

Summary
Histone-modifying enzymes regulate transcription and are sensitive to availability of endogenous small-molecule metabolites, allowing chromatin to respond to changes in environment. The gut microbiota produces a myriad of metabolites that affect host physiology and susceptibility to disease, however the underlying molecular events remain largely unknown. Here we demonstrate that microbial colonization regulates global histone acetylation and methylation in multiple host tissues in a diet-dependent manner: consumption of a “Western-type” diet prevents many of the microbiota-dependent chromatin changes that occur in a polysaccharide rich diet. Finally, we demonstrate that supplementation of germ-free mice with short-chain fatty acids, major products of gut bacterial fermentation, is sufficient to recapitulate chromatin modification states and transcriptional responses of colonization on host epigenetic programming. These findings have profound implications for understanding the complex functional interactions between diet, gut microbiota, and host health.
Introduction
The eukaryotic genome is organized into a highly compressed nucleoprotein structure known as chromatin. Histone proteins (H2A, H2B, H3 and H4) are major components of chromatin and act as spools, wrapping DNA into fundamental nucleosome units that can fold into higher order structures. Histones undergo a myriad of covalent post-translational modifications (PTMs), and along with histone variant replacement, comprise what is known as the “histone code,” wherein the local PTM-state dictates whether chromatin is repressive or activating toward transcription (Jenuwein and Allis, 2001). Enzymes that add or remove PTMs are sensitive to the availability of endogenous metabolites (Fan et al., 2015). For example, acetyl-coenzyme A (acetyl-CoA) is a necessary substrate for histone acetyltransferases (HATs), and increased availability can increased HAT activity.
The gut microbiota produces a variety of metabolites detectable in host circulation, including small organic acids, bile acids, vitamins, choline metabolites, and lipids (reviewed in (Nicholson et al., 2012)). Dietary poly- and oligosaccharides resistant to digestion by the mammalian host pass to the distal gut where they serve as a source of carbon and energy for gut bacteria. Through fermentative reactions, the gut microbiota can metabolize complex carbohydrates to produce small organic acids, the majority of which are comprised of the short chain fatty acids (SCFAs) acetate, propionate, and butyrate (≥95%) (Besten et al., 2013). SCFAs have been implicated in both disease promoting and therapeutic effects (Perry et al., 2016; Tan et al., 2014), prompting a need for increased understanding of the underlying molecular mechanisms. Robust associations between gut microbiota and host metabolic outcomes include cardiovascular disease (Karlsson et al., 2012), metabolic syndrome (Cabreiro et al., 2013; Chassaing et al., 2015), obesity (Bäckhed et al., 2004; Ley et al., 2005; Zhao, 2013), diabetes mellitus (Amar et al., 2011), and inflammatory bowel disorders and malignancy (Chassaing et al., 2015; Donohoe et al., 2012; 2014).
Eukaryotic histone modifying enzymes have evolved to sense and integrate environmental signals, ultimately programming gene expression patterns and mediating phenotype. Given the sensitivity of these enzymes to endogenous metabolites, we hypothesized that gut microbial metabolites absorbed and metabolized by the host may exert similar control. Here, we explore whether the gut microbiota affects host epigenetic programming in a variety of tissues and how this relationship is affected by host diet. We provide evidence of gut microbiota-mediated changes in global histone acetylation and methylation not only in colon, which is in direct contact with microbes and their metabolites, but in tissues outside the gut. We demonstrate that this regulatory relationship is sensitive to host diet, wherein a “Western-type” diet limits microbial SCFA production, abolishes the effects of microbiota on host chromatin states, and results in functionally relevant alterations in hepatic gene expression. Finally, we identify an underlying mechanism that reveals SCFA supplementation of germ-free mice is sufficient to recapitulate the epigenetic phenotype associated with gut colonization.
Results and Discussion
Gut microbiota affect host tissue epigenetic states
To investigate whether gut microbes and their metabolites affect host chromatin states, we examined histone PTM states as a function of colonization. We focused our analysis on proximal colon, liver, and white adipose tissue (WAT). The experimental workflow is described in Fig. 1A. Briefly, mice were either maintained germ-free (GF) throughout the experiment, allowed to acquire a microbiota from birth to adulthood (conventionally raised, ConvR), or colonized with a complete (uncultured) microbiota (conventionalized, ConvD) harvested from ConvR donors. Use of a ConvD mouse model allows for the determination of whether the phenotype observed in ConvR animals is transferrable via the gut microbiota alone. Additionally, since ConvR animals experience different environmental exposure early in life and have developmental differences (K. Smith et al., 2007) that may exhibit phenotypic differences vs. their GF controls, the use of ConvD mice for relatively short time periods allows for dissection of effects more directly related to differences in microbial metabolism. Histones were extracted from tissues and prepared for mass spectrometry analysis using an in-house workflow (Krautkramer et al., 2015).
Figure 1.
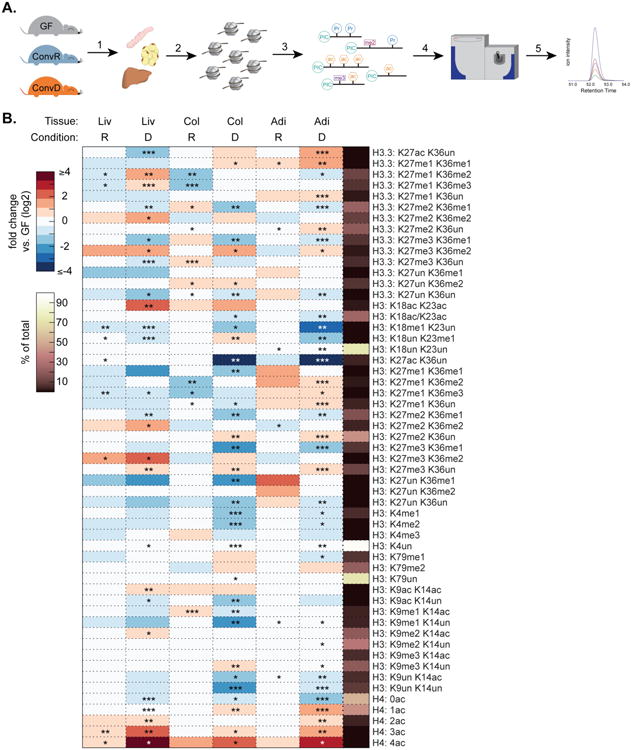
Gut microbiota affect host tissue epigenetic states. (A) Experimental design: 1. tissues harvested from germ-free (GF), conventionally raised (ConvR), and conventionalized (ConvD) mice. 2-3. Histones extracted, chemically derivatized and trypsinized. 4-5. Histone peptides injected onto mass spectrometer and data acquired on >60 unique histone PTM states. (B) Relative abundance of histone PTMs on H3, H3.3, and H4. Values are reported as a fold change vs. GF controls (log2). Mean % of peptide family total across all samples (right-most column). *p<0.05, **p<0.01, ***p<0.001, n=4 mice per condition.
We surveyed 55 unique and combinatorial acetylated and methylated histone PTM states in proximal colon, liver, and WAT (Dataset S1). Colonization induced robust increases in H4 acetylation in all three tissues (Fig. 1B). This peptide includes the first 4 lysines (K5, K8, K12, and K16) on the H4 N-terminal tail. Thus H4: 0ac indicates peptides where no lysine residues are acetylated, whereas H4:1ac-4ac indicates peptides where any 1-4 of the 4 lysines are acetylated. In ConvR animals, there was a significant 2.1-fold increase in both triply and quadruply acetylated H4 (H4: 3ac and H4:4ac, respectively). Similarly, ConvR animals showed a 3-fold and 1.30-fold increase in the highly acetylated H4: 4ac peptide in proximal colon and adipose tissue, respectively, relative to GF mice (Fig. 1B). The effects of colonization on H4 acetylation were even more robust in ConvD mice, with a 4.5-, 6.0-, and 12.0-fold increase in H4: 4ac of proximal colon, adipose, and liver, respectively (Fig. 1B). Triply acetylated H4 peptides also increased 2.1- to 4.2-fold in proximal colon, adipose, and liver (Fig. 1B). It is noteworthy that these two H4 states collectively account for just over 1% of the total H4 states, suggesting that this open chromatin state is confined to very specific loci along the genome. Consistent with the conversion of unacetylated states to higher acetylation, the completely unmodified form decreased significantly (H4: 0ac, 1.33 to 3.3-fold across tissues surveyed) in colonized animals.
Microbes similarly induced acetylation of canonical H3 and the variant H3.3. The doubly acetylated canonical H3 K9ac+K14ac and H3 K18ac+K23ac peptides increased significantly in ConvD mouse livers and trended toward a similar magnitude increase in proximal colon of ConvR and ConvD mice (Fig. 1B). These two doubly acetylated canonical H3 peptides account for roughly 2% or less of total histone PTM states observed in each peptide family, again supporting a more loci-specific role for these modified nucleosomes along the genome (Fig. 1B). Interestingly, the singly acetylated peptides K9ac+K14un and K9un+K14ac decrease concomitantly with an increase in the doubly acetylated K9ac + K14ac peptide, and a similar pattern occurs on the singly acetylated and coeluting K18ac/K23ac peptides (Fig. 1B). These results are consistent with a shift away from a singly acetylated state toward a maximally acetylated state.
H3 methylation patterns are also altered as a function of gut colonization status. There is a modest, yet statistically significant increase in H3 K27me3+K36un in proximal colon, liver, and adipose tissues from ConvD mice vs. their GF controls (1.4- to 1.5-fold increase, Fig. 1B), accounting for ∼12% of total PTM states and suggesting more broad regulatory effects in comparison to highly acetylated states of H3 and H4. There were increases in peptides containing highly methylated forms of K27 and K36 (i.e. me2 and me3) on both the canonical H3 and the variant H3.3, however these effects were not present consistently across all three tissues, suggesting some tissue-specificity in the response to colonization (Fig. 1B). Notably, H3 K18me1 decreased across all three ConvD tissues (Fig. 1B, 2.1 to 8.3-fold across liver, proximal colon, and adipose tissue). A similar pattern was present for the combinatorial K27me2+K36me1 peptide on H3 and variant H3.3 (Fig. 1B). Together, these results demonstrate that gut microbiota affect host tissue acetylated and methylated chromatin states in a site-specific and combinatorial fashion, strongly supporting a role for the gut microbiota as a driver of host tissue chromatin regulation. While some histone PTM states appear to be similarly regulated across all tissues surveyed, other changes are unique within a tissue.
The ability to detect histone PTMs within the context of neighboring modifiable sites allowed us to uncover combinatorial PTM states that account for a very small percentage of the total and that are not necessarily resolvable by orthologous techniques such as western blot analysis. Indeed, we performed several western blots and found no statistically significant differences in H3 K9ac, H3 K27me3, or pan-acetyl(K) detectable via western blot in histone extracts from GF, ConvR, and ConvD mouse livers (Fig. S1A-B). To compare methods, we summed all possible permutations of peptides containing K9ac and K27me3 on canonical histone H3 to obtain single western blot-like estimates of K9ac and K27me3 abundance for GF, ConvR, and ConvD mouse liver histone PTM states (see Supp. Methods). Calculating fold changes relative to GF, both K9ac and K27me3, as a total among all combinatorial forms quantified, remained relatively unchanged between colonized and GF mice (Figure S1C), consistent with quantitative western blot results (Fig. S1B).
Gut microbiota-mediated changes in chromatin state are sensitive to host diet
Given the known affect of host diet on gut microbial community composition and metabolism (Turnbaugh et al., 2009b), we next evaluated the effects of host diet on microbiota-mediated regulation of host chromatin states (Dataset S1). ConvR and GF mice were fed a “Western-type” high fat, high sucrose diet (HF/HS), which contains low levels of fermentable substrate for the gut microbiota, for 16 weeks prior to sacrifice at 19 weeks of age. Tissues were harvested, and physiological parameters and histone PTM states were measured (Fig. 2A). As anticipated, ConvR mice fed a HF/HS diet weighed more than diet-matched GF controls (Fig. 2B). HF/HS-fed ConvR mice displayed higher hepatic total cholesterol and triglycerides vs. diet-matched GF controls and chow-fed mice (Fig. 2C-D). Thus, HF/HS feeding impacted host metabolic state in a microbiota-dependent manner. To determine whether HF/HS feeding altered SCFA production, we measured acetate, propionate and butyrate levels in cecal contents (i.e. principal site of fermentation) of ConvR, ConvD, and GF mice on both chow and HF/HS diets. Gut microbial colonization increased these metabolites in the ceca of mice, and this increase was more pronounced in mice fed a chow diet compared to a HF/HS diet low in fermentable complex polysaccharides (Fig. 2E). In HF/HS-fed ConvR mice, cecal acetate, propionate, and butyrate levels were lower relative to chow-fed ConvR mice (1.9-6.0 fold, Fig. 2E, Fig. S4A). Interestingly, chow-fed ConvD mice had greater cecal SCFAs than ConvR mice (Fig. 2E). This pattern in cecal SCFA levels is consistent with that of histone PTM changes in ConvD and ConvR mice on chow, wherein PTM states trend in the same direction, but the magnitude of change is larger in ConvD mice vs. ConvR (Fig. 1B). Thus, SCFA availability influences histone PTM states. In peripheral venous blood, levels of these SCFAs were not significantly different (Fig. S4B), consistent with these organic acids having undergone significant metabolism in the liver prior to reaching peripheral venous blood.
Figure 2.

Gut microbiota-mediated epigenetic changes are sensitive to diet. (A) Experimental design: 1. GF and ConvR mice on either chow or a HF/HS (“Westernized”) diet. 2-5. Tissues prepared and analyzed as described in Figure 1. (B) Weights of GF and ConvR mice fed a HF/HS diet at the time of sacrifice. (C) Hepatic total cholesterol, and (D) hepatic triglycerides in GF, ConvD, and ConvR mice on chow and HF/HS diets. (E) SCFA measurement in cecal contents from GF, ConvR, and ConvD animals on chow and HF/HS diet. *p<0.05, **p<0.01, error bars represent standard deviation, n = 4 mice per condition. (F-H) H4 (K5, K8, K12, and K16) acetylation in colonized liver (F), adipose tissue (G), and proximal colon (H) relative to GF controls (fold change, log2). (I-J) H3 K18 and K23 methylation and acetylation in colonized liver (I) and proximal colon (J) relative to GF controls (fold change, log2). *p<0.05, **p<0.01, ***p<0.001, error bars represent standard error from the mean, n=4 mice per condition
As predicted by cecal SCFA data, the gut microbiota-host epigenome relationship was altered in response to HF/HS feeding. While there was a microbiota-dependent increase in H4 acetylation in ConvR and ConvD tissues of chow-fed mice (Fig. 1B), HF/HS-feeding abolished the effects of gut colonization in liver and WAT (Fig. 2F and 2G). Interestingly, the microbiota-dependent effects on H4 acetylation were attenuated, but still significantly increased relative to GF controls in HF/HS-fed mouse proximal colon (Fig. 2H). This pattern of diet-dependence was also present in other histone PTM states. The response to gut microbiota on H3 K18 and K23 also trended as a function of diet: K18me1 and K23me1 peptides both decreased significantly in livers of both ConvR and ConvD chow fed mice, but remained unchanged in HF/HS-fed mice (Fig. 2I). The coeluting peptides K18ac and K23ac (i.e. K18ac/K23ac) were unchanged in response to gut microbiota in livers of chow-fed mice, yet decreased in HF/HS-fed mouse livers (Fig. 2I). However, in proximal colon this diet-dependency was again absent and histone PTM states trended in similar directions regardless of dietary conditions (Fig. 2J). There was also a diet-dependent, microbiota-independent increase in basal histone H4 acetylation in liver, proximal colon, and adipose tissue. In comparison to H4 acetylation in chow-fed ConvR and GF animals, there were significant increases in nearly all forms of 1ac-4ac H4 peptides in HF/HS-fed tissues ranging from 1.2 to 7.2-fold (Fig. 1B).
Although the direction of change in both acetylated and methylated histone PTM states was similar in ConvR and ConvD tissues, differences in magnitude (Figs. 1B, 2F-J) trended with cecal SCFA levels (Fig. 2E). To investigate whether these differences reflected alterations to microbial community composition, we performed 16S rRNA sequencing. Principal Coordinates Analysis (PCoA) of weighted UniFrac distances revealed that microbial community composition of ConvR and ConvD mice on a chow diet was more similar to each other than it was to the microbial community from mice fed HF/HS diet (Fig. S2A). ConvR mice fed a HF/HS diet had significantly fewer Bacteroidetes and a greater abundance of Firmicutes (Figs. S2B-C) than chow fed ConvR and ConvD mice, consistent with previous observations that diet and obesity alter this ratio in the gut (Ley et al., 2005). Relative abundance of these two major phyla in chow-fed ConvD mice was intermediate between chow- and HF/HS-fed ConvR mice (Figs. S2B-D). Together, these data suggest that gut microbial community composition and metabolite production are important factors that connect gut microbiota and host chromatin states.
Functional impact: Co-regulation of hepatic genes associates with altered chromatin states
To assess whether microbiota-dependent changes in host chromatin state affected tissue gene expression, we performed RNA-seq analyses on livers of colonized (both ConvR and ConvD) and GF mice on the two diets described above. Each group of colonized mice was compared to its diet-matched GF control: i.e., chow-fed ConvR vs. GF, chow-fed ConvD vs. GF, and HF/HS-fed ConvR vs. GF. A total of 623 genes were differentially expressed (DE) among these three groups, as determined by an FDR cut-off of 0.05 (Dataset S2). K-means clustering of hepatic DE genes revealed 6 optimal clusters, each enriched for unique biological pathways (Fig. 3A, Dataset S2). When comparing ConvR mice on either chow or HF/HS diets to their respective GF controls, cluster 2 contained genes that are co-regulated as a function of both diet and microbiota (Fig. 3A-B). This group of genes was enriched for processes involved in insulin, SREBP, and PPAR signaling, and adaptive immunity. Additionally, this cluster contains a number of genes that may regulate histone PTM states via modulation of small molecule metabolite availability. Clusters 4 and 6 contain genes whose expression patterns differ in ConvR animals as a function of diet (Fig. 3A, C-D). Cluster 4 genes are enriched for pathways involved in cholesterol, retinol, and amino acid metabolism as well as host immunity, whereas cluster 6 contains a number of genes involved in lipid and amino acid metabolism as well as a group of genes involved in regulation of folate, which ultimately affects the availability of the one-carbon donor SAM to histone methyltransferases. Notably, 4% of DE genes are known hepatic targets of SREBP and PPAR.
Figure 3.
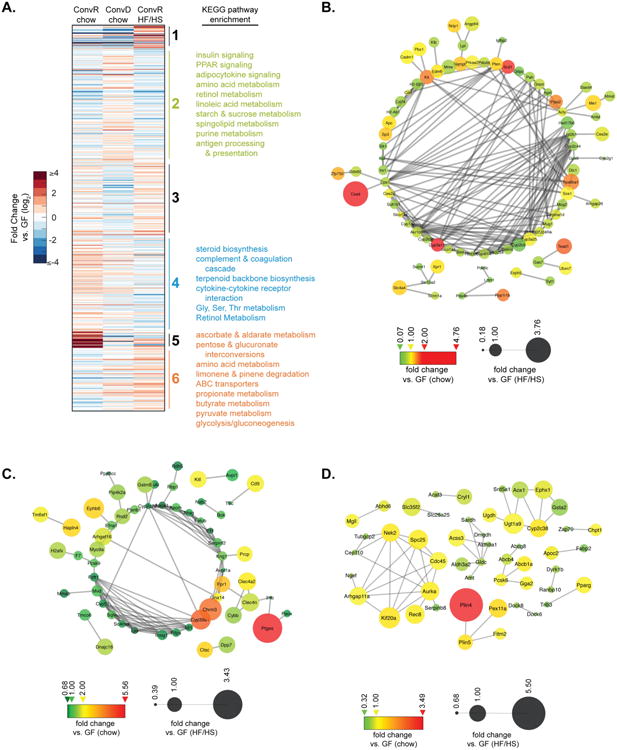
Hepatic genes are co-regulated in colonized mice as a function of diet or colonization status. (A) K-means clustering of differentially expressed hepatic genes (left, fold change vs. GF, log2) and KEGG pathway enrichment terms (right). (B-D) Interaction network for cluster 2 (B), cluster 4 (C), and cluster 6 (D). Only genes with at least one reported interaction are graphed. Edges indicate interaction. Node size indicates relative expression in HF/HS-fed mouse livers. Node color indicates relative expression in chow-fed mouse livers.
To assess the effects of diet alone, we compared GF and ConvR mice across dietary conditions (i.e. HF/HS vs. chow). Of the 868 differentially expressed genes, 413 increased and 455 decreased in expression in HF/HS-fed mice relative to chow-fed controls (Fig. S3A-B). Although a fraction of DE up (Fig. S3A) and DE down (Fig. S3B) genes are regulated in both ConvR and GF mice, the fact that there are 1.8-fold more total DE genes in response to diet in ConvR mice vs. GF mice suggests that gut microbiota drive a significant portion of the response to HF/HS feeding in liver. KEGG pathway analysis of unique and overlapping DE genes in GF and ConvR mice revealed several oppositely regulated pathways as a function of diet (Figs. S3C-D). For example, while starch and sucrose metabolism is enriched in DE up genes of ConvR mice, this same pathway is enriched in DE down genes of GF mice. The same pattern is present for pathways involved in arachidonic acid metabolism, cytokine-cytokine receptor interaction, and endocytosis. Other key differences include DE up gene enrichment for processes involved in the TCA cycle and propionate metabolism in GF mice only, and butyrate metabolism in ConvR mice only (Fig. S3C). Importantly, genes involved in PPAR signaling, insulin signaling, and diabetes mellitus were enriched in DE up genes shared by both GF and ConvR mice (Fig. S3C). Pathways involved in de-novo cholesterol synthesis are significantly enriched in DE down genes shared by both GF and ConvR mice, suggesting that HF/HS feeding decreases host de-novo cholesterol synthesis irrespective of gut colonization status.
Gut microbiota altered expression of genes linked to metabolites that directly regulate histone PTM addition or removal. For example, expression of ATP citrate lyase (Acly) is decreased in ConvR vs. GF mice, under both chow and HF/HS feeding. Acly is essential for glucose-driven, but not acetate-driven, histone acetylation in mammalian cells (Wellen et al., 2009). Acly expression decreases most in HF/HS-fed ConvR mouse livers, suggesting that the presence of other sources of carbon and energy, such as bacterial SCFAs or highly energetic lipids from HF/HS-feeding, may suppress glucose-driven histone modification. Availability of NAD+, a necessary co-substrate for Class III HDACs, may also be modulated by gut microbiota. NAMPT, which catalyzes the rate-limiting step in NAD+ biosynthesis, was differentially expressed in colonized vs. GF livers. Lastly, there were four genes in cluster 6 linked to regulation of folate, which can affect the availability of the methyl donor SAM for histone methyltransferases: Sardh, Dmgdh, Amt, and Gldc (Fig. 3D). Loss of Gldc has been associated with neural tube defects, growth retardation, and decreased levels of one carbon-carrying folates (Pai et al., 2015). Thus, decreased expression of these enzymes may affect tissue SAM availability, via modulation of folate levels.
Finally, a number of genes associated with host immunity and inflammation were differentially expressed in colonized and GF+SCFA mice relative to their GF controls (Fig. 3A-D, 4F-G). Given that GF mice are naive to microbiota, it is not unexpected that colonized mice have altered expression of immunomodulatory genes relative to GF mice. Gut microbiota are important for the development and function of host immunity (Belkaid and Hand, 2014), and SCFAs play an important role in both adaptive and innate immunity (Belkaid and Hand, 2014; P. M. Smith et al., 2013). More detailed information about specific gene cluster membership, expression, and pathway enrichment among clusters is described in Datasets S2-S3, Fig. S3, and the Supp. Information.
Figure 4.
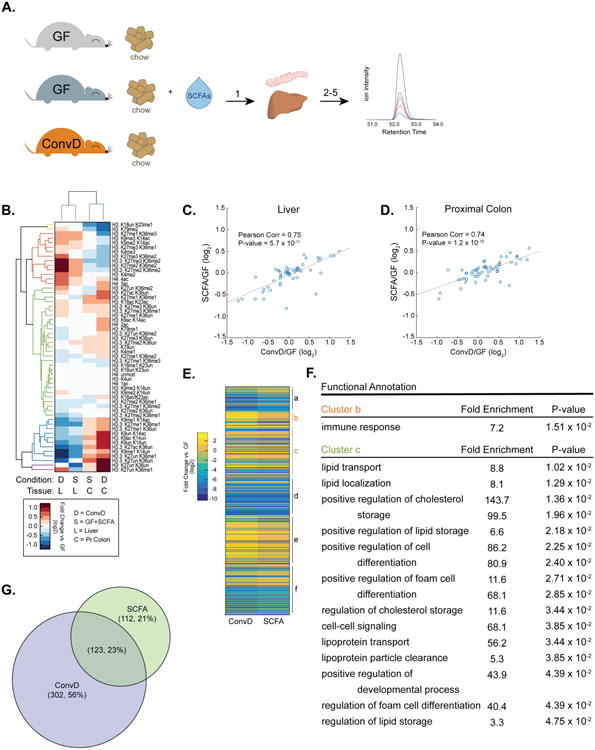
SCFA-supplementation mimics colonization-induced epigenetic programming. (A) Experimental design: 1. Germ-free mice supplemented with SCFAs (GF+SCFA) or colonized (ConvD) and tissues were harvested. 2-5. Histone extracts prepared as described in figure 1. (B) Hierarchical clustering of histone PTMs in colonized and GF+SCFA mouse tissues (fold change vs. GF, log2). (C-D) Pearson's correlation of ConvD and GF+SCFA mouse tissue histone PTM states in liver (C) and proximal colon (D). (E) K-means clustering of differentially expressed hepatic genes in ConvD and GF+SCFA mice. FDR cutoff for differential expression = 0.05, n = 3 mice per condition. (F) GO-term enrichment in clusters b and c. (G) Overlap of differentially expressed genes between ConvD and GF+SCFA mice.
SCFA-supplementation partially phenocopies the effects of colonization on host epigenetic programming
We hypothesized that SCFAs acetate, propionate, and butyrate were key mediators of systemic microbiota-induced changes in host chromatin states. To explore this idea, we supplemented germ-free mice with acetate, propionate, and butyrate (GF+SCFA) and harvested liver and proximal colon for histone PTM and gene expression analyses (Fig. 4A). These GF+SCFA mice were then compared to GF and ConvD mice, as negative and positive controls, respectively. Hierarchical clustering revealed that histone PTM states of GF+SCFA and ConvD mice were strikingly similar across both acetylated and methylated peptides (Fig. 4B). Further, GF+SCFA and ConvD histone PTM signatures were highly correlated, with a Pearson's correlation coefficient of 0.74 - 0.75, for proximal colon and liver samples (p = 1.2 × 10-10 and 5.7 × 10-11, respectively; Fig. 4C-D). These data reveal that the global chromatin states induced by SCFAs mimic gut colonization.
While levels of acetate, propionate, and butyrate were significantly increased in the cecal contents of ConvD mice, there were no significant differences between GF+SCFA mice and GF controls (Fig. S4C). Similar to previous observations (Fig. S4B), peripheral blood levels of SCFAs were unchanged as a function of either gut colonization or SCFA-supplementation (Fig. S4D). Thus, while conventionalization results in increased local concentration of cecal SCFAs, oral supplementation of SCFAs did not, likely due to absorption along alimentary tract. Despite these potential differences, the physiological impacts are likely to be similar, given that venous blood from the small intestine also drains to the liver via the hepatic portal vein where it mixes with blood traveling from the colon.
To determine whether these highly similar global chromatin states elicited similar biological effects in ConvD and GF+SCFA mice, we used RNAseq analyses to examine hepatic gene expression in ConvD, GF+SCFA, and GF mice. Consistent with histone PTM observations, GF+SCFA mice had highly similar transcriptional profiles to ConvD mice (Fig. 4E & 4G). K-means clustering of 537 DE genes revealed 6 clusters of co-regulated genes that were enriched for a number of metabolic and immunological processes (Fig. 4E-F, Dataset S4). In particular, clusters b and c were enriched for GO-terms involved in immunity (cluster b) and regulation, storage, and metabolism of lipids and cholesterol (cluster c, Dataset S4). Finally, there was striking overlap of DE genes between GF+SCFA and ConvD mice, wherein >50% of DE genes in GF+SCFA livers overlapped with DE genes in ConvD mice (Fig. 4G). Together these data suggest that SCFAs are partially causative metabolites in the complex regulatory relationship between diet, gut microbiota, and host tissue epigenetic programming.
SCFAs play a dual role as both substrates for metabolism and as signaling molecules (Besten et al., 2013). These SCFAs can be either directly converted (acetate) or oxidized (propionate and butyrate) to acetyl-CoA, the substrate for HAT enzymes. As much as 70% of acetate and roughly 30% of propionate is taken up by the liver, where both serve as sources of energy. Further, butyrate is a known HDAC inhibitor. Both scenarios could increase histone acetylation. Acetate can also serve as a substrate for cholesterol, long-chain fatty acid, glutamine, and glutamate synthesis in the liver. The remainder is metabolized by other tissues, including white adipose. As ligands of the G-protein coupled receptors FFAR2 (GPR43) and FFAR3 (GPR41), SCFAs play a role in lipid and glucose metabolism. Thus, these metabolites play complex roles in regulation of host metabolic phenotype. How combinations of or specific SCFAs contribute to beneficial or pathogenic effects in the host remains unclear.
Though the link between histone acetylation and SCFAs may be direct, the link between SCFAs and alterations in histone methylation is less clear. Moreover, the effects of SCFA-supplementation broadly recapitulated both chromatin states and gene expression patterns of ConvD mice, however, it is notable that the magnitude of the effects were generally less in SCFA+GF tissues relative to ConvD (Fig. 4B, 4E). Collectively, these results suggests that while SCFAs alone can induce a “colonized”-like state in host chromatin, there are likely other bacterial metabolites affecting host epigenome, particular interesting are those directing linked to methyl-donor capacity for epigenetic methylation. We postulate that eukaryotic histone-modifying enzymes have evolved to “sense” not only endogenous metabolites, but those produced by commensal microbiota.
Experimental Procedures
Mouse husbandry
Animal care and study protocols were approved by the UW-Madison Animal Care and Use Committee. Mice were housed in the Microbial Sciences Building vivarium. Conventionally-raised (ConvR) and germ-free (GF) C57BL/6J mice were bred at UW-Madison. GF mice were housed in separate plastic flexible vinyl gnotobiotic isolators.
Mice were group housed by colonization status and diet (3-5 mice/cage) under standard conditions (12 h light:dark, temperature- and humidity-controlled conditions), and received ad libitum access to water and food. After 3 weeks of age, mice were maintained on either a control breeder chow (5021, Lab Diet, 23.7%-kcal fat, 53.2% carbohydrate, 23.1% protein) or a high-fat high-sucrose (HF/HS) diet (TD.08811, Envigo Teklad, 44.6%-kcal fat, 40.6% carbohydrate, 14.8% protein). Diets were sterilized by irradiation and autoclaving. Sterility of germ-free animals was assessed by incubating freshly collected fecal samples under aerobic and anaerobic conditions using standard microbiology methods. Final dissection and data collection were performed at 19-weeks of age.
Conventionalized (ConvD) mice were generated by colonizing GF C57BL/6J mice with fresh cecal contents were collected from 15-week old conventionally-raised C57BL/6J mice maintained on a control breeder chow diet (n = 2 mice per donor cecal microbiota sample per experiment; 5021, Lab Diet). Immediately after sacrifice, fresh cecal contents from donor mice were re-suspended in Mega Medium (1:100 w/v) in an anaerobic chamber (Romano et al., 2015). Suspensions were transferred into anaerobic sealed tubes and used to colonize mice in a sterile biological safety cabinet. Germ-free 15-week-old C57BL/6J male mice were inoculated via a single oral gavage with ∼0.2 ml of cecal inocula (Turnbaugh et al., 2009b) and kept in sealed filter-top gnotobiotic cages for 4 weeks.
SCFA Supplementation
Germ-free (GF) C57BL/6J male mice were maintained in sterile HEPA filter cages and fed water and autoclaved chow ad libitum. At 12 weeks of age a subset of GF mice were supplemented with a mixture of short chain fatty acids (SCFA) (acetate 67.5mM, butyrate 40mM, propionate 25.9mM (P. M. Smith et al., 2013)) via drinking water supplied ad libitum or conventionalized (ConvD) with a C57BL/6J intestinal microbial community by gavage. Inoculum for colonization was prepared by suspending freshly collected fecal pellets in Mega Medium. SCFA supplemented water was freshly prepared and changed every 5 days and again 24h before sacrifice. Sterility of germ-free animals was assessed by incubating freshly collected fecal samples under aerobic and anaerobic conditions using standard microbiology methods. Prior to sacrifice and tissue collection at 14 weeks of age all mice (GF, GF+SCFA, ConvD) were anesthetized with 1-5% inhalant isoflurane supplied in oxygen.
The methods used to measure histone PTMs, 16S rRNA sequencing, gene expression, hepatic total cholesterol and triglycerides, and cecal and peripheral blood SCFAs are described in the Supplemental Methods.
Supplementary Material
Acknowledgments
K.A.K. is supported by NIH F30 DK108494, J.A.D by NIH GM059789-15/P250VA. Additional support: Clinical and Translational Science Award program through the NIH National Center for Advancing Translational Sciences grants UL1TR000427 and KL2TR000428 (F.E.R.), NIH DK108259 (F.E.R.), DK101573 (A.D.A.). An extensive personal thanks is provided in Supplemental.
Footnotes
Author contributions: K.A.K. conceived of the project, performed experiments, interpreted results, prepared figures, and wrote the manuscript. J.H.K., K.A.R, and G.A.B. performed experiments, interpreted results, and contributed to writing the manuscript. E.I.V. and M.E.R. performed experiments. A.D.A. and M.P.K. interpreted results and contributed to writing the manuscript. J.M.D and F.E.R conceived of the project, interpreted results, and wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amar J, Serino M, Lange C, Chabo C, Iacovoni J, Mondot S, Lepage P, Klopp C, Mariette J, Bouchez O, Perez L, Courtney M, Marre M, Klopp P, Lantieri O, Doré J, Charles MA, Balkau B, Burcelin R D.E.S.I.R. Study Group. Involvement of tissue bacteria in the onset of diabetes in humans: evidence for a concept. Diabetologia. 2011;54:3055–3061. doi: 10.1007/s00125-011-2329-8. [DOI] [PubMed] [Google Scholar]
- Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, Robertson CL, Serova N, Davis S, Soboleva A. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Research. 2013;41:D991–5. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA. 2004;101:15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014 doi: 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besten den G, van Eunen K, Groen AK, Venema K, Reijngoud DJ, Bakker BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res. 2013;54:2325–2340. doi: 10.1194/jlr.R036012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Canadian Journal of Biochemistry and Physiology. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Cabreiro F, Au C, Leung KY, Vergara-Irigaray N, Cochemé HM, Noori T, Weinkove D, Schuster E, Greene NDE, Gems D. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell. 2013;153:228–239. doi: 10.1016/j.cell.2013.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010a;26:266–267. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nat Chem Biol. 2010b;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassaing B, Koren O, Goodrich JK, Poole AC, Srinivasan S, Ley RE, Gewirtz AT. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature. 2015;519:92–96. doi: 10.1038/nature14232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:3. [PubMed] [Google Scholar]
- DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and Environmental Microbiology. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohoe D, Collins L, Wali A, Bigler R, Sun W, Bultman S. The Warburg Effect Dictates the Mechanism of Butyrate-Mediated Histone Acetylation and Cell Proliferation. Molecular Cell. 2012;48:612–626. doi: 10.1016/j.molcel.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohoe DR, Holley D, Collins LB, Montgomery SA, Whitmore AC, Hillhouse A, Curry KP, Renner SW, Greenwalt A, Ryan EP, Godfrey V, Heise MT, Threadgill DS, Han A, Swenberg JA, Threadgill DW, Bultman SJ. A Gnotobiotic Mouse Model Demonstrates that Dietary Fiber Protects Against Colorectal Tumorigenesis in a Microbiota- and Butyrate-Dependent Manner. Cancer Discovery. 2014 doi: 10.1158/2159-8290.CD-14-0501. CD–14–0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Fan J, Krautkramer KA, Feldman JL, Denu JM. Metabolic regulation of histone post-translational modifications. ACS Chem Biol. 2015;10:95–108. doi: 10.1021/cb500846u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, Bork P, Mering von C. STRING 8-a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Research. 2009;37:D412–D416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Johnson DG, Dent SYR. Chromatin: receiver and quarterback for cellular signals. Cell. 2013;152:685–689. doi: 10.1016/j.cell.2013.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson FH, Fåk F, Nookaew I, Tremaroli V, Fagerberg B, Petranovic D, Bäckhed F, Nielsen J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat Commun. 2012;3:1245. doi: 10.1038/ncomms2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessner D, Chambers M, Burke R, Agus D, Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24:2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Applied and Environmental Microbiology. 2013;79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krautkramer KA, Reiter L, Denu JM, Dowell JA. Quantification of SAHA-Dependent Changes in Histone Modifications Using Data-Independent Acquisition Mass Spectrometry. J Proteome Res. 2015;14:3252–3262. doi: 10.1021/acs.jproteome.5b00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng N, Dawson JA, Thomson JA, Ruotti V, Rissman AI, Smits BMG, Haag JD, Gould MN, Stewart RM, Kendziorski C. EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics. 2013;29:1035–1043. doi: 10.1093/bioinformatics/btt087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maile TM, Izrael-Tomasevic A, Cheung T, Guler GD, Tindell C, Masselot A, Liang J, Zhao F, Trojer P, Classon M, Arnott D. Mass Spectrometric Quantification of Histone Post-translational Modifications by a Hybrid Chemical Labeling Method. Mol Cell Proteomics. 2015;14:1148–1158. doi: 10.1074/mcp.O114.046573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6:610–618. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S. Host-gut microbiota metabolic interactions. Science. 2012;336:1262–1267. doi: 10.1126/science.1223813. [DOI] [PubMed] [Google Scholar]
- Pai YJ, Leung KY, Savery D, Hutchin T, Prunty H, Heales S, Brosnan ME, Brosnan JT, Copp AJ, Greene NDE. Glycine decarboxylase deficiency causes neural tube defects and features of non-ketotic hyperglycinemia in mice. Nat Commun. 2015;6:6388. doi: 10.1038/ncomms7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Peng L, Barry NA, Cline GW, Zhang D, Cardone RL, Petersen KF, Kibbey RG, Goodman AL, Shulman GI. Acetate mediates a microbiome-brain-β-cell axis to promote metabolic syndrome. Nature. 2016;534:213–217. doi: 10.1038/nature18309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano KA, Vivas EI, Amador-Noguez D, Rey FE. Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine-N-oxide. MBio. 2015;6:e02481. doi: 10.1128/mBio.02481-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K, McCoy KD, Macpherson AJ. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin Immunol. 2007;19:59–69. doi: 10.1016/j.smim.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, Glickman JN, Garrett WS. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, McKenzie C, Potamitis M, Thorburn AN, Mackay CR, Macia L. The role of short-chain fatty acids in health and disease. Adv Immunol. 2014;121:91–119. doi: 10.1016/B978-0-12-800100-4.00003-9. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI. A core gut microbiome in obese and lean twins. Nature. 2009a;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The Effect of Diet on the Human Gut Microbiome: A Metagenomic Analysis in Humanized Gnotobiotic Mice. Sci Transl Med. 2009b;1:6ra14. doi: 10.1126/scitranslmed.3000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L. The gut microbiota and obesity: from correlation to causality. Nat Rev Micro. 2013;11:639–647. doi: 10.1038/nrmicro3089. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.