

Abstract
Nonhomologous end joining (NHEJ) is the major DNA double-strand break (DSB) repair pathway in mammalian cells. A critical step in this process is DNA ligation, involving the Xrcc4–DNA ligase IV complex. DNA end processing is often a prerequisite for ligation, but the coordination of these events is poorly understood. We show that polynucleotide kinase (PNK), with its ability to process ionizing radiation-induced 5′-OH and 3′-phosphate DNA termini, functions in NHEJ via an FHA-dependent interaction with CK2-phosphorylated Xrcc4. Analysis of the PNK FHA–Xrcc4 interaction revealed that the PNK FHA domain binds phosphopeptides with a unique selectivity among FHA domains. Disruption of the Xrcc4–PNK interaction in vivo is associated with increased radiosensitivity and slower repair kinetics of DSBs, in conjunction with a diminished efficiency of DNA end joining in vitro. Therefore, these results suggest a new role for Xrcc4 in the coordination of DNA end processing with DNA ligation.
Keywords: DNA double-strand breaks, DNA repair, FHA domain, nonhomologous end joining, Xrcc4
Introduction
DNA double-strand breaks (DSBs) pose a major threat to cell survival and genome stability. If left unrepaired, DSBs can result in the loss of genetic material or cell death. Moreover, mutations or gross genetic aberrations can occur if DSB repair (DSBR) is inaccurate (Mills et al, 2003). Great efforts are therefore made by the cell to prevent, detect, signal and repair DSBs.
In vertebrates, a major DSBR pathway is nonhomologous end joining (NHEJ). NHEJ is active throughout the cell cycle, although it predominates during G0/G1 (Lieber et al, 2003). In addition, NHEJ plays a key role in the processing and repair of the developmentally regulated DSBs that occur during V(D)J recombination, the process by which T-cell receptor and antibody diversity is established (Gellert, 2002). NHEJ has several known components: the DNA-PK holoenzyme (composed of the DNA end-binding complex Ku70/Ku80 and the DNA-PKcs serine/threonine kinase; Smith and Jackson, 1999), the Artemis nuclease and the Xrcc4–DNA ligase IV complex (Lieber et al, 2003). Mammalian cells deficient in these NHEJ components share phenotypes that include sensitivity to ionizing radiation (IR) and impaired V(D)J recombination, underscoring their critical role in DSBR (Lieber et al, 2003).
Most models of mammalian NHEJ do not take into account the requirement for DNA end processing prior to ligation. Indeed, most sources of DNA damage either produce nonligatable chemical modifications or generate protein–DNA adducts at DNA termini that must require processing prior to DNA ligation. For example, IR produces nonligatable 5′-OH and 3′-phosphate DNA termini among other types of ends (Coquerelle et al, 1973). Therefore, the recruitment and regulation of specific DNA end processors is critical for NHEJ depending on the type of terminal groups present at DSBs.
The Artemis protein was recently shown to possess overhang endonucleolytic activity and is perhaps the best characterized example of a DNA end processor associated with NHEJ (Lieber et al, 2003). Artemis forms a complex with DNA-PKcs and its interaction with, along with its phosphorylation by, DNA-PKcs is required to convert Artemis from an exonuclease to an endonuclease (Ma et al, 2002). These findings link DSB detection by DNA-PK to DNA end processing by Artemis. However, Artemis cannot process every type of nonligatable ends and therefore other types of end-processing enzymes must act during NHEJ. The mammalian polynucleotide kinase (PNK) enzyme is a good candidate for an NHEJ DNA end processor as it contains both 5′-DNA kinase and 3′-DNA phosphatase activities that could specifically act during the repair of 5′-OH and 3′-phosphate-modified DNA termini (Karimi-Busheri et al, 1999). Interestingly, recent studies from Chappell and colleagues, using an in vitro NHEJ system, identified PNK as a key factor for the processing of DSBs with 5′-OH DNA termini in vitro (Chappell et al, 2002).
In addition to its potential role in NHEJ, PNK has a well-established role in single-strand break repair (SSBR), where it interacts with a multi-protein complex that includes the protein Xrcc1 (Whitehouse et al, 2001). In metazoans, from Drosophila to humans, PNK orthologs also harbor a putative FHA domain N-terminal to their catalytic domains. The FHA domain is found in a number of proteins of diverse functions, where it acts as a phosphothreonine-binding module (Durocher and Jackson, 2002). The presence of an FHA domain on PNK therefore suggests that PNK engages in phosphorylation-dependent interactions and indicates that the assembly of the SSBR or DSBR machineries may be in part controlled by phosphorylation.
In this study, we sought to identify phosphorylation-dependent PNK-interacting proteins, identify the kinase(s) responsible for these interactions, as well as characterize the functional importance of these interactions in DNA repair. Here, we show that PNK physically interacts with Xrcc4 in a phosphorylation- and FHA-dependent manner. Disruption of the Xrcc4–PNK interaction is associated with increased sensitivity to IR, concomitant with a diminished efficiency of DNA end joining in vitro. This study therefore identifies the mechanism by which PNK participates in NHEJ and uncovers a new role for Xrcc4 in the coordination of DNA end processing with DNA ligation.
Results
PNK interacts with Xrcc4 in an FHA-dependent manner
To identify proteins that specifically interact with the PNK FHA domain, we purified the PNK FHA domain as a GST fusion protein (PNKFHA) and separately purified a mutated version of the PNK FHA domain substituting a conserved arginine residue (Arg35) to alanine (PNKFHA-R35A), a mutation shown in other FHA domains to be essential for phosphothreonine binding (Durocher and Jackson, 2002). Both purified fusion proteins were immobilized on glutathione–sepharose beads and incubated with whole-cell extracts (WCEs) from human HEK293T cells. We excised a total of 18 bands that displayed selective interaction with the PNKFHA domain and prepared them for tandem mass spectrometry. As a control to ensure identification of proteins retrieved in a PNKFHA-dependent fashion, corresponding gel slices in the PNKFHA-R35A lane were also excised and analyzed by mass spectrometry. As shown in Table I, the PNKFHA protein, in contrast to PNKFHA-R35A, selectively retrieved SSBR components known to interact with PNK (Xrcc1, DNA ligase III and PARP; Whitehouse et al, 2001), indicating that PNK may interact with Xrcc1 in an FHA domain-dependent manner, a result recently confirmed by the Caldecott group (Loizou et al, 2004; see Table I). We also identified a number of novel PNK interactors such as NHEJ components (Xrcc4, the Ku70/Ku80 heterodimer and DNA-PKcs), as well as other regulators of DNA repair and chromatin remodeling such as components of the FACT complex (Table I). The interaction between PNK and NHEJ components was of particular interest, given the recent report by Chappell et al (2002) that suggests a role for PNK during NHEJ. The physical and functional association of PNK with NHEJ components was therefore further investigated.
Table 1.
Summary of MS data
| Banda | Protein | Peptides identified | Accession numberb | Functionc |
|---|---|---|---|---|
| a | Xrcc4 | 10 | gi∣12408647 | NHEJ |
| HnRNP H1 | 5 | gi∣5764101 | RNA metabolism | |
| b | AdoHcyase | 9 | gi∣6094223 | Metabolism |
| c | DKFZp451l037 | 6 | gi∣34364737 | Unknown |
| TRIM26 | 3 | gi∣4508005 | Unknown | |
| NASP | 3 | gi∣27262628 | Binds histone H1 | |
| d | Ku70 | 30 | gi∣4503841 | NHEJ |
| e | Nohitswith>2 peptides | |||
| f | Ku80 | 33 | gi∣10863945 | NHEJ |
| Xrcc1 | 8 | gi∣40226177 | SSBR | |
| SSRP1 | 3 | gi∣4507241 | Part of FACT complex; DNA repair | |
| g | MCM5 | 19 | gi∣1232079 | DNA replication |
| Xrcc1 | 10 | gi∣5454172 | SSBR | |
| i | MCM3 | 22 | gi∣1552242 | DNA replication |
| DNA ligase III | 25 | gi∣7710126 | SSBR | |
| j | Gemin 4 | 11 | gi∣10503982 | Protein of ‘Gem bodies' |
| PITSLRE | 8 | gi∣3978441 | Protein kinase | |
| DNA ligase III | 8 | gi∣7710126 | SSBR | |
| k | PARP | 10 | gi∣190167 | SSBR |
| DDB1 | 3 | gi∣13435359 | NER (UV-binding protein) | |
| HnRNP U | 3 | gi∣14141161 | Matrix-associated protein | |
| l | NOPP140 | 3 | gi∣12804871 | Nucleolar protein |
| m | FACT140 | 10 | gi∣6005757 | Chromatin remodeling |
| NRD-C | 6 | gi∣29840826 | Protease | |
| NASP | 4 | gi∣27262628 | Binds histone H1 | |
| n | KIAA1035 | 7 | gi∣20521740 | Unknown |
| TCOF1 | 3 | gi∣15079955 | Treacher–Collins syndrome | |
| o | TCOF1 | 7 | gi∣4507411 | Treacher-Collins syndrome |
| VprBP | 3 | gi∣7662316 | Binds HIV-1 Vpr | |
| p | E2-230K | 12 | gi∣12698013 | Ubiquitin conjugation |
| TCOF1 | 3 | gi∣4507411 | Treacher–Collins syndrome | |
| q | TCOF1 | 17 | gi∣1778432 | Treacher–Collins syndrome |
| RBAF600 | 4 | gi∣4557447 | Rb-associated factor | |
| CHD1 | 7 | gi∣24416002 | Chromatin remodeling | |
| r | DNA-PKcs | 11 | gi∣13654237 | NHEJ |
| RBAF600 | 13 | gi∣24416002 | Rb-associated factor | |
| ATRX | 8 | gi∣20336207 | Alpha-thalassemia/mental retardation syndrome/chromatin remodeling | |
| s | RBAF600 | 56 | gi∣24416002 | Rb-associated factor |
| aRefer to Figure 1A. | ||||
| bAccession number retrieved by the Mascot search engine. | ||||
| cNHEJ: nonhomologous end joining; SSBR: single-strand break repair; NER: nucleotide excision repair. | ||||
We first sought to determine whether the NHEJ components identified by mass spectrometry interacted with full-length PNK in an FHA-dependent manner. Full-length, epitope-tagged, PNK or PNKR35A were expressed in human HEK293T cells and tested for their ability to physically interact with NHEJ proteins in co-immunoprecipitation experiments. As shown in Figure 1B, PNK co-immunoprecipitates with Xrcc4 and DNA ligase IV, and these interactions are dependent on the phosphothreonine-binding activity of the FHA domain, as PNKR35A fails to interact with either protein. However, the potential interaction between PNK and the DNA-PK holoenzyme was not confirmed by co-immunoprecipitation and therefore was not investigated further. The PNK–DNA ligase IV interaction is also detected when the direction of the co-immunoprecipitation is reversed (Figure 1C) and, importantly, the interaction between PNK and the Xrcc4-DNA ligase IV complex is not bridged by DNA since the interaction is resistant to 50 μg/ml of the DNA intercalating agent ethidium bromide (Figure 1D). Finally, co-immunoprecipitation experiments using anti-PNK antibodies confirmed that the interaction between endogenous Xrcc4 and PNK occurs in vivo (Figure 1E). These results indicate that PNK interacts with the Xrcc4–DNA ligase IV complex in human cells and that their association is dependent on a functional PNK FHA domain.
Figure 1.
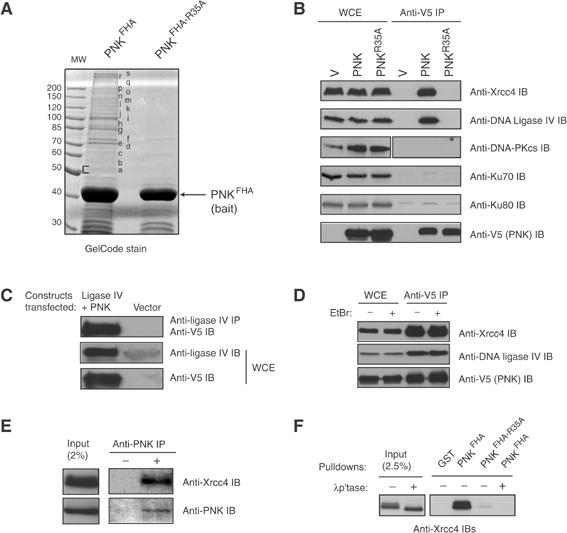
PNK physically interacts with Xrcc4 in an FHA-dependent manner. (A) Recombinant GST-PNKFHA and -PNKFHA-R35A proteins were incubated with HEK293T WCEs, the interacting proteins were resolved on SDS–PAGE, stained with GelCode, and prepared for mass spectrometry. The identities of the proteins found in gel slices are found in Table I. (B) WCE from HEK293T cells transfected with V5-tagged PNK, PNKR35A or empty vector (V) were immunoprecipitated with an anti-V5 antibody. The precipitates or WCEs were separated on SDS–PAGE, and probed to detect PNK, Xrcc4, DNA ligase IV, DNA-PKcs and Ku70/Ku80 as indicated. (C) WCEs from HEK293T cells transfected with DNA ligase IV, V5-tagged PNK or empty vector were immunoprecipitated with anti-DNA ligase IV antibody, followed by anti-V5 immunoblotting (top panel). WCEs were also probed with anti-DNA ligase IV antibody (middle panel) and anti-V5 antibody (bottom panel). We note that, for the Xrcc4–PNK interaction, the immunoprecipitation cannot be reversed for technical reasons. (D) WCEs from (A) treated with or without ethidium bromide were immunoprecipitated with anti-V5 antibody and probed with anti-Xrcc4 and DNA ligase IV antibodies. (E) HEK293T WCEs were immunoprecipitated with anti-PNK antibody, followed by immunoblotting with anti-Xrcc4 antibody or anti-PNK antibody. (F) Recombinant GST, PNKFHA, or PNKFHA-R35A were mixed in PD assays with HEK293T WCEs treated with or without λ protein phosphatase (λp'tase) as indicated. As a control, 2.5% of input WCE was loaded. The samples were resolved on SDS–PAGE and probed with anti-Xrcc4 antibody.
Since FHA domains are phosphothreonine recognition modules, we examined if protein phosphorylation was required for the PNK–Xrcc4 interaction. We used the PNKFHA pull-down (PD) assay used in Figure 1A, as a means to rapidly monitor the PNKFHA–Xrcc4 interaction. As shown in Figure 1F, PNKFHA efficiently retrieves Xrcc4 from WCEs and the interaction is essentially abolished by the Arg35Ala mutation. Furthermore, λ protein phosphatase treatment of cell extracts prior to incubation with PNKFHA completely abolishes the interaction between Xrcc4 and PNKFHA. These results support the notion that the interaction is phosphorylation-dependent and indicate that the PNK FHA domain is both sufficient and essential to promote an interaction with Xrcc4.
Threonine 233 (Thr233) of Xrcc4 is required for binding to PNK
Next, we sought to determine the region of Xrcc4 required for its association with PNK. A series of epitope-tagged Xrcc4 constructs were generated (Figure 2A), expressed in HEK293T cells and tested for their ability to interact with PNKFHA in PD assays. As shown in Figure 2B, PNKFHA efficiently retrieves Xrcc4 and Xrcc4250Δ from WCEs, but is unable to interact with Xrcc4213Δ or Xrcc4179Δ. From these observations, we conclude that the PNK–Xrcc4 interaction requires a region of Xrcc4 encompassing amino-acid residues 213–250.
Figure 2.
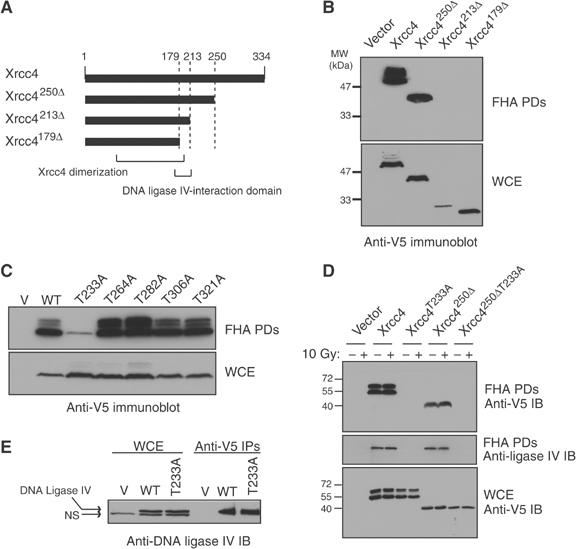
The Xrcc4 Thr233 residue is required for interaction with PNK. (A) Schematic representation of Xrcc4 proteins analyzed for interaction with PNK: wild-type Xrcc4; Xrcc4250Δ (residues 1–250); Xrcc4 213Δ (residues 1–213); Xrcc4 179Δ (residues 1–179). Also indicated is the DNA-ligase IV Xrcc4-interacting region (residues 179–213), and the portion of Xrcc4 important for mediating homodimerization (residues 115–204). (B) WCEs from HEK293T cells transfected with V5-tagged Xrcc4 constructs (or empty vector as control) were incubated with recombinant GST-PNKFHA in PD assays. PNKFHA-interacting proteins were resolved by SDS–PAGE, and detected with an anti-V5 antibody (top panel). As a control, 20 μg of WCE was analyzed by immunoblotting (bottom panel). (C) Conserved threonine residues of Xrcc4 were mutated to alanine, and the resulting mutants were expressed in HEK293T cells. PD assays were carried out with PNKFHA, and Xrcc4 was detected by anti-V5 immunoblotting (top panel). In the bottom panel, 20 μg of WCE was immunoblotted with anti-V5 antibody to control for protein expression. (D) CHO Xrcc4-deficient (XR-1) cell lines stably transfected with Xrcc4WT, Xrcc4T233A, Xrcc4250Δ, Xrcc4250ΔT233A, or an empty vector were treated with or without 10 Gy of X-rays and assessed for their ability to interact with PNKFHA in PD assays and detected by immunoblotting (IB) with anti-V5 antibody (top panel). The blot was stripped and immunoblotted with anti-DNA ligase IV antibody (middle panel). Examination of WCEs (bottom panel) by anti-V5 immunoblotting revealed similar expression levels between the cell lines. (E) WCEs from XR-1 lines transfected with Xrcc4WT, Xrcc4T233A or a control vector were immunoprecipitated (IPs) with the anti-V5 antibody and probed for the presence of DNA ligase IV by anti-DNA ligase IV immunoblotting. DNA ligase IV was expressed at similar levels in both cell lines, as revealed by anti-DNA ligase IV immunoblotting from WCEs. DNA-ligase IV is indicated by the top arrow, and the bottom arrow represents a faster migrating nonspecific (NS) band.
Since FHA domains are phosphothreonine-binding modules, we next assessed whether conserved threonine residues on Xrcc4 are required for the Xrcc4–PNK interaction. Five highly conserved threonine residues in Xrcc4 were mutated to alanine (Thr233, 264, 282, 306 and 321; see Supplementary Figure 1A). Of these, Thr321 is known to be phosphorylated by DNA-PK (Yu et al, 2003), making it a particularly interesting candidate. The resulting Xrcc4 mutants were expressed in HEK293T cells and assessed for binding to PNKFHA in PD assays (Figure 2C). We found that only the Thr233Ala mutation resulted in the impairment of the Xrcc4–PNKFHA interaction. Notably, the location of this residue in Xrcc4 is in agreement with the regional mapping data presented in Figure 2B, which located the PNK-binding site between amino-acid residues 213 and 250, and suggests that the PNK FHA domain recognizes an epitope encompassing Thr233 on Xrcc4.
Since Xrcc4 is oligomeric in nature (dimeric or tetrameric; Junop et al, 2000; Lee et al, 2000), we sought to ensure that multimerization with endogenous Xrcc4 present in HEK293T extracts did not enable some Xrcc4 mutants to interact with the PNK FHA domain. We therefore generated stable cell lines expressing V5-epitope-tagged Xrcc4, Xrcc4250Δ, Xrcc4T233A and Xrcc4250ΔT233A in the Xrcc4-deficient CHO cell line XR-1, and tested their ability to be bound by PNKFHA in PD assays (Figure 2D). Again, we confirmed that the Xrcc4 Thr233 residue is critical for association with the PNK FHA domain, both in the context of full-length Xrcc4 and Xrcc4250Δ proteins (Figure 2D). Interestingly, the association between Xrcc4 and PNK is not modulated by DNA damage as the interaction is observed at similar levels after 10 Gy of X-rays (Figure 2D). In addition, the binding of DNA ligase IV to PNKFHA also depends on the Xrcc4 Thr233 residue (Figure 2D). This observation raises the possibility that DNA ligase IV interacts with PNK via a bridging interaction with Xrcc4. To rule out the possibility that the loss of the DNA ligase IV–PNK interaction in cells expressing Xrcc4T233A was due to a disruption of the Xrcc4–DNA ligase IV complex, Xrcc4 or Xrcc4T233A proteins were immunoprecipitated from XR-1 stable transfectants and examined for association with DNA ligase IV (Figure 2E). We found that DNA ligase IV interacts equally well with wild-type Xrcc4 or with Xrcc4T233A. As DNA ligase IV stability is dependent on its association with Xrcc4 (Bryans et al, 1999), we also note that Xrcc4T233A stabilizes endogenous DNA ligase IV to the same extent as wild-type Xrcc4. Collectively, these results are consistent with the formation of a tripartite complex comprising PNK, Xrcc4 and DNA ligase IV and that the PNK–Xrcc4 interaction requires Thr233 on Xrcc4.
PNK FHA domain displays a novel phosphothreonine-binding mode
The results presented above suggest that Thr233 may be phosphorylated and bound by the PNK FHA domain. We thus tested if peptides derived from Xrcc4 and containing the sequence surrounding Thr233 could interact with PNKFHA or PNKFHA-R35A proteins in peptide PD assays. Biotinylated, Thr233-phosphorylated and unphosphorylated Xrcc4-derived peptides (designated as T233P and T233 peptides, respectively; Figure 3A) were employed in peptide-binding assays to assess whether the interaction is phosphorylation-dependent. Both peptides were coupled to streptavidin magnetic beads and incubated with PNK and PNKR35A. As shown in Figure 3B, only the phosphorylated peptide (T233P) is able to bind appreciably to PNK. Conversely, when PNK and PNKR35A were compared for their ability to bind to T233P, only PNK bound efficiently (Figure 3B). These results indicate that the FHA domain of PNK is required to directly interact with an epitope on Xrcc4 surrounding the phosphorylated threonine at position 233.
Figure 3.
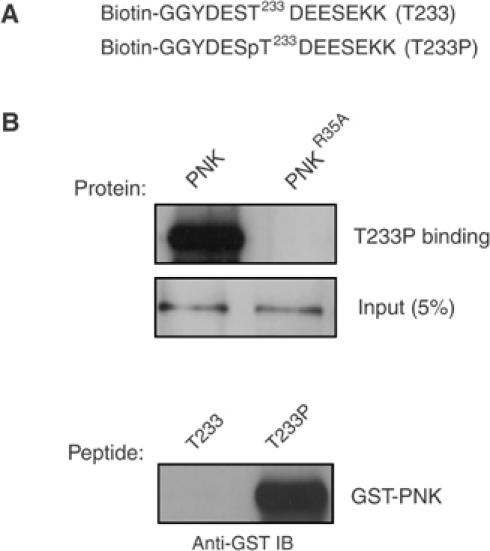
The PNK FHA domain binds to phosphorylated Thr233. (A) Sequence (in single-letter amino acids) of the biotinylated unphosphorylated (T233) and phosphorylated (T233P) peptides from Xrcc4 corresponding to amino acids 229–238. Two C-terminal lysines and two N-terminal glycine residues were also added. (B) The immobilized T233P peptide was mixed with recombinant GST fusion proteins (PNK or PNKR35A proteins), and peptide binding was detected by anti-GST immunoblotting (top panel). In the bottom panel, PNK was incubated with the T233 or T233P coupled peptides, and association was assessed by anti-GST immunoblotting. Input amounts of the recombinant proteins (for both the upper and lower panels) are indicated in the middle panel, and have also been detected by probing with anti-GST antibody.
The PNK FHA domain is part of a newly identified class of FHA domains comprising the FHA domain of aprataxin (Caldecott, 2003). Interestingly, iterative BLAST searches with the FHA domain of PNK identify a third member of this subfamily of FHA domains (MGC47799, Supplementary Figure 2). In order to test whether this FHA domain subfamily may display a unique mode of phosphopeptide recognition, we characterized the binding properties of the PNK FHA domain in more detail. First, we determined the dissociation constant for the interaction between purified PNKFHA and the T233P peptide using isothermal titration calorimetry (ITC; Figure 4A) and fluorescence polarization (FP), using a fluorescein-labeled version of the T233P peptide (Figure 4B). We obtained a KD of 4.1 μM using ITC and 4.2 μM by FP. A similar dissociation constant is also obtained when full-length recombinant PNK is used in ITC experiments (KD of 2.5 μM; Mark Glover, personal communication). The low micromolar affinity of the T233P peptide–PNKFHA interaction is within the range of dissociation constants calculated for other FHA-peptide-binding interactions (Durocher and Jackson, 2002).
Figure 4.
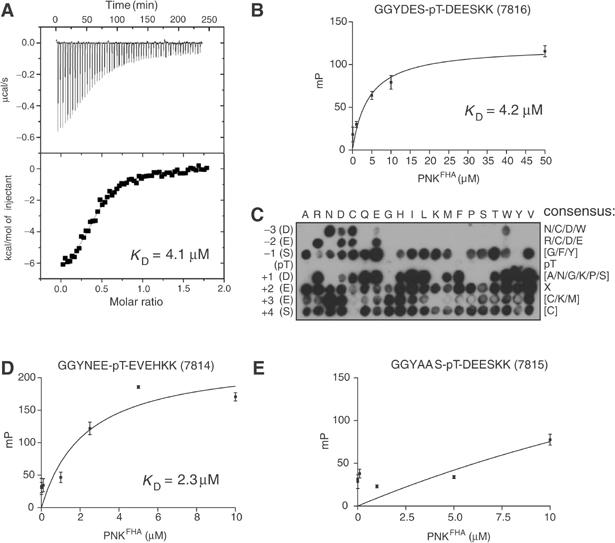
The PNK FHA domain demonstrates unique phosphopeptide-binding selectivity. (A) Peptide-binding affinity determined by ITC. A representative ITC trace is shown outlining the binding of the T233P phosphopeptide to recombinant PNKFHA. (B) PNKFHA binding to fluorescein-labeled 7816 peptide was quantitated by FP. The calculated dissociation constant is indicated. (C) PNKFHA domain binding to a filter array of peptides. The PNKFHA domain was detected by immunoblotting with anti-GST antibody. The resulting consensus binding motif is indicated on the right column. ‘X' indicates no dominant selection, and letters enclosed in square brackets are specifically counter-selected. The left row indicates the amino acid that was substituted in the Xrcc4 peptide sequence and the top row indicates to which amino acid the residue was substituted for. (D, E) PNKFHA binding to 7814 (D) or 7815 peptides (E) was quantitated by FP. A dissociation constant could not be calculated for the 7815 peptide.
To further characterize the binding specificity of the PNK FHA domain, we synthesized a peptide array based on the sequence encompassing Thr233 of Xrcc4 on a cellulose membrane, using a SPOT peptide synthesizer. We tested the ability of PNKFHA to interact with the peptide array using Southwestern blotting. As shown in Figure 4C, substitution of residues located C-terminal to the phosphothreonine does not, in large part, affect binding to PNKFHA. However, substitution at the -2 or -3 positions relative to pThr can only be tolerated by a few amino acids (Arg/Asp/Cys/Glu at -2, and Asn/Asp/Cys/Trp at -3), indicating that the binding selectivity of the PNK FHA domain resides N-terminal to the phosphothreonine residue. This result was unexpected given that every FHA domain characterized to date possesses major binding selectivity determinants C-terminal to the phosphothreonine (Durocher and Jackson, 2002). To confirm this unique binding selectivity, we generated two additional fluorescein-labeled peptides (Figure 4D and E), designed to either disrupt or maintain PNKFHA binding, and examined the interactions using FP. As predicted from the peptide array data, the peptide containing alanine substitutions at the -2 and -3 positions abolished PNKFHA binding (Figure 4E). In contrast, substitution of five out of nine amino acids in the native Xrcc4 sequence for residues expected to maintain binding did in fact augment PNKFHA-binding affinity relative to the wild-type peptide (KD of 2.3 versus 4.2 μM, respectively; Figure 4B and D). These results are consistent with the notion that positions N-terminal to the phosphothreonine site, in particular the -2 and -3 positions, are important for determining PNKFHA-binding specificity. Therefore, among FHA domains, PNKFHA recognizes phosphopeptides with a unique mode of binding selectivity.
Xrcc4 Thr233 is a CK2 phosphorylation site
The amino-acid sequence surrounding Thr233 (Asp–Glu–Ser–Thr233–Asp–Glu–Glu) is highly acidic and suggests that Thr233 may be a target for acidophilic kinases. Indeed, analysis of the Xrcc4 sequence in Scansite (Yaffe et al, 2001) reveals that the sequence encompassing Thr233 conforms to a consensus CK2 phosphorylation site (along with Ser232). We therefore tested if the Xrcc4-derived peptide containing Thr233 (T233 peptide, and see above) could act as a substrate for CK2. As shown in Figure 5A, the T233 peptide is efficiently phosphorylated by recombinant CK2. As a control, we also assessed if the phosphorylated T233P peptide could act as a substrate for CK2. Figure 5A shows that the T233P peptide is a much poorer substrate for recombinant CK2 relative to the T233 peptide. The residual phosphorylation of the phosphopeptide may be explained by some phosphorylation at the serine equivalent to Ser232 on Xrcc4, which also conforms to a CK2 consensus phosphorylation site.
Figure 5.
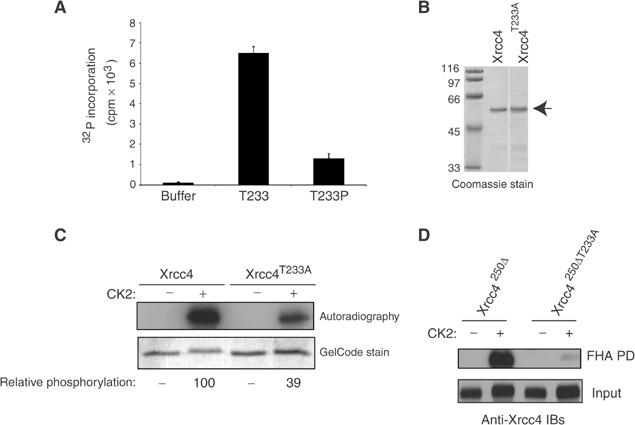
The Thr233 residue of Xrcc4 is phosphorylated by CK2. (A) The T233 and T233P peptides were used in CK2 protein kinase assays. The histogram values represent the corrected means of at least two independent experiments done in duplicate or quadruplicate and the error bars represent standard deviations. (B) Coomassie Blue staining of His-tagged Xrcc4WT or Xrcc4T233A proteins used in the kinase assay in (C). (C) An amount of 1 μg of each recombinant protein shown in (B) was used in CK2 kinase assays. A typical experiment is shown. An autoradiograph is shown in the upper panel and normalized, relative phosphorylation values were determined using NIH image software. Input protein was assessed by GelCode staining. (D) Recombinant Xrcc4250Δ and Xrcc4250ΔT233A proteins phosphorylated (+) or not (−) by CK2 were incubated with PNKFHA in PD assays (top panel). Input from the kinase reactions was also assessed by anti-Xrcc4 immunoblotting (bottom panel).
Next, we assayed the ability of recombinant CK2 to phosphorylate recombinant Xrcc4. To do so, protein kinase assays were carried out with CK2 on recombinant Xrcc4 and Xrcc4T233A proteins (Figure 5B). Wild-type Xrcc4 is efficiently phosphorylated by CK2, as illustrated by the incorporation of radiolabeled phosphate into Xrcc4, with a concomitant reduction in mobility on SDS–PAGE (Figure 5C, bottom panel). By contrast, when Xrcc4T233A is used as a CK2 substrate, incorporation of radio-labeled phosphate is reduced by approximately three-fold, along with a less pronounced mobility shift compared to Xrcc4. Collectively, these results indicate that Thr233 can be a substrate for CK2 phosphorylation.
To determine if CK2-phosphorylated recombinant Xrcc4 can be efficiently bound by the PNK FHA domain, recombinant Xrcc4 proteins were phosphorylated by CK2 in vitro, incubated with PNKFHA, and binding was assessed in PD assays. Figure 5D reveals that only CK2-phosphorylated Xrcc4250Δ binds efficiently to the PNK FHA domain. This finding contrasts to the severely impaired association of CK2-phosphorylated Xrcc4250ΔT233A with PNKFHA. These results strongly suggest that Xrcc4 can directly bind to PNK and that Thr233 phosphorylation is important for their association. Moreover, it indicates that CK2 can act as the kinase responsible for directing the Xrcc4–PNK interaction.
In vivo phosphorylation of Xrcc4 Thr233 is sensitive to CK2 inhibition
To determine whether Thr233 is phosphorylated in vivo, we immunoprecipitated Xrcc4250Δ from stably transfected XR-1 cells and employed an anti-phosphothreonine (anti-pThr) antibody to probe the phosphorylation status of Thr233. Figure 6A shows that the Xrcc4250Δ protein is recognized by the anti-pThr antibody, whereas the Xrcc4250ΔT233A is not, strongly suggesting that the Thr233 residue is phosphorylated in vivo. In parallel, in order to assess whether CK2 is responsible for the observed anti-pThr reactivity, we treated cells with the highly selective CK2 inhibitor, 4,5,6,7-tetrabromo-2-azabenzamidazole (TBB; Sarno et al, 2002) and probed the phosphorylation status of Thr233 with the anti-pThr antibody. As shown in Figure 6A, TBB treatment of cells prior to cell extraction essentially abolishes the anti-pThr reactivity of Xrcc4250Δ. Since the observed anti-pThr reactivity is dependent on the Thr233 residue, we conclude that Thr233 phosphorylation is dependent on CK2 activity.
Figure 6.
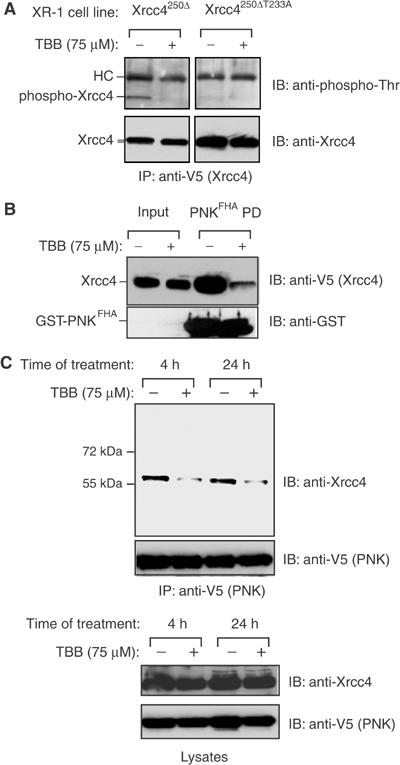
Phosphorylation of Xrcc4 Thr233 is sensitive to CK2 inhibition in vivo. (A) XR-1 cells stably expressing Xrcc4250Δ and Xrcc4250ΔT233A were incubated in the presence of 75 μM TBB or DMSO for 4 h. WCEs were then examined by anti-phosphothreonine (upper panels) or anti-Xrcc4 immunoblotting (lower panels). (B) WCEs from the Xrcc4250Δ XR-1 cell line treated as in (A) were mixed with PNKFHA in a PD assay, followed by immunoblotting with anti-V5 antibody (to detect V5-tagged Xrcc4250Δ) and anti-GST antibody. (C) HEK293T cells transiently expressing V5-tagged PNK were treated with 75 μM TBB for 4 or 24 h, or DMSO as a control, harvested and the WCEs were immunoprecipitated with anti-V5 antibody. The WCEs and immunoprecipitates were then examined by anti-Xrcc4, anti-V5 (PNK) immunoblotting as indicated.
Next, we tested whether CK2 inhibition by TBB alters the ability of PNK to interact with Xrcc4. As shown in Figure 6B, the ability of the PNKFHA domain to interact with Xrcc4250Δ is severely decreased when cells are treated with TBB prior to lysis. Furthermore, TBB treatment of HEK293T cells results in a decrease of greater than 50% in the amount of endogenous Xrcc4 immunoprecipitated by PNK, while the overall protein levels remained unchanged (Figure 6C). Although our data do not exclude the possibility that other acidophilic kinases might also phosphorylate Xrcc4 at the Thr233 site, these results are in agreement with the notion that CK2 phosphorylates Xrcc4 at Thr233 in vivo and thereby controls the Xrcc4–PNK interaction.
PNK stimulates end joining of 5′-hydroxyl ends by Xrcc4-DNA ligase IV
The PNK–Xrcc4 interaction suggests that Xrcc4 may recruit PNK to DSBs in order to facilitate processing of DNA termini with 5′-OH or 3′-phosphate ends. Recombinant Xrcc4 purified from insect cells is phosphorylated and can be readily bound by PNKFHA (Figure 7A), indicating that it can be used as a source of Xrcc4 to study the impact of the PNK–Xrcc4 interaction in vitro. We therefore tested whether PNK can stimulate the ligation of dephosphorylated, linearized plasmid DNA catalyzed by the Xrcc4–DNA ligase IV complex. As expected, the Xrcc4-DNA ligase IV complex alone is unable to ligate the dephosphorylated plasmid substrate (Figure 7B). However, inclusion of PNK in the end-joining reaction efficiently stimulates Xrcc4-DNA ligase IV-mediated end joining of 5′-OH DNA termini, as measured by the generation of linear DNA multimers. To test whether this requirement for PNK is dependent on its FHA domain, we sought to compare PNKWT versus PNKR35A in the end-joining assay. As shown in Figure 7C, PNKR35A, despite having robust 5′-DNA kinase activity (data not shown), only poorly stimulates end joining by Xrcc4–DNA ligase IV. Finally, we examined whether inhibiting the PNK–Xrcc4 interaction with the Xrcc4-derived peptides T233 and T233P would affect the outcome of the end-joining assay. Figure 7D shows that increasing amounts of the T233P peptide, in contrast to the T233 peptide, impairs in vitro end joining, without affecting PNK 5′-kinase activity (Figure 7E). Collectively, these results indicate that the FHA- and phospho-dependent interaction between PNK and the Xrcc4–DNA ligase IV complex is important for efficient in vitro end joining of 5′-OH DNA termini.
Figure 7.
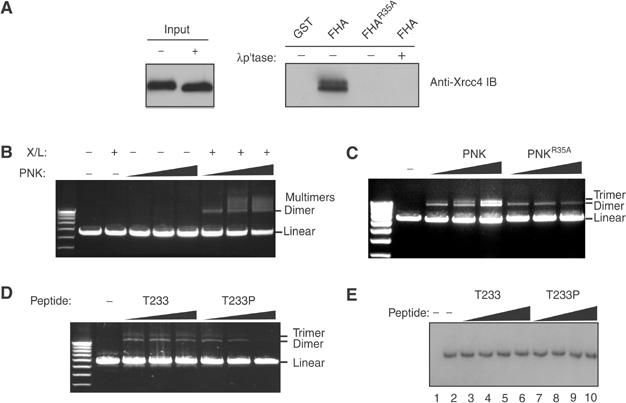
PNK stimulates the efficiency of end joining by Xrcc4-DNA ligase IV via an FHA- and phospho-dependent interaction with Xrcc4. (A) Recombinant Xrcc4–DNA ligase IV complex purified from insect cells was assessed for its ability to interact with PNKFHA in PD assays. The interaction is dependent on the phosphorylation of the complex and the FHA domain of PNK. (B) The effect of increasing amounts of PNK was examined in end joining assays using linearized, phosphatased DNA as a substrate and in the presence (+) and absence (−) of the Xrcc4-DNA ligase IV complex. (C) Increasing amounts of PNKFHA or PNKFHA-R35A proteins were added to a constant amount of Xrcc4–DNA ligase IV complex in end joining assays, and analyzed as described in (B). (D) End joining reactions were performed in the absence (−) or presence of increasing amounts (0.1, 1 and 10 μM final concentration) of the T233 and T233P peptides. Reaction products were visualized as above. (E) Increasing amounts (0.1, 1, 10, 100 μM final concentration) of the T233 (lanes 3–6) and T233P peptides (lanes 7–10) were added in PNK 5′-DNA kinase assays and visualized by autoradiography. In lane 1 no PNK has been added, and in lane 2 the kinase reaction was performed in the absence of peptide.
Xrcc4–PNK interaction is required for optimal DSBR in vivo
We next examined whether the PNK–Xrcc4 interaction is important for DNA end joining in vivo. To investigate this possibility, we assessed whether mutation of Thr233 in Xrcc4 leads to radiosensitivity in clonogenic survival assays. XR-1 cell lines were stably transfected with plasmids encoding for Xrcc4250Δ (the C-terminal residues 250–334 of Xrcc4 are not required to complement the IR sensitivity of XR-1 cells; Grawunder et al, 1998), Xrcc4250ΔT233A or with an empty vector control. The resulting cell lines were exposed to increasing doses of X-irradiation, plated at different dilutions and the number of viable colonies was counted 7 days later. As shown in Figure 8A, a dose of 2 Gy results in a reduction in clonogenic survival of approximately 30% in cells expressing Xrcc4250ΔT233A compared to Xrcc4250Δ. The observed decrease in clonogenic survival of the cells expressing Xrcc4250ΔT233A cannot be explained by differences in protein expression as the Xrcc4250ΔT233A and Xrcc4250Δ proteins are expressed at similar levels in both cell lines and both proteins are predominantly nuclear (Figure 8B and C). This result therefore strongly suggests that Thr233 is important for mediating full resistance to X-irradiation.
Figure 8.
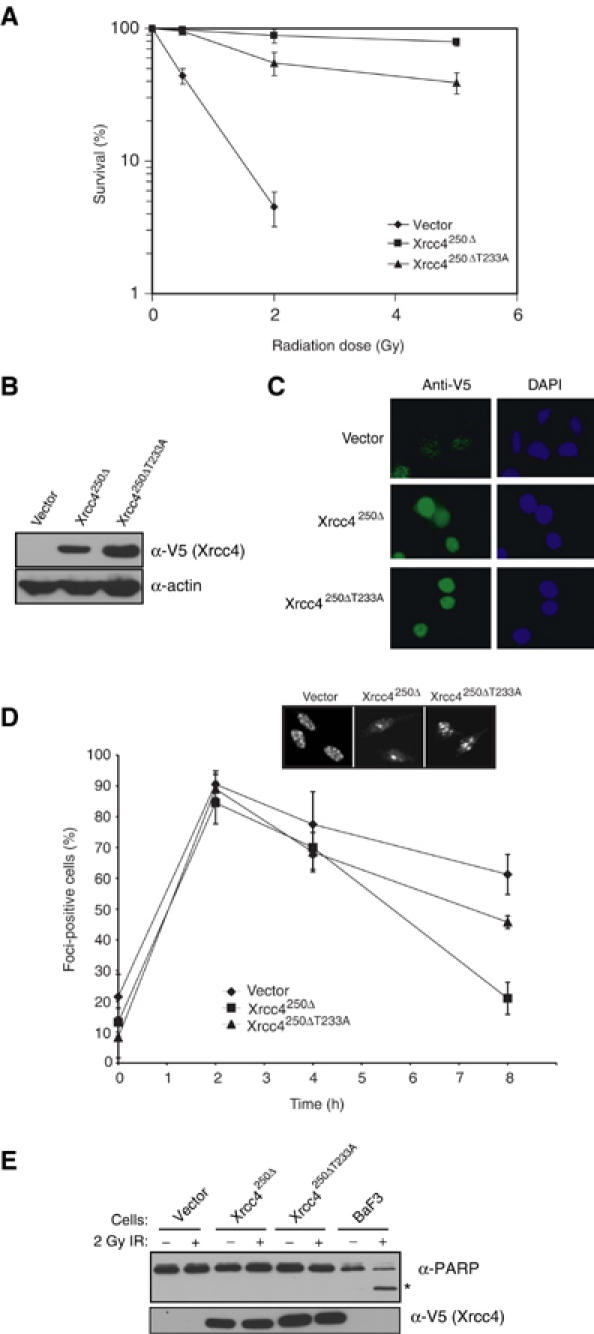
Thr233 of Xrcc4 is required for DSB repair in vivo. (A) XR-1 cell lines stably expressing Xrcc4250Δ, Xrcc4250ΔT233A and empty vector were treated with 2 Gy of X-irradiation and viability was assessed by clonogenic assays. The values represent at least three experiments carried out in triplicate and the data are presented as the percentage of surviving colonies compared to unirradiated controls. Error bars represent standard deviations. (B) XR-1 cells (used in A) stably expressing empty vector, Xrcc4250Δ and Xrcc4250ΔT233A were examined with anti-V5 (Xrcc4) immunoblotting (upper panel). An anti-actin immunoblot was used as a protein-loading control (bottom panel). (C) The cell lines in (A) were examined using indirect immunofluorescence, staining with anti-V5 antibody and DAPI. (D) The cell lines in (A) were exposed to 2 Gy of X-rays, and examined for γ-H2AX foci formation at different time points using fluorescence microscopy. Each data point represents the mean of four independent readings from two independent experiments. Error bars represent standard deviations. (E) Apoptosis in the cell lines from (A) and the lymphoid BaF3 line was assessed 8 h following 0 or 2 Gy of X-rays. Formation of the 89 kDa PARP apoptotic cleavage fragment (*) was detected by anti-PARP immunoblotting (top panel). The WCEs were also examined by anti-V5 (Xrcc4) immunoblotting (bottom panel).
To test whether the increased radiosensitivity of Xrcc4250ΔT233A-expressing cells is correlated with a defect in DSBR, we monitored the kinetics of irradiation-induced DSBs by γ-H2AX immunofluorescence. XR-1 cell lines stably transfected with Xrcc4250Δ, Xrcc4250ΔT233A or vector control were irradiated with 2 Gy of X-rays, and the presence of γ-H2AX foci was assessed by immunofluorescence at different time points (Figure 8D). As shown in Figure 8D, the initial appearance of γ-H2AX foci-positive cells was similar between the tested cell lines up to 4 h post-irradiation. However, at 8 h post-irradiation, we noted a difference in the percentage of foci-positive cells (Figure 8D). Specifically, when compared to cells expressing Xrcc4250Δ, Xrcc4250ΔT233A-expressing cells demonstrated a persistence of γ-H2AX foci. Notably, this difference was not attributable to an increase in apoptosis, as judged by PARP cleavage (Figure 8E). These results suggest that the presence of the Xrcc4 Thr233 residue impacts on the kinetics of DSBR and are consistent with the Xrcc4–PNK interaction being important for DSBR in vivo.
Discussion
A PNK–Xrcc4–DNA ligase IV complex
As a means to understanding the role of phosphorylation-dependent interactions imparted by the PNK FHA domain in DNA repair, we employed a proteomic strategy to identify proteins and pathways that physically interact with PNK. In this study, we report the characterization of a physical and functional interaction between PNK and the NHEJ factor Xrcc4, in human cells. This interaction occurs via the FHA domain of PNK and an epitope on Xrcc4 comprising the phosphorylated Thr233 residue. In addition, DNA ligase IV is found in PNK immunoprecipitates, indicating that a tripartite complex consisting of Xrcc4, PNK and DNA ligase IV exists in human cells. Our results indicate that Xrcc4 bridges the PNK–DNA ligase IV interaction, but we cannot exclude the possibility that Xrcc4 and PNK can associate independently of DNA ligase IV under certain conditions. Indeed, it has been recently shown that the Xrcc4–DNA ligase IV complex is in equilibrium with a tetrameric form of Xrcc4 that excludes DNA ligase IV (Modesti et al, 2003). Whether PNK can bind to Xrcc4 tetramers remains to be tested.
The elegant studies of Chappell et al (2002) first identified a role for PNK in NHEJ in vitro. Our work supports their findings and describes the mechanism by which PNK acts during NHEJ. The phospho-dependent Xrcc4–PNK interaction is functionally important for NHEJ since its disruption reduces the efficiency of DNA ligase IV-dependent end joining of plasmids with 5′-OH termini (Figure 7). The possible additional role of PNK in the stimulation of in vitro end joining of 5′-phosphorylated or 3′-OH termini, not expected to require PNK activity, is intriguing (Supplementary Figure 3). Given that PNK possesses DNA-binding activity, it is possible that PNK may assist in recruiting Xrcc4-DNA ligase IV to DNA ends. However, since deletion of the PNK-interacting site (Thr233) does not affect V(D)J recombination (Modesti et al, 1999), this activity may only play a minor role in the function of the Xrcc4-DNA ligase IV complex on 5′-phosphorylated ends.
Interestingly, whereas the first 200 amino-acid residues of Xrcc4 have been described to be sufficient for its interaction with DNA ligase IV and complementation of the V(D)J recombination defects in CHO-derived Xrcc4-deficient cells (XR-1 cells; Modesti et al, 1999), this region of Xrcc4 exhibits an approximate 20% reduction in radioresistance compared to wild-type Xrcc4 in XR-1 cells with 2 Gy of IR (Leber et al, 1998). By contrast, deletion of Xrcc4 residues 250–334 does not impact IR sensitivity in this system (Grawunder et al, 1998). These observations suggest that the Xrcc4–DNA ligase IV interaction does not account for all the activities of Xrcc4 and that a putative additional activity may lie between amino acids residues 200–250. Our results (Figure 8) are consistent with these observations and suggest that the PNK–Xrcc4 interaction is required for NHEJ in vivo.
A role for basal Xrcc4 phosphorylation in NHEJ
In human cells, Xrcc4 exists as a phosphoprotein whether or not cells have experienced DNA damage. The role of this ‘basal' post-translational modification has remained, until now, unclear. Based on the observation that Xrcc4 can act as an efficient substrate for DNA-PK (Critchlow et al, 1997; Leber et al, 1998), most studies have focused on the impact of DNA-PK-mediated phosphorylation on Xrcc4 function (Modesti et al, 1999; Yu et al, 2003). However, DNA-PK-dependent phosphorylation of Xrcc4 does not appear to play a role in mediating resistance to IR or V(D)J recombination (Yu et al, 2003), and the modest DNA-PK-dependent phosphorylation of Xrcc4 observed in vivo is only seen when cells are treated with doses approaching 100 Gy (Matsumoto et al, 2000). We have also observed that pretreatment of HEK293T cells with wortmannin does not affect the Xrcc4–PNK interaction, and the interaction is still detected in DNA-PK-deficient cells (Supplementary Figure 4). Although these observations indicate that Xrcc4 must be phosphorylated by kinases other than DNA-PK to mediate the FHA-dependent interaction with PNK, they do not exclude a role for DNA-PK in regulating some aspects of Xrcc4 function, such as its DNA-binding activity (Modesti et al, 1999). Our data are consistent with a role for CK2 in phosphorylating Xrcc4 based on the following observations: (i) the Thr233 residue of Xrcc4 is part of a consensus CK2 site, (ii) Thr233 can be efficiently phosphorylated by CK2 in vitro, (iii) CK2-phosphorylated Xrcc4 can be bound directly by PNK, (iv) Thr233 is phosphorylated in vivo as measured by anti-phosphothreonine immunoreactivity, (v) the anti-phosphothreonine immunoreactivity imparted by Thr233 is sensitive to the highly specific CK2 inhibitor TBB, and (vi) treatment of cells with TBB strikingly decreases the PNK–Xrcc4 interaction as measured by co-immunoprecipitations and PD assays. Therefore, given the functional requirement of the Xrcc4–PNK interaction for the repair of a subset of DSBs, our results suggest that CK2 phosphorylation of Xrcc4 plays a role in NHEJ. Interestingly, CK2 has been described as a constitutively active enzyme (Pinna, 2002) that may explain, at least in part, how basal phosphorylation of Xrcc4 is accomplished. Constitutive phosphorylation of Xrcc4 by CK2 is consistent with our observation that the PNK–Xrcc4–DNA ligase IV complex exists under undamaged conditions. Interestingly, a remarkably similar mechanism of interaction between Xrcc1 and PNK has been recently described, suggesting that the SSBR protein Xrcc1 may be constitutively phosphorylated by CK2 as well and that phosphorylated Xrcc1 complexes with PNK under undamaged conditions (Loizou et al, 2004). Based on our observations, we suggest that the PNK–Xrcc4–DNA ligase IV complex may be pre-formed, rather than sequentially assembled, and that other signals or protein activities are required to direct this complex to DSBs.
The PNK FHA domain has a novel mode of phosphopeptide recognition
The FHA domain is an ancient signal transduction module that recognizes linear phosphothreonine epitopes on proteins (Durocher and Jackson, 2002). The PNK FHA domain is a divergent member of this family (Caldecott, 2003; Supplementary Figure 1) and is not readily identified as an FHA domain by common domain recognition algorithms. Thus, a goal of this study was to uncover if the apparent divergence in sequence impacted on the way PNK recognizes phosphopeptides. Remarkably, and in contrast to other FHA domains, sequences N-terminal to the phosphorylated threonine residue are important determinants for PNK FHA binding, demonstrating flexibility in FHA domain sequence recognition. The capacity to recognize residues either N- or C-terminal of the phosphoresidue appears to be a novel feature of the FHA domain not shared by other phosphopeptide-binding domains such as the SH2 or the PTB domains (Pawson et al, 2001). The structural basis for this selectivity may be due to the way the phosphopeptide sits atop the loops connecting the β strands of the FHA domain (Durocher and Jackson, 2002). An immediate implication of this study is that FHA domains may recognize a wider repertoire of phosphothreonine sequences than previously anticipated. It will be informative to determine the three-dimensional structure of the PNK FHA domain complexed to an optimal phosphopeptide in order to understand the basis of this selectivity.
Xrcc4 and Xrcc1 as DNA repair scaffolds linking DNA end processing to DNA ligation
Since its discovery in 1995 (Li et al, 1995), the exact role of Xrcc4 in NHEJ has remained poorly understood. Xrcc4 does not appear to have any catalytic activity, but Xrcc4 was found to bind, stabilize and stimulate DNA ligase IV (Critchlow et al, 1997; Grawunder et al, 1997, 1998; Bryans et al, 1999; Modesti et al, 1999). Furthermore, Xrcc4 is able to bind DNA and to recruit DNA ligase IV to DNA in vitro (Modesti et al, 1999; Chen et al, 2000). These results suggest that Xrcc4 recruits DNA ligase IV to DSBs, a model supported by chromatin-immunoprecipitation studies on the budding yeast homolog of Xrcc4, Lif1 (Teo and Jackson, 2000). Based on our observation that Xrcc4 controls the formation of a tripartite complex consisting of Xrcc4, DNA ligase IV and PNK, we propose that Xrcc4 acts as an NHEJ scaffold, targeting PNK and DNA ligase IV to DSBs, and thereby physically linking the enzymatic reactions of DNA end processing and DNA ligation.
Scaffolds are a common theme in signal transduction pathways, where they alter signaling by physically ordering catalytic steps or by controlling subcellular colocalization of signaling components (Burack and Shaw, 2000). We propose that Xrcc4 acts in a fashion analogous to signal transduction scaffolds, where it could function to recruit PNK and DNA ligase IV to DSBs and/or ensure that the DNA end processing activities of PNK are physically linked to the DNA ligase activity of DNA ligase IV. The coordination of these catalytic steps by Xrcc4 may be important for reasons other than simply increasing the rate of NHEJ. For example, one can envision that linking end processing to DNA ligation may promote more accurate and less mutagenic repair.
The complex assembled by Xrcc4 is remarkably analogous to the Xrcc1-dependent protein complex assembled during SSBR. Xrcc4 and Xrcc1 both interact with a DNA ligase (DNA ligase IV and DNA ligase III, respectively) and also interact with at least one common factor, PNK. The human tyrosyl-DNA phosphodiesterase Tdp1 has been shown to process DSBs bearing 3′-phosphoglycolates (Inamdar et al, 2002) and also interacts with Xrcc1 to repair camptothecin-induced SSBs (Plo et al, 2003). Therefore, it is reasonable to predict that Tdp1 may also play a role in NHEJ and it will be interesting to test whether Tdp1 can interact with Xrcc4. Interestingly, the parallels seen between these two repair complexes also seem to extend to their modes of regulation. In particular, our preliminary results have defined three putative CK2 threonine phosphorylation sites in Xrcc1 that appear to play a critical role in regulating the FHA-dependent PNK–Xrcc1 interaction, extending the work of Loizou et al (Supplementary Figure 2; Loizou et al, 2004). Therefore, the physical linking of DNA end processing to DNA ligation appears to be a common theme of both SSBR and DSBR in metazoans.
Materials and methods
Detailed description of Molecular Biology, Peptides and Mass Spectrometry methods can be found in the Supplementary data.
Expression and purification of fusion proteins
PNK and Xrcc4 recombinant proteins were produced in Escherichia coli BL21(DE3)/pLysS (Novagen), according to the manufacturer's protocol. See Supplementary Methods for more detailed information.
Cell culture
All cell lines were grown at 37°C in a humidified atmosphere containing 5% CO2. HEK293T cells were grown in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum (FBS) and 50 U of penicillin and 50 μg of streptomycin antibiotics per ml. The Xrcc4-deficient Chinese hamster ovary (CHO) cell line, XR-1 (Coriell Cell Repositories), was grown in F-12 media supplemented with 5% FBS and antibiotics. The murine pre-B cell line BaF3 was grown in RPMI containing 10% FBS and 5% WEHI-3 conditioned media.
Supplementary Material
Supplementary Methods
Supplementary Figures
Supplementary Data
Acknowledgments
We are thankful to the Durocher laboratory helpful discussions. In particular, we thank Pamela Kanellis, Lea Harrington and Mark Glover for their comments on the manuscript. We thank Steve Jackson for reagents and advice on γ-H2AX staining. We are also grateful to Bruce Seet and Steve Orlicky for their assistance with ITC and FP experiments. CAK is supported by the Princess Margaret Hospital (UHN). DD is a recipient of the Hitchings-Elion Fellowship of the Burroughs-Wellcome Fund and a Canada Research Chair (tier II) in Proteomics, Bioinformatics and Functional Genomics. This work was supported by grant MOP49435 from the Canadian Institutes of Health Research to DD.
References
- Bryans M, Valenzano MC, Stamato TD (1999) Absence of DNA ligase IV protein in XR-1 cells: evidence for stabilization by XRCC4. Mutat Res 433: 53–58 [DOI] [PubMed] [Google Scholar]
- Burack WR, Shaw AS (2000) Signal transduction: hanging on a scaffold. Curr Opin Cell Biol 12: 211–216 [DOI] [PubMed] [Google Scholar]
- Caldecott KW (2003) DNA single-strand break repair and spinocerebellar ataxia. Cell 112: 7–10 [DOI] [PubMed] [Google Scholar]
- Chappell C, Hanakahi LA, Karimi-Busheri F, Weinfeld M, West SC (2002) Involvement of human polynucleotide kinase in double-strand break repair by non-homologous end joining. EMBO J 21: 2827–2832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Trujillo K, Sung P, Tomkinson AE (2000) Interactions of the DNA ligase IV–XRCC4 complex with DNA ends and the DNA-dependent protein kinase. J Biol Chem 275: 26196–26205 [DOI] [PubMed] [Google Scholar]
- Coquerelle T, Bopp A, Kessler B, Hagen U (1973) Strand breaks and K′ end-groups in DNA of irradiated thymocytes. Int J Radiat Biol Relat Stud Phys Chem Med 24: 397–404 [DOI] [PubMed] [Google Scholar]
- Critchlow SE, Bowater RP, Jackson SP (1997) Mammalian DNA double-strand break repair protein XRCC4 interacts with DNA ligase IV. Curr Biol 7: 588–598 [DOI] [PubMed] [Google Scholar]
- Durocher D, Jackson SP (2002) The FHA domain. FEBS Lett 513: 58–66 [DOI] [PubMed] [Google Scholar]
- Gellert M (2002) V(D)J recombination: RAG proteins, repair factors, and regulation. Annu Rev Biochem 71: 101–132 [DOI] [PubMed] [Google Scholar]
- Grawunder U, Wilm M, Wu X, Kulesza P, Wilson TE, Mann M, Lieber MR (1997) Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature 388: 492–495 [DOI] [PubMed] [Google Scholar]
- Grawunder U, Zimmer D, Kulesza P, Lieber MR (1998) Requirement for an interaction of XRCC4 with DNA ligase IV for wild-type V(D)J recombination and DNA double-strand break repair in vivo. J Biol Chem 273: 24708–24714 [DOI] [PubMed] [Google Scholar]
- Inamdar KV, Pouliot JJ, Zhou T, Lees-Miller SP, Rasouli-Nia A, Povirk LF (2002) Conversion of phosphoglycolate to phosphate termini on 3′ overhangs of DNA double strand breaks by the human tyrosyl-DNA phosphodiesterase hTdp1. J Biol Chem 277: 27162–27168 [DOI] [PubMed] [Google Scholar]
- Junop MS, Modesti M, Guarne A, Ghirlando R, Gellert M, Yang W (2000) Crystal structure of the Xrcc4 DNA repair protein and implications for end joining. EMBO J 19: 5962–5970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi-Busheri F, Daly G, Robins P, Canas B, Pappin DJ, Sgouros J, Miller GG, Fakhrai H, Davis EM, Le Beau MM, Weinfeld M (1999) Molecular characterization of a human DNA kinase. J Biol Chem 274: 24187–24194 [DOI] [PubMed] [Google Scholar]
- Leber R, Wise TW, Mizuta R, Meek K (1998) The XRCC4 gene product is a target for and interacts with the DNA-dependent protein kinase. J Biol Chem 273: 1794–1801 [DOI] [PubMed] [Google Scholar]
- Lee KJ, Huang J, Takeda Y, Dynan WS (2000) DNA ligase IV and XRCC4 form a stable mixed tetramer that functions synergistically with other repair factors in a cell-free end-joining system. J Biol Chem 275: 34787–34796 [DOI] [PubMed] [Google Scholar]
- Li Z, Otevrel T, Gao Y, Cheng HL, Seed B, Stamato TD, Taccioli GE, Alt FW (1995) The XRCC4 gene encodes a novel protein involved in DNA double-strand break repair and V(D)J recombination. Cell 83: 1079–1089 [DOI] [PubMed] [Google Scholar]
- Lieber MR, Ma Y, Pannicke U, Schwarz K (2003) Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol 4: 712–720 [DOI] [PubMed] [Google Scholar]
- Loizou JI, El-Khamisy SF, Zlatanou A, Moore DJ, Chan DW, Qin J, Sarno S, Meggio F, Pinna LA, Caldecott KW (2004) The protein kinase CK2 facilitates repair of chromosomal DNA single-strand breaks. Cell 117: 17–28 [DOI] [PubMed] [Google Scholar]
- Ma Y, Pannicke U, Schwarz K, Lieber MR (2002) Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell 108: 781–794 [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Suzuki N, Namba N, Umeda N, Ma XJ, Morita A, Tomita M, Enomoto A, Serizawa S, Hirano K, Sakaia K, Yasuda H, Hosoi Y (2000) Cleavage and phosphorylation of XRCC4 protein induced by X-irradiation. FEBS Lett 478: 67–71 [DOI] [PubMed] [Google Scholar]
- Mills KD, Ferguson DO, Alt FW (2003) The role of DNA breaks in genomic instability and tumorigenesis. Immunol Rev 194: 77–95 [DOI] [PubMed] [Google Scholar]
- Modesti M, Hesse JE, Gellert M (1999) DNA binding of Xrcc4 protein is associated with V(D)J recombination but not with stimulation of DNA ligase IV activity. EMBO J 18: 2008–2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modesti M, Junop MS, Ghirlando R, van de Rakt M, Gellert M, Yang W, Kanaar R (2003) Tetramerization and DNA ligase IV interaction of the DNA double-strand break repair protein XRCC4 are mutually exclusive. J Mol Biol 334: 215–228 [DOI] [PubMed] [Google Scholar]
- Pawson T, Gish GD, Nash P (2001) SH2 domains, interaction modules and cellular wiring. Trends Cell Biol 11: 504–511 [DOI] [PubMed] [Google Scholar]
- Pinna LA (2002) Protein kinase CK2: a challenge to canons. J Cell Sci 115: 3873–3878 [DOI] [PubMed] [Google Scholar]
- Plo I, Liao ZY, Barcelo JM, Kohlhagen G, Caldecott KW, Weinfeld M, Pommier Y (2003) Association of XRCC1 and tyrosyl DNA phosphodiesterase (Tdp1) for the repair of topoisomerase I-mediated DNA lesions. DNA Repair (Amst) 2: 1087–1100 [DOI] [PubMed] [Google Scholar]
- Sarno S, Moro S, Meggio F, Zagotto G, Dal Ben D, Ghisellini P, Battistutta R, Zanotti G, Pinna LA (2002) Toward the rational design of protein kinase casein kinase-2 inhibitors. Pharmacol Ther 93: 159–168 [DOI] [PubMed] [Google Scholar]
- Smith GC, Jackson SP (1999) The DNA-dependent protein kinase. Genes Dev 13: 916–934 [DOI] [PubMed] [Google Scholar]
- Teo SH, Jackson SP (2000) Lif1p targets the DNA ligase Lig4p to sites of DNA double-strand breaks. Curr Biol 10: 165–168 [DOI] [PubMed] [Google Scholar]
- Whitehouse CJ, Taylor RM, Thistlethwaite A, Zhang H, Karimi-Busheri F, Lasko DD, Weinfeld M, Caldecott KW (2001) XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell 104: 107–117 [DOI] [PubMed] [Google Scholar]
- Yaffe MB, Leparc GG, Lai J, Obata T, Volinia S, Cantley LC (2001) A motif-based profile scanning approach for genome-wide prediction of signaling pathways. Nat Biotechnol 19: 348–353 [DOI] [PubMed] [Google Scholar]
- Yu Y, Wang W, Ding Q, Ye R, Chen D, Merkle D, Schriemer D, Meek K, Lees-Miller SP (2003) DNA-PK phosphorylation sites in XRCC4 are not required for survival after radiation or for V(D)J recombination. DNA Repair (Amst) 2: 1239–1252 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods
Supplementary Figures
Supplementary Data