024. [DOI] [PubMed] [Google Scholar]
- 114.Kurtzhals P, Havelund S, Jonassen I, Kiehr B, Larsen UD, Ribel U, Markussen J. Biochem J. 1995;312:725–731. doi: 10.1042/bj3120725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nagaraja TN, Croxen RL, Panda S, Knight RA, Keenan KA, Brown SL, Fenstermacher JD, Ewing JR. J Neurosci Methods. 2006;157:238–245. doi: 10.1016/j.jneumeth.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 116.Schwitter J, Saeed M, Wendland MF, Derugin N, Canet E, Brasch RC, Higgins CB. J Am Coll Cardiol. 1997;30:1086–1094. doi: 10.1016/s0735-1097(97)00245-3. [DOI] [PubMed] [Google Scholar]
- 117.McMurry TJ, Parmelee DJ, Sajiki H, Scott DM, Ouellet HS, Walovitch RC, Tyeklár Z, Dumas S, Bernard P, Nadler S, Midelfort K, Greenfield M, Troughton J, Lauffer RB. J Med Chem. 2002;45:3465–3474. doi: 10.1021/jm0102351. [DOI] [PubMed] [Google Scholar]
- 118.Reuben J. J Phys Chem. 1971;75:3164–3167. [Google Scholar]
- 119.McDonald MA, Watkin KL. Invest Radiol. 2003;38:305–310. [PubMed] [Google Scholar]
- 120.Aime S, Chiaussa M, Digilio G, Gianolio E, Terreno E. JBIC, J Biol Inorg Chem. 1999;4:766–774. doi: 10.1007/s007750050349. [DOI] [PubMed] [Google Scholar]
- 121.Caravan P, Parigi G, Chasse JM, Cloutier NJ, Ellison JJ, Lauffer RB, Luchinat C, McDermid SA, Spiller M, McMurry TJ. Inorg Chem. 2007;46:6632–6639. doi: 10.1021/ic700686k. [DOI] [PubMed] [Google Scholar]
- 122.Reuben J. Biochemistry. 1971;10:2834–2838. doi: 10.1021/bi00791a005. [DOI] [PubMed] [Google Scholar]
- 123.Fasano M, Curry S, Terreno E, Galliano M, Fanali G, Narciso P, Notari S, Ascenzi P. IUBMB Life. 2005;57:787–796. doi: 10.1080/15216540500404093. [DOI] [PubMed] [Google Scholar]
- 124.Prasad PV, Cannillo J, Chavez DR, Pinchasin ES, Dolan RP, Walovitch R, Edelman RR. Invest Radiol. 1999;34:566. doi: 10.1097/00004424-199909000-00003. [DOI] [PubMed] [Google Scholar]
- 125.Krause MHJ, Kwong KK, Xiong J, Gragoudas ES, Young LHY. Magn Reson Imaging. 2003;21:725–732. doi: 10.1016/s0730-725x(03)00100-0. [DOI] [PubMed] [Google Scholar]
- 126.Rapp JH, Wolff SD, Quinn SF, Soto JA, Meranze SG, Muluk S, Blebea J, Johnson SP, Rofsky NM, Duerinckx A, Foster GS, Kent KC, Moneta G, Middlebrook MR, Narra VR, Toombs BD, Pollak J, Yucel EK, Shamsi K, Weisskoff RM. Radiology. 2005;236:71–78. doi: 10.1148/radiol.2361040148. [DOI] [PubMed] [Google Scholar]
- 127.Caravan P, Cloutier NJ, Greenfield MT, McDermid SA, Dunham SU, Bulte JWM, Amedio JC, Looby RJ, Supkowski RM, Horrocks WD, McMurry TJ, Lauffer RB. J Am Chem Soc. 2002;124:3152–3162. doi: 10.1021/ja017168k. [DOI] [PubMed] [Google Scholar]
- 128.Zobel K, Koehler MFT, Beresini MH, Caris LD, Combs D. Bioorg Med Chem Lett. 2003;13:1513–1515. doi: 10.1016/s0960-894x(03)00209-9. [DOI] [PubMed] [Google Scholar]
- 129.Goyen M. Vasc Health Risk Manage. 2008;4:1–9. doi: 10.2147/vhrm.2008.04.01.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Raymond KN, Pierre VC. Bioconjugate Chem. 2005;16:3–8. doi: 10.1021/bc049817y. [DOI] [PubMed] [Google Scholar]
- 131.Caravan P, Ellison JJ, McMurry TJ, Lauffer RB. Chem Rev. 1999;99:2293–2352. doi: 10.1021/cr980440x. [DOI] [PubMed] [Google Scholar]
- 132.Yang C-T, Chuang K-H. MedChemComm. 2012;3:552–565. [Google Scholar]
- 133.Michaely HJ, Attenberger UI, Dietrich O, Schmitt P, Nael K, Kramer H, Reiser MF, Schoenberg SO, Walz M. Invest Radiol. 2008;43:635–641. doi: 10.1097/RLI.0b013e31817ee53a. [DOI] [PubMed] [Google Scholar]
- 134.Haneder S, Attenberger UI, Biffar A, Dietrich O, Fink C, Schoenberg SO, Michaely HJ. Invest Radiol. 2011;46:678–685. doi: 10.1097/RLI.0b013e31822428ad. [DOI] [PubMed] [Google Scholar]
- 135.Anzidei M, Napoli A, Marincola BC, Kirchin MA, Neira C, Geiger D, Zaccagna F, Catalano C, Passariello R. Invest Radiol. 2009;44:784–792. doi: 10.1097/RLI.0b013e3181bfe38a. [DOI] [PubMed] [Google Scholar]
- 136.Schipper R-J, Smidt ML, van Roozendaal LM, Castro CJG, de Vries B, Heuts EM, Keymeulen KBMI, Wildberger JE, Lobbes MBI, Beets-Tan RGH. Invest Radiol. 2013;48:134–139. doi: 10.1097/RLI.0b013e318277f056. [DOI] [PubMed] [Google Scholar]
- 137.Rahbar H, Partridge SC, Javid SH, Lehman CD. Curr Probl Diagn Radiol. 2012;41:149–158. doi: 10.1067/j.cpradiol.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 138.O’Flynn E, deSouza N. Breast Cancer Res. 2011;13:204. doi: 10.1186/bcr2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rogers DF, Boschetto P, Barnes PJ. J Pharmacol Methods. 1989;21:309–315. doi: 10.1016/0160-5402(89)90068-5. [DOI] [PubMed] [Google Scholar]
- 140.Martínez-Palones JM, Gil-Moreno A, Pérez-Benavente MaA, Roca I, Xercavins J. Gynecol Oncol. 2004;92:845–850. doi: 10.1016/j.ygyno.2003.11.028. [DOI] [PubMed] [Google Scholar]
- 141.Patterson CE, Rhoades RA, Garcia JG. J Appl Physiol. 1992;72:865–873. doi: 10.1152/jappl.1992.72.3.865. [DOI] [PubMed] [Google Scholar]
- 142.Verrecchia T, Huve P, Bazile D, Veillard M, Spenlehauer G, Couvreur P. J Biomed Mater Res. 1993;27:1019–1028. doi: 10.1002/jbm.820270807. [DOI] [PubMed] [Google Scholar]
- 143.Pardridge WM, Eisenberg J, Cefalu WT. Am J Physiol: Endocrinol Metab. 1985;249:E264–E267. doi: 10.1152/ajpendo.1985.249.3.E264. [DOI] [PubMed] [Google Scholar]
- 144.Desai NP, Hubbell JA. J Biomed Mater Res. 1991;25:829–843. doi: 10.1002/jbm.820250704. [DOI] [PubMed] [Google Scholar]
- 145.Bedrosian I, Scheff AM, Mick R, Callans LS, Bucky LP, Spitz FR, Helsabeck C, Elder DE, Alavi A, Fraker DF, Czerniecki BJ. J Nucl Med. 1999;40:1143–1148. [PubMed] [Google Scholar]
- 146.Albertini JJ, Lyman GH, Cox C, et al. J Am Med Assoc. 1996;276:1818–1822. [PubMed] [Google Scholar]
- 147.Veronesi U, Paganelli G, Viale G, Galimberti V, Luini A, Zurrida S, Robertson C, Sacchini V, Veronesi P, Orvieto E, De Cicco C, Intra M, Tosi G, Scarpa D. J Natl Cancer Inst. 1999;91:368–373. doi: 10.1093/jnci/91.4.368. [DOI] [PubMed] [Google Scholar]
- 148.Maccauro M, Lucignani G, Aliberti G, Villano C, Castellani M, Solima E, Bombardieri E. Eur J Nucl Med Mol Imaging. 2005;32:569–574. doi: 10.1007/s00259-004-1709-4. [DOI] [PubMed] [Google Scholar]
- 149.Wilhelm AJ, Mijnhout GS, Franssen EJF. Eur J Nucl Med. 1999;26:S36–S42. doi: 10.1007/pl00014793. [DOI] [PubMed] [Google Scholar]
- 150.Ahrén B, Schmitz O. Horm Metab Res. 2004;36:867–876. doi: 10.1055/s-2004-826178. [DOI] [PubMed] [Google Scholar]
- 151.Nauck MA, Vardarli I, Deacon CF, Holst JJ, Meier JJ. Diabetologia. 2011;54:10–18. doi: 10.1007/s00125-010-1896-4. [DOI] [PubMed] [Google Scholar]
- 152.Baggio LL, Huang Q, Brown TJ, Drucker DJ. Diabetes. 2004;53:2492–2500. doi: 10.2337/diabetes.53.9.2492. [DOI] [PubMed] [Google Scholar]
- 153.Kim J-G, Baggio LL, Bridon DP, Castaigne J-P, Robitaille MF, Jetté L, Benquet C, Drucker DJ. Diabetes. 2003;52:751–759. doi: 10.2337/diabetes.52.3.751. [DOI] [PubMed] [Google Scholar]
- 154.Meier JJ. Nat Rev Endocrinol. 2012;8:728–742. doi: 10.1038/nrendo.2012.140. [DOI] [PubMed] [Google Scholar]
- 155.Kreymann B, Ghatei MA, Williams G, Bloom SR. Lancet. 1987;330:1300–1304. doi: 10.1016/s0140-6736(87)91194-9. [DOI] [PubMed] [Google Scholar]
- 156.Turton MD, O’Shea D, Gunn I, Beak SA, Edwards CMB, Meeran K, Choi SJ, Taylor GM, Heath MM, Lambert PD, Wilding JPH, Smith DM, Ghatei MA, Herbert J, Bloom SR. Nature. 1996;379:69–72. doi: 10.1038/379069a0. [DOI] [PubMed] [Google Scholar]
- 157.Drucker DJ, Nauck MA. Lancet. 2006;368:1696–1705. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- 158.Hou S, Li C, Huan Y, Liu S, Liu Q, Sun S, Jiang Q, Jia C, Shen Z. J Diabetes Res. 2015;2015:817839. doi: 10.1155/2015/817839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Pollaro L, Heinis C. MedChemComm. 2010;1:319–324. [Google Scholar]
- 160.Rosenstock J, Reusch J, Bush M, Yang F, Stewart M G for the Albiglutide Study. Diabetes Care. 2009;32:1880–1886. doi: 10.2337/dc09-0366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Rosenstock J, Reusch J, Bush M, Yang F, Stewart M ftAS Group. Diabetes Care. 2009;32:1880–1886. doi: 10.2337/dc09-0366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Cai Y, Yue P. J Chromatogr A. 2011;1218:6953–6960. doi: 10.1016/j.chroma.2011.07.107. [DOI] [PubMed] [Google Scholar]
- 163.Cortes J, Saura C. Eur J Cancer Suppl. 2010;8:1–10. [Google Scholar]
- 164.Gradishar WJ, Tjulandin S, Davidson N, Shaw H, Desai N, Bhar P, Hawkins M, O’Shaughnessy J. J Clin Oncol. 2005;23:7794–7803. doi: 10.1200/JCO.2005.04.937. [DOI] [PubMed] [Google Scholar]
- 165.Lluch A, Álvarez I, Muñoz M, Á Seguí M, Tusquets I, García-Estévez L. Crit Rev Oncol Hematol. 2014;89:62–72. doi: 10.1016/j.critrevonc.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 166.Cucinotto I, Fiorillo L, Gualtieri S, Arbitrio M, Ciliberto D, Staropoli N, Grimaldi A, Luce A, Tassone P, Caraglia M, Tagliaferri P. J Drug Delivery. 2013;2013:10. doi: 10.1155/2013/905091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chen H, Huang X, Wang S, Zheng X, Lin J, Li P, Lin L. Chin J Cancer Res. 2015;27:190–196. doi: 10.3978/j.issn.1000-9604.2014.12.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Dennis MS, Jin H, Dugger D, Yang R, McFarland L, Ogasawara A, Williams S, Cole MJ, Ross S, Schwall R. Cancer Res. 2007;67:254–261. doi: 10.1158/0008-5472.CAN-06-2531. [DOI] [PubMed] [Google Scholar]
- 169.John TA, Vogel SM, Tiruppathi C, Malik AB, Minshall RD. Am J Physiol: Lung Cell Mol Physiol. 2003;284:L187–L196. doi: 10.1152/ajplung.00152.2002. [DOI] [PubMed] [Google Scholar]
- 170.Riehemann K, Schneider SW, Luger TA, Godin B, Ferrari M, Fuchs H. Angew Chem, Int Ed. 2009;48:872–897. doi: 10.1002/anie.200802585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Sahay G, Alakhova DY, Kabanov AV. J Controlled Release. 2010;145:182–195. doi: 10.1016/j.jconrel.2010.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Shi Q, Bao S, Song L, Wu Q, Bigner DD, Hjelmeland AB, Rich JN. Oncogene. 2007;26:4084–4094. doi: 10.1038/sj.onc.1210181. [DOI] [PubMed] [Google Scholar]
- 173.Neuzillet C, Tijeras-Raballand A, Cros J, Faivre S, Hammel P, Raymond E. Cancer Metastasis Rev. 2013;32:585–602. doi: 10.1007/s10555-013-9439-3. [DOI] [PubMed] [Google Scholar]
- 174.Desai N, Trieu V, Damascelli B, Soon-Shiong P. Transl Oncol. 2009;2:59–64. doi: 10.1593/tlo.09109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Haber CL, Gottifredi V, Llera AS, Salvatierra E, Prada F, Alonso L, Helene SE, Podhajcer OL. Int J Cancer. 2008;122:1465–1475. doi: 10.1002/ijc.23216. [DOI] [PubMed] [Google Scholar]
- 176.Huang Y, Zhang J, Zhao Y-Y, Jiang W, Xue C, Xu F, Zhao H-Y, Zhang Y, Zhao L-P, Hu Z-H, Yao Z-W, Liu Q-Y, Zhang L. Chin J Cancer. 2012;31:541–548. doi: 10.5732/cjc.012.10212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Schnitzer JE, Oh P. Am J Physiol: Heart Circ Physiol. 1992;263:H1872–H1879. doi: 10.1152/ajpheart.1992.263.6.H1872. [DOI] [PubMed] [Google Scholar]
- 178.Lane TF, Sage EH. FASEB J. 1994;8:163–173. [PubMed] [Google Scholar]
- 179.Vandelli MA, Rivasi F, Guerra P, Forni F, Arletti R. Int J Pharm. 2001;215:175–184. doi: 10.1016/s0378-5173(00)00681-5. [DOI] [PubMed] [Google Scholar]
- 180.Weber C, Coester C, Kreuter J, Langer K. Int J Pharm. 2000;194:91–102. doi: 10.1016/s0378-5173(99)00370-1. [DOI] [PubMed] [Google Scholar]
- 181.Crisante F, Francolini I, Bellusci M, Martinelli A, D’Ilario L, Piozzi A. Eur J Pharm Sci. 2009;36:555–564. doi: 10.1016/j.ejps.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 182.Yang L, Cui F, Cun D, Tao A, Shi K, Lin W. Int J Pharm. 2007;340:163–172. doi: 10.1016/j.ijpharm.2007.03.028. [DOI] [PubMed] [Google Scholar]
- 183.Qi J, Yao P, He F, Yu C, Huang C. Int J Pharm. 2010;393:177–185. doi: 10.1016/j.ijpharm.2010.03.063. [DOI] [PubMed] [Google Scholar]
- 184.Lee SH, Heng D, Ng WK, Chan H-K, Tan RBH. Int J Pharm. 2011;403:192–200. doi: 10.1016/j.ijpharm.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 185.Gong J, Huo M, Zhou J, Zhang Y, Peng X, Yu D, Zhang H, Li J. Int J Pharm. 2009;376:161–168. doi: 10.1016/j.ijpharm.2009.04.033. [DOI] [PubMed] [Google Scholar]
- 186.Xie J, Wang J, Niu G, Huang J, Chen K, Li X, Chen X. Chem Commun. 2010;46:433–435. doi: 10.1039/b917195a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kirchner C, Liedl T, Kudera S, Pellegrino T, Muñoz Javier A, Gaub HE, Stölzle S, Fertig N, Parak WJ. Nano Lett. 2005;5:331–338. doi: 10.1021/nl047996m. [DOI] [PubMed] [Google Scholar]
- 188.Kratz F, Ehling G, Kauffmann H-M, Unger C. Hum Exp Toxicol. 2007;26:19–35. doi: 10.1177/0960327107073825. [DOI] [PubMed] [Google Scholar]
- 189.Kratz F. Expert Opin Invest Drugs. 2007;16:855–866. doi: 10.1517/13543784.16.6.855. [DOI] [PubMed] [Google Scholar]
- 190.Sanchez E, Li M, Wang C, Nichols CM, Li J, Chen H, Berenson JR. Clin Cancer Res. 2012;18:3856–3867. doi: 10.1158/1078-0432.CCR-11-3130. [DOI] [PubMed] [Google Scholar]
- 191.Kratz F, Fichtner I, Graeser R. Invest New Drugs. 2012;30:1743–1749. doi: 10.1007/s10637-011-9686-5. [DOI] [PubMed] [Google Scholar]
- 192.Sudimack J, Lee RJ. Adv Drug Delivery Rev. 2000;41:147–162. doi: 10.1016/s0169-409x(99)00062-9. [DOI] [PubMed] [Google Scholar]
- 193.Wiener EC, Konda S, Shadron A, Brechbiel M, Gansow O. Invest Radiol. 1997;32:748–754. doi: 10.1097/00004424-199712000-00005. [DOI] [PubMed] [Google Scholar]
- 194.Parker N, Turk MJ, Westrick E, Lewis JD, Low PS, Leamon CP. Anal Biochem. 2005;338:284–293. doi: 10.1016/j.ab.2004.12.026. [DOI] [PubMed] [Google Scholar]
- 195.Smith-Jones PM, Pandit-Taskar N, Cao W, O’Donoghue J, Philips MD, Carrasquillo J, Konner JA, Old LJ, Larson SM. Nucl Med Biol. 2008;35:343–351. doi: 10.1016/j.nucmedbio.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Müller C, Vlahov IR, Santhapuram HKR, Leamon CP, Schibli R. Nucl Med Biol. 2011;38:715–723. doi: 10.1016/j.nucmedbio.2010.12.013. [DOI] [PubMed] [Google Scholar]
- 197.Fani M, Wang X, Nicolas G, Medina C, Raynal I, Port M, Maecke H. Eur J Nucl Med Mol Imaging. 2011;38:108–119. doi: 10.1007/s00259-010-1597-8. [DOI] [PubMed] [Google Scholar]
- 198.Liu Z, Amouroux G, Zhang Z, Pan J, Hundal-Jabal N, Colpo N, Lau J, Perrin DM, Bénard F, Lin K-S. Mol Pharmaceutics. 2015;12:974–982. doi: 10.1021/acs.molpharmaceut.5b00003. [DOI] [PubMed] [Google Scholar]
- 199.Port M, Corot C, Rousseaux O, Raynal I, Devoldere L, Idée J-M, Dencausse A, Greneur S, Simonot C, Meyer D. Magn Reson Mater Phys, Biol Med. 2001;12:121–127. doi: 10.1007/BF02668093. [DOI] [PubMed] [Google Scholar]
- 200.Kowalski J, Henze M, Schuhmacher J, Mäcke HR, Hofmann M, Haberkorn U. Mol Imaging Biol. 2003;5:42–48. doi: 10.1016/s1536-1632(03)00038-6. [DOI] [PubMed] [Google Scholar]
- 201.Liu Z, Pourghiasian M, Bénard F, Pan J, Lin K-S, Perrin DM. J Nucl Med. 2014;55:1499–1505. doi: 10.2967/jnumed.114.137836. [DOI] [PubMed] [Google Scholar]
- 202.Velikyan I, Sundberg ÅL, Lindhe Ö, Höglund AU, Eriksson O, Werner E, Carlsson J, Bergström M, Långström B, Tolmachev V. J Nucl Med. 2005;46:1881–1888. [PubMed] [Google Scholar]
- 203.Ugur Ö, Kothari PJ, Finn RD, Zanzonico P, Ruan S, Guenther I, Maecke HR, Larson SM. Nucl Med Biol. 2002;29:147–157. doi: 10.1016/s0969-8051(01)00290-6. [DOI] [PubMed] [Google Scholar]
- 204.Liu Z, Pourghiasian M, Radtke MA, Lau J, Pan J, Dias GM, Yapp D, Lin K-S, Bénard F, Perrin DM. Angew Chem, Int Ed. 2014;53:11876–11880. doi: 10.1002/anie.201406258. [DOI] [PubMed] [Google Scholar]
- 205.Selhub J, Franklin WA. J Biol Chem. 1984;259:6601–6606. [PubMed] [Google Scholar]
- 206.Selhub J, Emmanouel D, Stavropoulos T, Arnold R. Am J Physiol: Renal, Fluid Electrolyte Physiol. 1987;252:F750–F756. doi: 10.1152/ajprenal.1987.252.4.F750. [DOI] [PubMed] [Google Scholar]
- 207.Birn H, Selhub J, Christensen EI. Am J Physiol: Cell Physiol. 1993;264:C302–C310. doi: 10.1152/ajpcell.1993.264.2.C302. [DOI] [PubMed] [Google Scholar]
- 208.Weitman SD, Lark RH, Coney LR, Fort DW, Frasca V, Zurawski VR, Kamen BA. Cancer Res. 1992;52:3396–3401. [PubMed] [Google Scholar]
- 209.Hjelle JT, Christensen EI, Carone FA, Selhub J. Am J Physiol: Cell Physiol. 1991;260:C338–C346. doi: 10.1152/ajpcell.1991.260.2.C338. [DOI] [PubMed] [Google Scholar]
- 210.Müller C, Struthers H, Winiger C, Zhernosekov K, Schibli R. J Nucl Med. 2013;54:124–131. doi: 10.2967/jnumed.112.107235. [DOI] [PubMed] [Google Scholar]
- 211.Kim TH, Park CW, Kim HY. Biol Pharm Bull. 2012;35:1076–1083. doi: 10.1248/bpb.b12-00029. [DOI] [PubMed] [Google Scholar]