

Abstract
This study compared three cryopreservation protocols on sperm functions, IVF outcomes, and embryo development. Epididymal spermatozoa cryopreserved using slow-cooling (18% w/v raffinose, RS-C) were compared with spermatozoa vitrified using 0.25 M sucrose (SV) or 18% w/v raffinose (RV). The motility, vitality, and DNA damage (TUNEL assay) of fresh control (FC) spermatozoa were compared with post-thawed or warmed RS-C, RV, and SV samples. Mouse oocytes (n = 267) were randomly assigned into three groups for insemination: RV (n = 102), RS-C (n = 86), and FC (n = 79). The number and the proportion of two-cell embryos and blastocysts from each treatment were assessed. Sperm motility (P < 0.01) and vitality (P < 0.05) were significantly reduced after vitrification compared with slow-cooled spermatozoa. However, DNA fragmentation was significantly reduced in spermatozoa vitrified using sucrose (15 ± 1.8% [SV] vs 26 ± 2.8% [RV] and 27 ± 1.2% [RS-C]; P < 0.01). Although the number of two-cell embryos produced by RS-C, RV, and FC spermatozoa was not significantly different, the number of blastocysts produced from two-cell embryos using RV spermatozoa was significantly higher than FC spermatozoa (P = 0.0053). This simple, small volume vitrification protocol and standard insemination method allows successful embryo production from small numbers of epididymal spermatozoa and may be applied clinically to circumvent the need for ICSI, which has the disadvantage of bypassing sperm selection.
Keywords: cryopreservation, epididymal sperm, in vitro fertilization/intracytoplasmic sperm injection, reproductive techniques, sperm DNA damage, vitrification
INTRODUCTION
Successful cryopreservation allows cells and tissues to be stored for long periods of time without significant change. Sperm cryopreservation was first reported in the early 20th Century by Polge et al.1 who discovered that after the addition of glycerol to the diluents, spermatozoa could regain their motility after long periods of freezing. Since that time, a plethora of papers have reported successful cryopreservation in different species, including humans.2,3 Clinical applications of cryopreservation include the preservation of male fertility before radiotherapy and/or chemotherapy,4 the use of frozen partner and donor spermatozoa for artificial insemination (AI), and in vitro fertilization (IVF) in infertility treatments.5 Furthermore, the ability to successfully cryopreserve epididymal and testicular sperm samples is important and useful for treating patients with obstructive and nonobstructive azoospermic who require the use of assisted reproductive technologies (ARTs).
Sperm diluents were originally designed for the use with slow-cooling methods. Initially, the whole ejaculate was added to the diluents, which contained high viscosity components and at least one permeable cryoprotectant (CPA). A popular initial diluent developed for animal and human sperm cryopreservation was glycerol egg yolk tris-citrate (GEYC). Later, more defined and commercially available diluents (e.g., human sperm preservation media, HSPM) were designed for cryopreservation of whole human ejaculates and washed human semen. These diluents have also been used with some success for slow-cooling human epididymal and testicular spermatozoa,6,7,8 but recovery rates were often low, due to initial poor quality of the samples and reduced survival of spermatozoa in samples with low concentration.9 Epididymal spermatozoa are routinely used in mouse IVF and slow-cooling methods have been optimized for epididymal sperm in this species. For mouse sperm cryopreservation, the most widely used diluent combines the non-permeating raffinose with skimmed milk powder.10 More recently, vitrification of spermatozoa has been proposed as a novel method for cryopreservation, using a nonpermeable CPA such as sucrose and high cooling rates (>40–1000°C min−1).11 However, very few studies have tested if slow-cooling diluents are effective for the vitrification. The aims of this study were to compare the use of different cryopreservation protocols on the functions of mouse epididymal spermatozoa and examine the effects of vitrification of epididymal spermatozoa on IVF outcomes and embryo development.
METHODOLOGY
Experimental design
Two experiments were designed to compare the effects of three different cryopreservation methods on various parameters of sperm function, fertilization ability, and embryo development.
Experiment 1: assessment of basic sperm parameters
Sperm motility, sperm vitality, and sperm DNA damage were compared in five replicates before and after cryopreservation using three different cryopreservation methods: conventional slow-cooling with raffinose (RS-C), two vitrification protocols using raffinose (RV), and sucrose (SV) (method previously described for human spermatozoa12 and modified for mouse spermatozoa).
Experiment 2: assessment of embryo production following standard IVF
Sperm samples (n = 5) were obtained from the cauda epididymis of F1 mice, and oocytes (n = 267, five replicates) were flushed from the F1 mouse oviducts for IVF. The effects of RS-C and RV on sperm motility, vitality, and DNA fragmentation, and on embryo production and development, were compared against results using fresh control sperm samples.
Mice
Ethics approval for this study was obtained from the Monash Medical Centre Animal Ethics Committee (AEC Approval No. MMCA2011/84). A 12-week-old F1 C57BL × CBA hybrid males were used as a source of epididymal spermatozoa for both experiments (n = 10). Six to seven weeks old F1 C57BL × CBA hybrid female mice were used for IVF (Experiment 2, n = 5). All mice were obtained from Monash University Animal Services and housed in a conventional animal house under a 12 h light-dark cycle with a relative humidity of 30%–60% and a temperature ranging from 21°C to 24°C. All mice had free access to food and water.
Superovulation and oocyte collection
Female mice were superovulated by administration of 5 IU of Pregnant Mare Serum Gonadotropin (PMSG, Folligon®, Bendigo, VIC, Australia) 48 h before collection, followed by 5 IU of human Chorionic Gonadotropin (hCG, Intervet®, Bendigo, VIC, Australia) 16–18 h before collection. The superovulated female mice were killed by cervical dislocation. Oviducts were cleaned of fat and connective tissue, and the cumulus-oocyte complexes (COCs) released by dissecting the ampulla. The COCs were used in IVF experiments with cryopreserved spermatozoa.
Media and vitrification solutions for spermatozoa
KSOM handling medium (KSOMH, adapted medium13) with 0.3% w/v Bovine serum albumin (BSA, Sigma®, Melbourne, VIC, Australia) was used as the handling medium for all gametes. KSOMH with 1% w/v BSA (KSOMH+) was used as the base for the sucrose vitrification (SV) solution (0.25 mol l−1 sucrose in KSOMH+). The raffinose-based cryoprotectant solution used for vitrification (RV) and slow-cooling (RS-C) consisted of 18% w/v raffinose diluted in handling medium with 3% w/v skim milk (adapted medium11). Modified Tyrode's solution (MT614) was used for insemination and embryos were cultured in LifeGlobal Total medium with 10% v/v HSA (LifeGlobal®, Guliford, Connecticut, United States).
Sperm collection and swim up
Male mice were killed by cervical dislocation and the epididymides were located, exposed, and isolated from the testes. The epididymides were cleaned of fat, and the caudal region was identified and excised. A cut was made through the convoluted duct in the cauda, and the incised tissue placed into 5 ml Falcon™ tubes (BD biosciences, San Jose, CA, USA) containing either 135 μl prewarmed Raffinose CPA (for RV and RS-C) or 135 μl prewarmed KSOMH (for SV). Spermatozoa were allowed to swim-up into the medium for 30 min at 37°C.
Sperm cryopreservation methodology
Conventional slow-cooling (RS-C)
After 30 min, 10 μl of the sperm sample was drawn into straws which were sealed with PVA, placed in liquid nitrogen vapor (10 cm above the liquid nitrogen) for 10 min and then plunged into liquid nitrogen.
Thawing (RS-C)
For motility, vitality, and DNA integrity assays, straws were exposed to air (10 s) and 37°C water (30 s) before expelling the thawed sperm sample into 50 μl KSOMH. For IVF, straws were directly plunged into the warmed, equilibrated MT6 droplets, and approximately 15 μl moved into droplets containing the COCs.
Vitrification procedures
Vitrification with raffinose (RV)
After swim-up into raffinose CPA for 30 min, the top 500 μl of medium containing motile spermatozoa was removed and mixed well. Samples with the same concentration (5 × 106 ml−1) were vitrified by loading 3 μl on to fiberplugs and placing them for 3 s onto a metal block platform (CVM, Cryologic®, Blackburn, VIC, Australia) cooled to −196°C. The vitrified samples on fiberplugs were then inserted into straws cooled to −196°C on the metal block platform and finally plunged into liquid nitrogen as a closed system.
Vitrification with 0.25 mol l−1 sucrose (SV)
This methodology was an adaptation from Isachenko et al.12 Following swim-up into KSOMH for 30 min, the top 500 μl was removed, centrifuged (300 g, 10 min), and resuspended in KSOMH to the desired minimum concentration of 5 × 106 ml−1, then diluted 1:1 with the SV just prior to vitrification, as described by the Isachenko group.12 From this step, vitrification was conducted as described above.
Warming procedure for SV and RV
For motility, vitality, and DNA integrity assays, the vitrified samples were warmed by directly placing the fiberplugs into 50 μl warmed KSOMH at 37°C. For IVF, fiberplugs were directly plunged into the warmed, equilibrated MT6 droplets containing the COCs.
Evaluation of sperm motility and vitality
Sperm motility and vitality were determined before and after cryopreservation. Motility was assessed by scoring the numbers of motile (progressive and nonprogressive) and immotile spermatozoa in a series of randomly selected microscope fields. In addition, sperm recovery rates (SSR) were calculated to compare sperm motility before and after cryopreservation for each method using the formula: SSR = ([% thawed motile spermatozoa/% fresh motile spermatozoa] ×100). Hypo-osmotic swelling (HOS) test was used to assess sperm vitality.15 This was determined by scoring the number of spermatozoa with swollen midpieces or tails as live spermatozoa (membrane intact) and those which showed no swelling as dead spermatozoa. The thawed/warmed sperm motility and vitality rates were analyzed. For both methods, at least 200 sperm cells were assessed by two different observers and a mean value calculated.
Sperm DNA fragmentation
The presence of DNA strand breaks was evaluated by the TdT (terminal deoxynucleotidyl transferase)-mediated dUDP nick-end labeling (TUNEL) assay using In Situ Cell Death Detection kit with fluorescein (Roche Diagnostics®, Mannheim, Germany), with some modifications to the manufacturer's protocol described below.
Mouse spermatozoa were loaded on to silane-coated slides (Strafrost®, Braunschweig, Germany), air dried, and then fixed with 3.5% v/v formaldehyde in phosphate-buffered saline (PBS) for 1 h at room temperature. Sperm preparations were washed three times in PBS to remove the fixative and then permeabilized with 0.1% v/v Triton X-100 in 0.1% w/v sodium citrate for 5 min on ice. Slides were washed twice with PBS, incubated with 50 μl of labeling solution (In Situ Cell Death Detection Kit) containing TdT for 1 h at 37°C in the dark, and rinsed three times in PBS. Spermatozoa were counterstained with propidium iodide (PI, Santa Cruz Biotechnology®, Dallas, TX, United States) at a final concentration of 50 μg l−1 and slides were coverslipped and sealed with nail polish. TUNEL assays were analyzed on a compound fluorescence microscope. Green fluorescence (TUNEL positive) from fluorescein was detected with two filters (TRITC: EX540/25, DM565, BA605/55, and FITC: EX465-495, DM505, BA515-555). Positive controls were prepared by incubating spermatozoa in 5U DNAse I (Roche®, Mannheim, Germany) for 45 min at 37°C before the labeling reaction and negative control spermatozoa were incubated without the TdT enzyme. At least 400 cells were assessed for each sample (two observers) at ×1000 magnification using a ×100 oil-immersion objective.
IVF, embryo production, and development
Mature mouse oocytes (n = 267) collected from the oviduct as cumulus-oocyte-complex (COCs) were randomly divided into three IVF groups: two groups using either RV or RS-C cryopreserved spermatozoa (RV: n = 102; RS-C: n = 86) and one group using fresh control spermatozoa (FC: n = 79). The COCs were transferred into preequilibrated MT6 drops (50 μl under oil), and dishes returned to the incubator at 37°C in 90% humidity and 5% CO2 in air.
Within 1 h, fresh, frozen-thawed, and vitrified-warmed spermatozoa were prepared by swim up (FC) or as described in the thawing (RS-C) and warming (RV) procedures giving a total of approximately 250 000 motile spermatozoa per ml in the insemination droplets.
The time of insemination was recorded for each group and 2–3 h after insemination oocytes were cleaned of cumulus cells and excess spermatozoa and moved to a 30 μl droplet of preequilibrated culture medium (LifeGlobal®, Guliford, Connecticut, United States) under mineral oil (Sigma®, St. Louis, Missouri, United States) for culture overnight.
Embryo development was observed every 24 h after insemination, and the culture medium was changed every 48 h. The number of two-cell embryos, blastocysts, and expanded blastocysts were recorded for each treatment.
Statistical analyses
The data were analyzed using Prism 5 software (GraphPad Prism 5, version 5.0c, GraphPad Software, Inc): for sperm parameters, one-way ANOVA was used to compare data between the experimental and control groups and the Bonferroni test was used as post hoc analysis. A paired Chi-square test (Fishers exact) was used to compare embryo production between groups. Statistical significance was defined as P < 0.05 and results are expressed as mean ± standard error (s.e.m.).
RESULTS
Motility and vitality
RS-C had significantly higher motility (46 ± 3.2%) than SV (14 ± 1.4%, P < 0.05) and RV (30 ± 1.6%, P < 0.05) (Figure 1a) as well as vitality (RS-C: 57 ± 2.0%; SV: 23 ± 4.2%; RV: 39 ± 0.6%, P < 0.05) (Figure 1b).
Figure 1.

Comparison of sperm motility, vitality and DNA fragmentation between fresh samples, conventional and vitrification methods. (a) Sperm motility: Motile spermatozoa percentage. (b) Sperm vitality: Viable spermatozoa percentage. (c) Sperm DNA fragmentation: Percentage of sperm DNA fragmentation. Fresh samples (Fresh), conventional (RS-C: raffinose slow-cooling) and vitrification methods (RV: raffinose vitrification; SV: sucrose vitrification). *Statistically significant differences (P < 0.05).
Between vitrification methods, RV spermatozoa had significantly higher motility (30 ± 1.6%) and vitality (39 ± 0.6%) than SV (14 ± 1.4%, P < 0.05 and 23 ± 4.2%, P < 0.05, respectively) (Figure 1a and 1b). Comparison of sperm motility recovery showed that RS-C spermatozoa (73 ± 2.7%, P < 0.05) recovered better from cryopreservation than RV (47 ± 1.2%, P < 0.05) or SV spermatozoa (21 ± 1.8%, P < 0.05) and that recovery was significantly better with RV than SV spermatozoa.
Sperm DNA damage
Spermatozoa from all three experimental groups showed significantly higher levels of DNA damage compared with the fresh sample (3.2 ± 0.2%, P < 0.05). In the three cryopreservation groups, significantly higher levels of damage were also seen between spermatozoa from RS-C (26 ± 2.8%) and RV (27 ± 1.2%), compared to SV spermatozoa (15 ± 1.8%, P < 0.05) (Figure 1c).
Sperm DNA fragmentation essays were performed on mouse sperm samples using TUNEL (Figure 2a). Positive control spermatozoa all showed DNA damage (Figure 2b). Negative controls showed no signals for sperm DNA fragmentation (Figure 2c).
Figure 2.
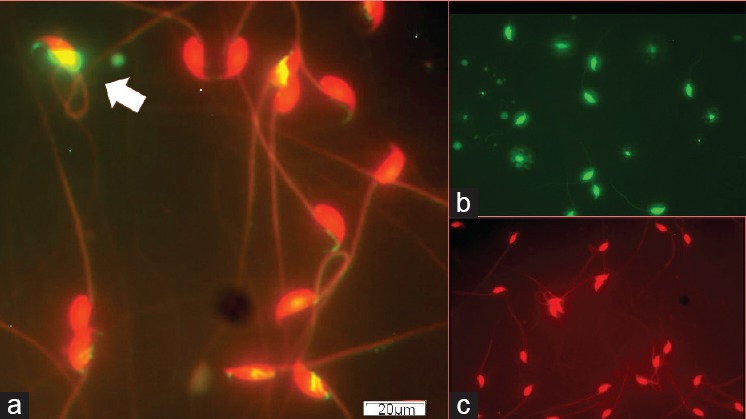
Micrograph showing sperm DNA fragmentation using TUNEL essay of mouse spermatozoa. (a) Sample from a prefreeze treatment. (b) Positive control. (c) Negative control. Arrow, spermatozoon with DNA fragmentation.
IVF and embryo development
The percentage of two-cell embryos produced using RV (n = 68/108, 66.7%), RS-C (n = 53/86, 61.6%), and FC (n = 41/79, 51.9%) spermatozoa was not significantly different (P > 0.05) (Table 1). The percentage of blastocysts produced from two-cell embryos was significantly higher in the FC group (n = 27/41, 65.9%) than RV group (n = 25/68, 36.8%) (2c/Blast; P = 0.0053). However, there were statistical differences in the number of expanded blastocyst in the RV group (n = 23/68, 33.8%) compared to the RS-C group (n = 9/53, 17%) (2c/HgB; P = 0.04) (Figure 3). In other words, 92% (n = 23/25) of RV blastocyst reached expanded blastocysts stage compared to 33.3% (n = 9/27) in the RS-C group and 29.6% (n = 8/27) in the FC groups, respectively (Table 1).
Table 1.
Comparison of the number of two-cell embryos, blastocysts, and expanded blastocyst between the two cryopreservation methods and fresh control

Figure 3.
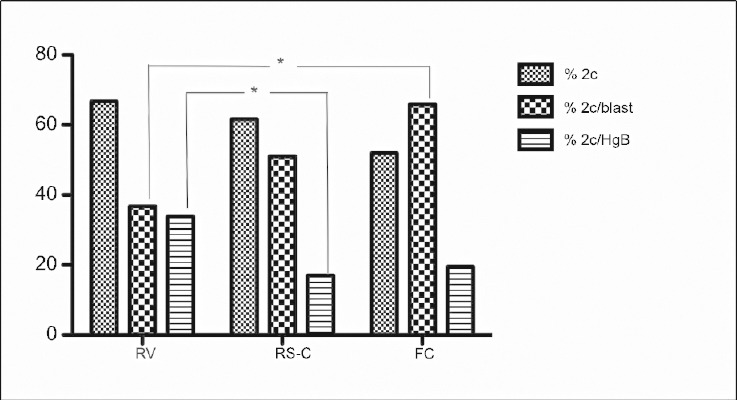
Comparison of number of two-cell embryos, blastocysts and expanded blastocyst among cryopreservation methods. % 2c: percentage of two-cell embryos; % 2c/blast: percentage of blastocysts from two-cell embryos; % 2c/HgB: percentage of expanded blastocyst from two-cell embryos. RV: raffinose vitrification; RS-C: raffinose slow-cooling; FC: fresh control. *Significant differences; P < 0.05.
DISCUSSION
Various methods of freezing spermatozoa have been previously described10,16,17,18,19,20,21,22 mostly using slow-cooling and different cryoprotectants.23 However, despite the absence of clear successful vitrification outcomes in the mouse, successful vitrification of human,12,24,25,26,27,28,29,30,31 canine,32 and fish (Oncorhynchus mykiss)33 spermatozoa has been reported using specific capillaries, low sample volume (3 μl), dropped directly into liquid nitrogen, and high cooling rates.
We developed a vitrification method based on the methodology described by Isachenko et al.12 testing the CPA proposed by this group and the same CPA widely used with mouse spermatozoa for conventional slow-cooling cryopreservation.10 Our results show that hybrid F1 mouse epididymal spermatozoa can be vitrified using fiber plugs as carriers and that sufficient motility and vitality are retained after warming to perform successful IVF, by direct transfer of these fiberplugs into the insemination droplet. It was interesting to note that the fresh spermatozoa resulted in the lowest number of two-cell embryos and also had proportionately fewer advanced embryos (hatching blastocysts) compared to the RV group. The fresh sperm outcomes may have been slightly compromised as we were trying to make the insemination protocol as “similar” as possible in all sperm groups by adding the same volume at the same time. Cryopreserving spermatozoa can artificially “accelerate” the processes leading to capacitation34 and this may explain the higher two-cell rates and slightly advanced embryo growth. We noticed that for both the RV and RS-C groups, motility declined very rapidly and it was therefore important that sperm-oocyte contact occurred immediately after warming.
In terms of motility and vitality, our data showed that vitrification using fiberplugs and 18% w/v raffinose as CPA resulted in better post-warming outcomes than 0.25 mol l−1 sucrose. However, both vitrification techniques resulted in lower sperm motility and vitality spermatozoa than conventional slow-cooling.
Despite CPAs having different cryoprotective properties, some studies have suggested that cryoprotection is independent of the concentration and the type of sugars used for cryopreservation.35 Although the molecular structure and concentration of sucrose and raffinose are different (raffinose is trisaccharide while sucrose is disaccharide), similar membrane fusion rates have been reported between thawed spermatozoa in 12% sucrose and 18% raffinose solutions.36 However, our use of these two CPAs with different concentrations of protein extenders (3% w/v skim milk vs 1% w/v BSA) may have improved our results using the protein extender to expand the osmotic tolerance limits of mouse spermatozoa.37
In contrast to the findings of Katkov et al.,38 our study showed that the highest post-thaw/warming motility and vitality resulted from cryopreservation using raffinose-based, rather than sucrose-based cryoprotectants. Also, with human spermatozoa,11,12,39 successful post-warming outcomes were achieved after vitrification with 0.25 mol l−1 sucrose. The difference may be explained by specific cryobiological characteristics of mouse spermatozoa, especially in the structure of the plasma membrane. In addition, our data from a pilot study (data not shown) correlated with previous reports that showed higher post-warming motility using 0.25 mol l−1 sucrose than with 0.50 mol l−1 and 0.75 mol l−1 sucrose, suggesting that these higher concentrations of sugars exceed the osmotic tolerance of mouse spermatozoa.35
An important factor affecting vitrification is the type of carrier used.23 As a novel approach, we loaded samples on to fiber plugs (Cryologic®) to provide the highest possible cooling rates using 3 μl volumes and no straw barriers. Other studies have used open-pulled straws (conventional method) or capillaries12 but resulted in unsuccessful cryopreservation of mouse spermatozoa. These carriers have a decreased cooling rate because samples are not in direct contact with liquid nitrogen due to the straw barriers. Fiberplugs, and also cryotops, recently used for vitrifying human samples,40 allow for an increase in cooling rates by avoiding the straw barriers in the vitrification process.
The lower motility outcomes that we report after warming compared with human studies11,12 may be explained by increased cryosensitivity of mouse spermatozoa due to their large size, asymmetric head structure, and higher sensitivity to manipulation. Cryobiological characteristics of mouse spermatozoa need to be taken into account to achieve successful vitrification.36,37 An important advantage of our method is that after warming, vitrified mouse spermatozoa can be used without additional treatment, such as centrifugation and density gradients; samples can be added directly into insemination (IVF) media at 37°C or media droplets intended for single sperm selection for ICSI. For practical purposes, this simplicity represents an advantage of this technology, because even with low motility, vitrification provides sufficient motile spermatozoa to perform successful IVF and ICSI.
In the mouse, sperm DNA damage did not show a close relationship with motility and vitality and embryo development as previously reported in human spermatozoa.12,41 On the contrary, cryopreservation of human sperm samples has been reported to affect crucial genes for fertilization and early embryo development.42 Moreover, increased sperm DNA fragmentation has been shown after cryopreservation but measured following a period of incubation at 37°C, and indeed, fresh samples also showed higher levels following incubation at 37°C. Thus, treatment post-cryopreservation, as well as the process itself, appears to affect sperm DNA.43 While raffinose both in slow-cooling and vitrification produced the best outcomes in terms of motility and viability, it resulted in significantly higher levels of sperm DNA damage than spermatozoa vitrified using sucrose as the CPA.
Although the effect of cryopreservation on DNA integrity is still not entirely understood, there is evidence that DNA damage or fragmentation is linked to the molecular weight, structure, concentration, and chemical features of the cryoprotectant used44,45,46 and the cryopreservation protocol.44,45,47,48 The use of different proteins and protein concentrations in the media in our study (3% w/v skim milk vs 1% w/v BSA) may also have contributed to the different levels of DNA damage due to different expansions of the osmotic tolerance limits of mouse spermatozoa.37 In addition, Paasch et al.49 showed that cryopreservation and thawing/warming are associated in human spermatozoa with varying degrees of activation of the apoptotic machinery in spermatozoa which may also trigger sperm DNA fragmentation in mouse spermatozoa.
Studies in humans have associated sperm DNA damage with decreased embryo development rate such as cleavage, blastulation and decreased implantation, and pregnancy rates.50,51 This has also been reported in the mouse. Yildiz et al.46 showed that sperm DNA damage caused a decreased in vitro embryo development rates to blastocyst using frozen–thawed C57BL/6 spermatozoa. The same group52 also showed that increased post-thaw sperm DNA fragmentation decreased embryo development rate to blastocyst using frozen–thawed spermatozoa from C3B6F1/J or B6129S1F1/J mouse strains when compared with fresh spermatozoa.
Although vitrification using sucrose showed the lowest values of sperm DNA damage, it also resulted in reduced levels of motility and vitality compared to mouse spermatozoa cryopreserved using raffinose. For the embryo development studies, as we were attempting IVF and not ICSI, we selected the vitrification group with the most motile sperm cells to enhance our chances of success when comparing these data with the fresh and slow-cooled groups.
In this study, we found no difference in the numbers of two-cell embryos from raffinose cryopreserved (slow-cooled and vitrified) and fresh control spermatozoa indicating that fresh and frozen spermatozoa had similar in vitro fertilization rates. This was not consistent with a previous study that reported a higher in vitro fertilization rate with fresh spermatozoa.52 In our experience, motility reduces very rapidly after warming/thawing and the short time between warming/thawing and insemination is critical. As reported in other studies,45,52,53 the number of two-cell embryos that developed into blastocysts from IVF, using fresh control spermatozoa was higher (65.9%) than those from slow-cooled spermatozoa. However, a higher rate of expanded blastocysts was observed using vitrified spermatozoa (33.8%) than slow-cooled spermatozoa (17.0%, P < 0.05).
CONCLUSION
Vitrification of mouse spermatozoa using raffinose and sucrose as CPA loaded on fiberplugs is possible; sperm motility and vitality are, however, lower after vitrification than after conventional slow-cooling and in fresh sperm samples. These outcomes produced acceptable rates of embryo production and development (raffinose) that are not inferior to those produced by the conventional slow-cooling method or indeed using fresh spermatozoa. Moreover, both vitrification methods were simpler and less time consuming, due to fewer steps in the protocols, than the conventional slow-cooling technique. However, further studies are now needed to examine the impact, if any, of minimal volume vitrification on sperm DNA damage and other sperm functions in spermatozoa of other species. In particularly, it will be important to consider these aspects after cryopreservation of small fluid volumes with low sperm concentrations that are often found in samples surgically retrieved from the human testis and epididymis.
AUTHORS CONTRIBUTION
FH carried out all sperm experiments, part of IVF experiments, its design, statistical analyses, and drafted the manuscript. HA carried out all sperm experiments, majority of IVF experiments, and part of statistical analyses. SJ performed all sperm experiment and part of statistical analyses. SC participated in its design, supervision, and coordination and helped draft the manuscript. PC participated in supervision and helped in experimental tasks. MP participated in coordination and provided extensive experimental advice during the project. PTS participated in its design, supervision, and coordination and helped draft the manuscript. All authors read and approved the final manuscript.
COMPETING INTERESTS
All authors declared no competing financial interests.
REFERENCES
- 1.Polge C, Smith AU, Parkes AS. Revival of spermatozoa after vitrification and dehydration at low temperatures. Nature. 1949;164:666. doi: 10.1038/164666a0. [DOI] [PubMed] [Google Scholar]
- 2.Bunge RG, Sherman JK. Fertilizing capacity of frozen human spermatozoa. Nature. 1953;172:767–8. doi: 10.1038/172767b0. [DOI] [PubMed] [Google Scholar]
- 3.Bunge RG. Clinical use of frozen semen: report of four cases. Fertil Steril. 1954;5:520–9. doi: 10.1016/s0015-0282(16)31802-7. [DOI] [PubMed] [Google Scholar]
- 4.Sanger WG, Olson JH, Sherman JK. Semen cryobanking for men with cancer – Criteria change. Fertil Steril. 1992;58:1024–7. doi: 10.1016/s0015-0282(16)55454-5. [DOI] [PubMed] [Google Scholar]
- 5.Clarke GN, Bourne H, Hill P, Johnston WI, Speirs A, et al. Artificial insemination and in-vitro fertilization using donor spermatozoa: a report on 15 years of experience. Hum Reprod. 1997;12:722–6. doi: 10.1093/humrep/12.4.722. [DOI] [PubMed] [Google Scholar]
- 6.Holden CA, Fuscaldo GF, Jackson P, Cato A, Southwick GJ, et al. Frozen-thawed epididymal spermatozoa for intracytoplasmic sperm injection. Fertil Steril. 1997;67:81–7. doi: 10.1016/s0015-0282(97)81860-2. [DOI] [PubMed] [Google Scholar]
- 7.Van Steirteghem A, Nagy P, Joris H, Janssenswillen C, Staessen C, et al. Results of intracytoplasmic sperm injection with ejaculated, fresh and frozen-thawed epididymal and testicular spermatozoa. Hum Reprod. 1998;13(Suppl 1):134–42. doi: 10.1093/humrep/13.suppl_1.134. [DOI] [PubMed] [Google Scholar]
- 8.Janzen N, Goldstein M, Schlegel PN, Palermo GD, Rosenwaks Z. Use of electively cryopreserved microsurgically aspirated epididymal sperm with IVF and intracytoplasmic sperm injection for obstructive azoospermia. Fertil Steril. 2000;74:696–701. doi: 10.1016/s0015-0282(00)01496-5. [DOI] [PubMed] [Google Scholar]
- 9.Cohen J, Garrisi GJ, Congedo-Ferrara TA, Kieck KA, Schimmel TW, et al. Cryopreservation of single human spermatozoa. Hum Reprod. 1997;12:994–1001. doi: 10.1093/humrep/12.5.994. [DOI] [PubMed] [Google Scholar]
- 10.Nakagata N. Cryopreservation of mouse spermatozoa. Mamm Genome. 2000;11:572–6. doi: 10.1007/s003350010109. [DOI] [PubMed] [Google Scholar]
- 11.Isachenko E, Rahimi G, Risopatròn J, Schulz M, Mallmann P, et al. Vitrification Technique – New Possibilities for Male Gamete Low-Temperature Storage. Germany: INTECH Open Access Publisher; 2012. pp. 41–76. [Google Scholar]
- 12.Isachenko V, Maettner R, Petrunkina A, Sterzik K, Mallmann P, et al. Vitrification of human ICSI/IVF spermatozoa without cryoprotectants: new capillary technology. J Androl. 2012;33:462–8. doi: 10.2164/jandrol.111.013789. [DOI] [PubMed] [Google Scholar]
- 13.Summers MC, Bhatnagar PR, Lawitts JA, Biggers JD. Fertilization in vitro of mouse ova from inbred and outbred strains: complete preimplantation embryo development in glucose-supplemented KSOM. Biol Reprod. 1995;53:431–7. doi: 10.1095/biolreprod53.2.431. [DOI] [PubMed] [Google Scholar]
- 14.Fraser LR. Mouse sperm capacitation in vitro involves loss of a surface associated inhibitory component. J Reprod Fertil. 1984;72:373–84. doi: 10.1530/jrf.0.0720373. [DOI] [PubMed] [Google Scholar]
- 15.Jeyendran RS, Van der Ven HH, Perez-Pelaez M, Crabo BG, Zaneveld LJ. Development of an assay to assess the functional integrity of the human sperm membrane and its relationship to other semen characteristics. J Reprod Fertil. 1984;70:219–28. doi: 10.1530/jrf.0.0700219. [DOI] [PubMed] [Google Scholar]
- 16.Penfold L, Moore H. A new method for cryopreservation of mouse spermatozoa. J Reprod Fertil. 1993;99:131–4. doi: 10.1530/jrf.0.0990131. [DOI] [PubMed] [Google Scholar]
- 17.Nakagata N, Takeshima T. Cryopreservation of mouse spermatozoa from inbred and F1 hybrid strains. Exp Anim. 1993;42:317–20. doi: 10.1538/expanim1978.42.3_317. [DOI] [PubMed] [Google Scholar]
- 18.Nakagata N, Ueda S, Yamanouchi K, Okamoto M, Matsuda Y, et al. Cryopreservation of wild mouse spermatozoa. Theriogenology. 1995;43:635–43. doi: 10.1016/0093-691x(94)00069-7. [DOI] [PubMed] [Google Scholar]
- 19.Nakagata N, Okamoto M, Ueda O, Suzuki H. Positive effect of partial zona-pellucida dissection on the in vitro fertilizing capacity of cryopreserved C57BL/6J transgenic mouse spermatozoa of low motility. Biol Reprod. 1997;57:1050–5. doi: 10.1095/biolreprod57.5.1050. [DOI] [PubMed] [Google Scholar]
- 20.Sztein JM, Farley JS, Young AF, Mobraaten LE. Motility of cryopreserved mouse spermatozoa affected by temperature of collection and rate of thawing. Cryobiology. 1997;35:46–52. doi: 10.1006/cryo.1997.2024. [DOI] [PubMed] [Google Scholar]
- 21.Songsasen N, Leibo S. Cryopreservation of mouse spermatozoa I. Effect of seeding on fertilizing ability of cryopreserved spermatozoa. Cryobiology. 1997;35:240–54. doi: 10.1006/cryo.1997.2048. [DOI] [PubMed] [Google Scholar]
- 22.Storey BT, Noiles EE, Thompson KA. Comparison of glycerol, other polyols, trehalose, and raffinose to provide a defined cryoprotectant media for mouse sperm cryopreservation. Cryobiology. 1998;37:46–58. doi: 10.1006/cryo.1998.2097. [DOI] [PubMed] [Google Scholar]
- 23.Moskovtsev SI, Lulat AG, Librach CL. Cryopreservation of Human Spermatozoa by Vitrification vs. Slow Freezing: Canadian Experience. Canada: INTECH Open Access Publisher; 2012. p. 80. [Google Scholar]
- 24.Isachenko V, Isachenko E, Katkov II, Montag M, Dessole S, et al. Cryoprotectant-free cryopreservation of human spermatozoa by vitrification and freezing in vapor: effect on motility, DNA integrity, and fertilization ability. Biol Reprod. 2004;71:1167–73. doi: 10.1095/biolreprod.104.028811. [DOI] [PubMed] [Google Scholar]
- 25.Isachenko V, Isachenko E, Montag M, Zaeva V, Krivokharchenko I, et al. Clean technique for cryoprotectant-free vitrification of human spermatozoa. Reprod Biomed Online. 2005;10:350–4. doi: 10.1016/s1472-6483(10)61795-6. [DOI] [PubMed] [Google Scholar]
- 26.Isachenko V, Maettner R, Petrunkina AM, Mallmann P, Rahimi G, et al. Cryoprotectant-free vitrification of human spermatozoa in large (up to 0.5 mL) volume: a novel technology. Clin Lab. 2010;57:643–50. [PubMed] [Google Scholar]
- 27.Isachenko V, Isachenko E, Petrunkina AM, Sanchez R. Human spermatozoa vitrified in the absence of permeable cryoprotectants: birth of two healthy babies. Reprod Fertil Dev. 2012;24:323–6. doi: 10.1071/RD11061. [DOI] [PubMed] [Google Scholar]
- 28.Sanchez R, Isachenko V, Petrunkina AM, Risopatron J, Schulz M, et al. Live birth after intrauterine insemination with spermatozoa from an oligoasthenozoospermic patient vitrified without permeable cryoprotectants. J Androl. 2012;3:559–62. doi: 10.2164/jandrol.111.014274. [DOI] [PubMed] [Google Scholar]
- 29.Mansilla MA, Merino O, Risopatrón J, Isachenko V, Isachenko E, et al. High temperature is essential for preserved human sperm function during the devitrification process. Andrologia. 2015;48:111–3. doi: 10.1111/and.12406. [DOI] [PubMed] [Google Scholar]
- 30.Kuznyetsov V, Moskovtsev SI, Crowe M, Lulat AG, Librach CL. Vitrification of a small number of spermatozoa in normozoospermic and severely oligozoospermic samples. Syst Biol Reprod Med. 2014;61:13–7. doi: 10.3109/19396368.2014.987855. [DOI] [PubMed] [Google Scholar]
- 31.Slabbert M, du Plessis SS, Huyser C. Large volume cryoprotectant-free vitrification: an alternative to conventional cryopreservation for human spermatozoa. Andrologia. 2014;47:594–9. doi: 10.1111/and.12307. [DOI] [PubMed] [Google Scholar]
- 32.Sánchez R, Risopatrón J, Schulz M, Villegas J, Isachenko V, et al. Canine sperm vitrification with sucrose: effect on sperm function. Andrologia. 2011;43:233–41. doi: 10.1111/j.1439-0272.2010.01054.x. [DOI] [PubMed] [Google Scholar]
- 33.Merino O, Risopatrón J, Sánchez R, Isachenko E, Figueroa E, et al. Fish (Oncorhynchus mykiss) spermatozoa cryoprotectant-free vitrification: stability of mitochondrion as criterion of effectiveness. Anim Reprod Sci. 2011;124:125–31. doi: 10.1016/j.anireprosci.2011.02.023. [DOI] [PubMed] [Google Scholar]
- 34.Gillan L, Evans G, Maxwell WM. Capacitation status and fertility of fresh and frozen-thawed ram spermatozoa. Reprod Fertil Dev. 1997;9:481–7. doi: 10.1071/r96046. [DOI] [PubMed] [Google Scholar]
- 35.Koshimoto C, Mazur P. Effects of cooling and warming rate to and from -70 degrees C, and effect of further cooling from -70 to -196 degrees C on the motility of mouse spermatozoa. Biol Reprod. 2002;66:1477–84. doi: 10.1095/biolreprod66.5.1477. [DOI] [PubMed] [Google Scholar]
- 36.Agca Y, Gilmore J, Byers M, Woods EJ, Liu J, et al. Osmotic characteristics of mouse spermatozoa in the presence of extenders and sugars. Biol Reprod. 2002;67:1493–501. doi: 10.1095/biolreprod.102.005579. [DOI] [PubMed] [Google Scholar]
- 37.Benson JD, Woods EJ, Walters EM, Critser JK. The cryobiology of spermatozoa. Theriogenology. 2012;78:1682–99. doi: 10.1016/j.theriogenology.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 38.Katkov I, Bolyukh V, Chernetsov O, Dudin P, Grigoriev A, et al. Kinetic Vitrification of Spermatozoa of Vertebrates: What Can We Learn from Nature. Current Frontiers in Cryobiology. Germany: INTECH Open Access Publisher; 2012. pp. 3–40. [Google Scholar]
- 39.Nawroth F, Isachenko V, Dessole S, Rahimi G, Farina M, et al. Vitrification of human spermatozoa without cryoprotectants. Cryo Letters. 2002;23:93–102. [PubMed] [Google Scholar]
- 40.Chen Y, Li L, Qian Y, Xu C, Zhu Y, et al. Small-volume vitrification for human spermatozoa in the absence of cryoprotectants by using cryotop. Andrologia. 2014;47:694–9. doi: 10.1111/and.12320. [DOI] [PubMed] [Google Scholar]
- 41.Isachenko E, Isachenko V, Katkov II, Rahimi G, Schöndorf T, et al. DNA integrity and motility of human spermatozoa after standard slow freezing versus cryoprotectant-free vitrification. Hum Reprod. 2004;19:932–9. doi: 10.1093/humrep/deh194. [DOI] [PubMed] [Google Scholar]
- 42.Valcarce DG, Cartón-García F, Riesco MF, Herráez MP, Robles V. Analysis of DNA damage after human sperm cryopreservation in genes crucial for fertilization and early embryo development. Andrology. 2013;5:723–30. doi: 10.1111/j.2047-2927.2013.00116.x. [DOI] [PubMed] [Google Scholar]
- 43.Gosálvez J, Núñez R, Fernández JL, López-Fernández C, Caballero P. Dynamics of sperm DNA damage in fresh versus frozen-thawed and gradient processed ejaculates in human donors. Andrologia. 2011;43:373–7. doi: 10.1111/j.1439-0272.2010.01022.x. [DOI] [PubMed] [Google Scholar]
- 44.Schuffener A, Morshedi M, Oehninger S. Cryopreservation of fractionated, highly motile human spermatozoa: effect on membrane phosphatidylserine externalization and lipid peroxidation. Hum Reprod. 2001;16:2148–53. doi: 10.1093/humrep/16.10.2148. [DOI] [PubMed] [Google Scholar]
- 45.Ngamwuttwong T, Kunathikom S. Evaluation of cryoinjury of sperm chromatin according to liquid nitrogen vapour method (I) J Med Assoc Thail. 2007;90:224. [PubMed] [Google Scholar]
- 46.Yildiz C, Ottaviani P, Law N, Ayearst R, Liu L, et al. Effects of cryopreservation on sperm quality, nuclear DNA integrity, in vitro fertilization, and in vitro embryo development in the mouse. Reproduction. 2007;133:585–95. doi: 10.1530/REP-06-0256. [DOI] [PubMed] [Google Scholar]
- 47.Vutyavanich T, Piromlertamorn W, Nunta S. Rapid freezing versus slow programmable freezing of human spermatozoa. Fertil Steril. 2010;93:1921–8. doi: 10.1016/j.fertnstert.2008.04.076. [DOI] [PubMed] [Google Scholar]
- 48.Twigg J, Irvine D, Aitken R. Oxidative damage to DNA in human spermatozoa does not preclude pronucleus formation at intracytoplasmic sperm injection. Hum Reprod. 1998;13:1864–71. doi: 10.1093/humrep/13.7.1864. [DOI] [PubMed] [Google Scholar]
- 49.Paasch U, Sharma RK, Gupta AK, Grunewald S, Mascha EJ, et al. Cryopreservation and thawing is associated with varying extent of activation of apoptotic machinery in subsets of ejaculated human spermatozoa. Biol Reprod. 2004;71:1828–37. doi: 10.1095/biolreprod.103.025627. [DOI] [PubMed] [Google Scholar]
- 50.Seli E, Sakkas D. Spermatozoal nuclear determinants of reproductive outcome: implications for ART. Hum Reprod Update. 2005;11:337–49. doi: 10.1093/humupd/dmi011. [DOI] [PubMed] [Google Scholar]
- 51.Sakkas D, Alvarez JG. Sperm DNA fragmentation: mechanisms of origin, impact on reproductive outcome, and analysis. Fertil Steril. 2010;93:1027–36. doi: 10.1016/j.fertnstert.2009.10.046. [DOI] [PubMed] [Google Scholar]
- 52.Yildiz C, Fleming C, Ottaviani P, McKerlie C. Fresh and frozen-thawed sperm quality, nuclear DNA integrity, in vitro fertility, embryo development, and live-born offspring of N-ethyl-N-nitrosourea (ENU) mice. Cryobiology. 2008;57:156–62. doi: 10.1016/j.cryobiol.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 53.Sztein JM, Farley JS, Mobraaten LE. In vitro fertilization with cryopreserved inbred mouse sperm. Biol Reprod. 2000;63:1774–80. doi: 10.1095/biolreprod63.6.1774. [DOI] [PubMed] [Google Scholar]