

Abstract
Centrosomes are the principal microtubule organising centres in somatic cells. Abnormal centrosome number is common in tumours and occurs after γ-irradiation and in cells with mutations in DNA repair genes. To investigate how DNA damage causes centrosome amplification, we examined cells that conditionally lack the Rad51 recombinase and thereby incur high levels of spontaneous DNA damage. Rad51-deficient cells arrested in G2 phase and formed supernumerary functional centrosomes, as assessed by light and serial section electron microscopy. This centrosome amplification occurred without an additional DNA replication round and was not the result of cytokinesis failure. G2-to-M checkpoint over-ride by caffeine or wortmannin treatment strongly reduced DNA damage-induced centrosome amplification. Radiation-induced centrosome amplification was potentiated by Rad54 disruption. Gene targeting of ATM reduced, but did not abrogate, centrosome amplification induced by DNA damage in both the Rad51 and Rad54 knockout models, demonstrating ATM-dependent and -independent components of DNA damage-inducible G2-phase centrosome amplification. Our data suggest DNA damage-induced centrosome amplification as a mechanism for ensuring death of cells that evade the DNA damage or spindle assembly checkpoints.
Keywords: ATM, centrosome, cell cycle checkpoint, mitosis, Rad51
Introduction
Accurate cell division requires DNA to be replicated without error and the replicated chromosomes to be segregated equally between the two newly forming daughter cells. To achieve this, the replicated chromosomes attach to microtubules emanating from the opposite poles of a bipolar mitotic spindle and congress to a metaphase plate. Upon anaphase initiation, the sister chromatids disjoin and move to the spindle poles. In most animal cells, each spindle pole is centred around a centrosome consisting of two cylindrical centrioles embedded in an amorphous matrix of pericentriolar material (for review, see Doxsey, 2001). The centrosome is duplicated in a remarkable semiconservative process that is completed in S phase, while it takes more than an entire cell cycle for the maturation of the daughter centriole (Rieder and Borisy, 1982; Vorobjev and Chentsov Yu, 1982; Hinchcliffe and Sluder, 2001; Lange, 2002). Because centrosome duplication is closely linked to the cell cycle under the control of Cdk2–cyclin A (in mammals), centrosome number is tightly correlated with the DNA content of a cell (Hinchcliffe et al, 1999; for reviews, see Karsenti, 1999; Meraldi et al, 1999).
It was suggested as long ago as 1914 that centrosome abnormalities might lead to mitotic and chromosomal defects characteristic of cancer (Boveri, 1914). This hypothesis has recently returned to prominence with the discovery that both centrosome amplification and structural abnormalities of the centrosomes are common characteristics of tumour cells (Lingle et al, 1998; Duensing et al, 2000; Pihan et al, 2001; D'Assoro et al, 2002). While centrosomal defects may be a cause or a consequence of cancerous transformation, it is clear that the formation of supernumerary centrosomes can lead to mitotic abnormalities, such as the formation of multipolar spindles. Such spindle abnormalities could, in turn, result in abnormal chromosome segregation and aneuploidy (Brinkley, 2001; Sluder and Nordberg, 2004). Given the serious consequences of centrosomal abnormalities for the cell and organism, it is clearly important to determine how those abnormalities arise in cancer cells.
Here, we investigate centrosome abnormalities in Rad51-deficient chicken DT40 cells. Rad51, which encodes the eukaryotic homologue of the Escherichia coli RecA recombinase, is an essential gene, and its loss causes an inability to repair endogenously generated DNA lesions, resulting in cell cycle arrest and death within 24 h, accompanied by high levels of chromosome aberrations (Sonoda et al, 1998). We also examine mitosis following ionising radiation (IR)-induced DNA damage and removal of the Rad54 recombinational repair protein by gene targeting (Bezzubova et al, 1997). Our analyses led us to the hypothesis that centrosomes can duplicate during a prolonged G2 arrest in the absence of additional rounds of DNA replication.
One of the principal signalling molecules involved in instigating a G2-to-M cell cycle arrest after DNA damage is ATM, the product of the gene responsible for the human genetic disease ataxia-telangiectasia, ATAXIA-TELANGIECTASIA, MUTATED (reviewed in Abraham, 2001; Shiloh, 2003). ATM is a member of a family of large serine-threonine kinases involved in DNA repair and checkpoint control that possess a carboxy-terminal sequence bearing significant homology to the catalytic domain of phosphatidylinositol 3-OH kinase, the PI3KK family. Other members of the PI3KK family include the catalytic subunit of the DNA-dependent protein kinase (DNA-PKcs) and the ATM- and Rad 3-related protein, ATR. We show that the G2-to-M arrest that permits centrosome amplification is partly controlled by ATM. Since a G2-phase arrest is a normal cellular response to DNA damage, our findings provide an explanation for the frequent observation of multiple centrosomes in cells with damaged DNA as a result of either mutations in DNA repair genes or exposure to IR. In addition, these results suggest a potential mechanism for aneuploidy resulting from those cells that escape the G2 DNA damage checkpoint and execute mitosis with supernumerary centrosomes.
Results
Loss of Rad51 induces the formation of supernumerary centrosomes
Previous studies of chicken DT40 cells revealed that conditional loss of Rad51 results in defects in recombinational repair of DNA damage, arrest in G2/M phase, as determined by FACS analysis, and subsequent cell death (Sonoda et al, 1998). To define more precisely the effect that Rad51 deficiency has on individual cells, we examined the microtubule network by immunofluorescence microscopy. Cells were fixed and stained for α- and γ-tubulin at various times after the shutoff of RAD51 transcription (‘Rad51OFF') in order to address the status of the centrosomes. Cells with multiple centrosomes were observed both in interphase and mitosis (Figure 1A). After 16 h of repression, the cells began to accumulate supernumerary centrosomes. By 24 h, 35% of the cells had more than two centrosomes and cells with up to eight γ-tubulin spots could be observed (Figure 1B).
Figure 1.
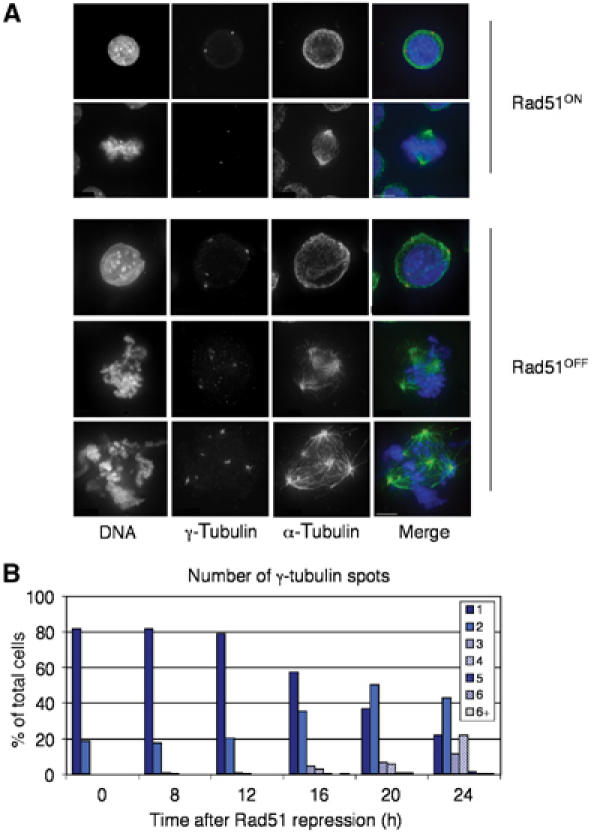
Centrosome amplification in Rad51-deficient cells. (A) Immunofluorescence microscopy of DT40 Rad51−/− cells before (Rad51ON) and after 24 h of doxycycline treatment (10 ng/ml) (Rad51OFF). Cells are stained for γ-tubulin (red), α-tubulin (green), and counterstained with DAPI (blue). Images are quick projections of Z (multiple focal planes) sections captured using the Deltavision deconvolution microscope. Scale bar, 5 μm. (B) Quantitation of supernumerary centrosomes. The number of γ-tubulin spots was counted at each of the time points shown after the addition of doxycycline. A total of 200 cells were counted at each time point.
The mitotic index in Rad51OFF cells decreases 12 h after repression of the Rad51 transgene, suggestive of a delay or arrest in the cell cycle prior to mitosis (Figure 2B). The proportion of the Rad51OFF population in G2–M, as determined by FACS analysis, increases up to 21 h postrepression (Figure 2A; Sonoda et al, 1998), apparently indicating a delay in G2 phase. This is not unexpected, as Rad51 deficiency causes DNA damage and the G2 DNA damage checkpoint is therefore activated. Ultimately, however, many of these cells eventually escape the checkpoint block and enter mitosis.
Figure 2.
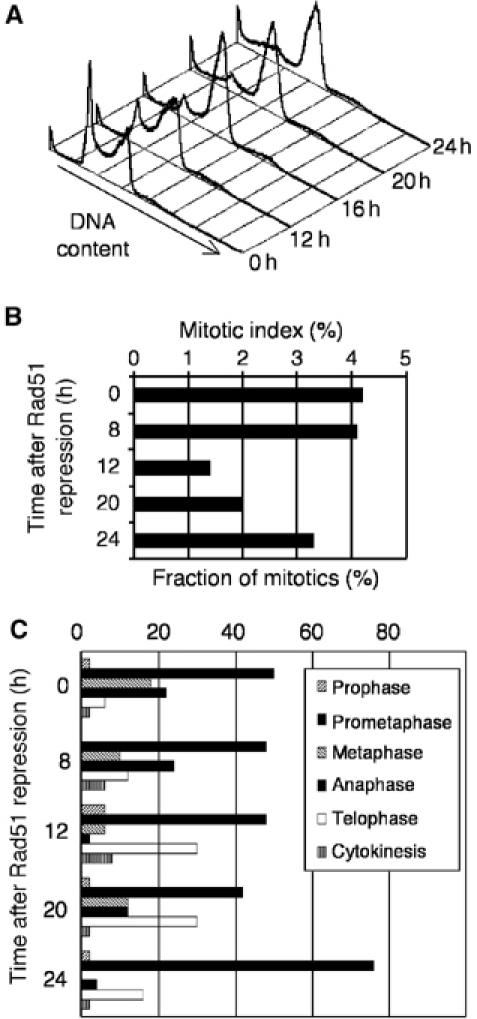
Entry into aberrant mitosis by Rad51-deficient cells with multiple centrosomes. (A) FACS analysis of DNA content of Rad51OFF cells at the times indicated at right after Rad51 repression. Note the accumulation in G2/M phase and the increase in dead cells without any increase in a >2N population. (B) The mitotic index of Rad51OFF cells, determined at various times after Rad51 repression. Data from at least 50 mitotic cells are shown per data point. (C) Distribution of Rad51OFF cells in mitotic phases at various times indicated after Rad51 repression, as judged by BubR1 staining (Supplementary Figure S1) and spindle staining.
Rad51-deficient mitotic cells examined 20 h after repression of the transgene appeared to traverse mitosis, albeit with a prolonged prometaphase. All stages of mitosis were observed (Figure 2C). The percentage of mitotic cells with multiple spindle poles increased with the length of time following the shutoff of Rad51 transcription, and by 24 h, 50% of all mitotic cells had spindle abnormalities (Figure 6B). Representative images of aberrant anaphase and telophase figures are shown in Supplementary Figure S1. These data indicate that the supernumerary centrosomes detected by γ-tubulin staining were functional and could organise additional spindle poles. Since a diploid complement of chromosomes is segregated to more than two poles, this increase in centrosome number therefore represents a potential mechanism of aneuploidy.
Figure 6.
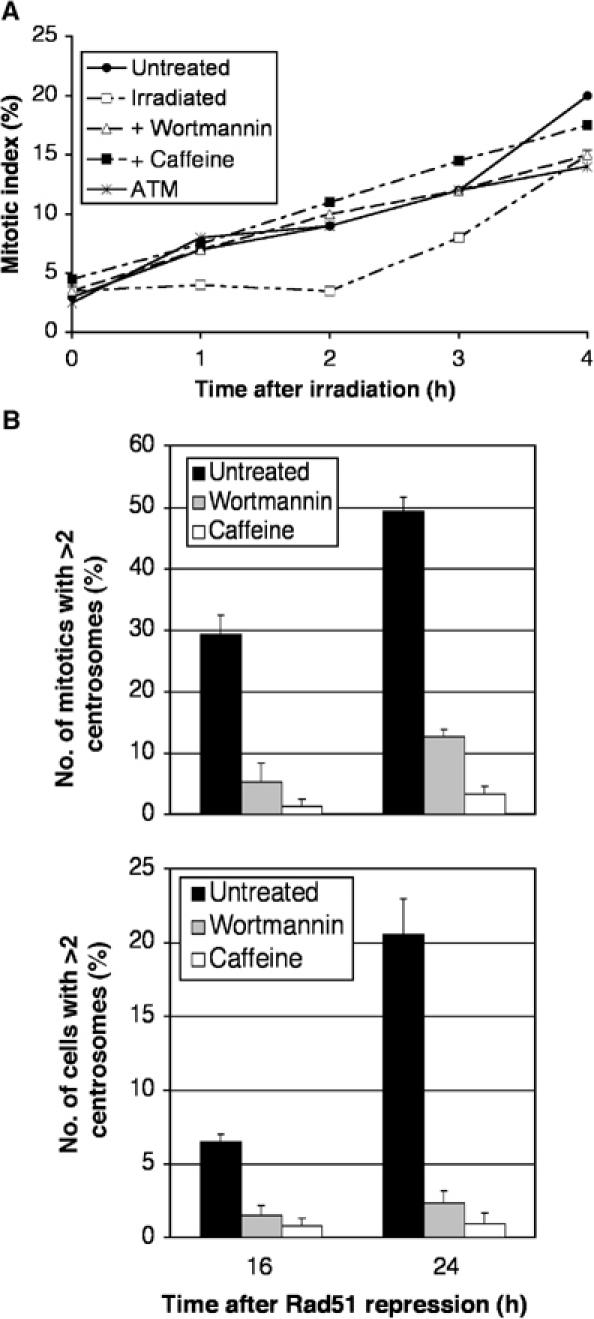
Suppression of centrosome amplification in Rad51OFF cells by G2-to-M checkpoint over-ride. (A) Cumulative mitotic indices of Rad51ON cells grown in the presence of colcemid for the times indicated after no treatment or after 1 Gy γ-irradiation, with or without preincubation with 1 μM wortmannin or 2 mM caffeine, as shown. A total of 200 cells were counted per time point. Similar results were obtained with wild-type DT40 cells in parallel experiments (data not shown). (B) Quantitation of cells with multiple centrosomes in the mitotic (upper) or entire cell population (lower) after the indicated length of time of Rad51 repression in the presence or absence of caffeine or wortmannin, as indicated. Centrosomes were counted by immunofluorescence microscopy of γ-tubulin spots. Data were obtained from 50 mitotic cells per experiment and histograms show the mean+standard deviation of results from three separate experiments, performed blind. Mean mitotic indices were not affected by the drug treatments and ranged from 2.76 to 3.04% over the entire experimental series shown.
Supernumerary centrosomes in Rad51OFF cells contain multiple centrioles
It has long been known that centrosomes in echinoderm eggs can split in response to a mitotic delay induced by β-mercaptoethanol (Sluder and Begg, 1983). More recently, it was shown that paired centrioles can separate from each other and migrate in the cytoplasm (Piel et al, 2000). Furthermore, centrosomes can fragment in response to release from cell cycle arrest induced by microtubule poisons (Keryer et al, 1984) and in response to impaired DNA integrity during mitosis (Hut et al, 2003).
In order to test whether the increase in γ-tubulin spots seen in Rad51OFF cells reflects a splitting or duplication of the centrosome structure, we stained Rad51-deficient cells with anti-centrin antibodies. Centrin has been used as a marker for the centriole structure in a number of studies (Paoletti et al, 1996; White et al, 2000). We observed that Rad51OFF cells contain multiple centrin spots associated with the centrosomes (Supplementary Figure S2). This suggests that the supernumerary centrosomes contain centrioles, and do not therefore arise by centrosome fragmentation. However, despite its striking localisation in immunofluorescence experiments, biochemical fractionation studies revealed that the bulk of centrin is not associated with centrioles in vertebrate cells (Paoletti et al, 1996), so additional proof was needed.
We therefore examined Rad51OFF cells by serial section electron microscopy 24 h after shutoff of transcription of the transgene. When cell pellets were embedded in resin and serially sectioned at 100 nm thickness, we observed interphase Rad51OFF cells that contained more than four centrioles, indicating that the centrosomes were duplicating rather than splitting or fragmenting (data not shown). This could be addressed most clearly in mitotic cells with multipolar spindles. To do this, Rad51OFF DT40 cells attached to glass coverslips were fixed with glutaraldehyde and stained for α-tubulin. Several cells (n=3) with multipolar spindles were photographed, and after flat embedding, relocated and serially sectioned for electron microscopy. Paired centrioles were found to be associated with each of the poles in these multipolar mitotic cells (Figure 3B). We therefore conclude that the supernumerary centrosomes contain paired centrioles and are a result of bona fide centrosome duplication, rather than centrosome splitting or fragmentation.
Figure 3.

Supernumerary centrosomes in Rad51OFF cells contain multiple centrioles. Electron microscopy was performed on (A) control or (B) Rad51OFF cells 24 h after the addition of doxycycline (10 ng/ml). Cells were processed for immunofluorescence microscopy of α-tubulin (green) and staining of the DNA (blue). These cells were then flat embedded, and ultrathin serial sections were cut. At each pole in both the Rad51ON and Rad51OFF cells, two centrioles can be seen. Numbers on each colour image indicate the positions of the centrioles shown in the corresponding electron micrographs. Scale bar, 0.5 μm.
Centrosomes are duplicated during a prolonged G2 phase
Current hypotheses on abnormal centrosome duplication in cancer cells suggest that this process can occur as a result of a prolonged S phase (Balczon et al, 1995; Wong and Stearns, 2003) or as a result of a failure in cytokinesis (Meraldi et al, 2002). However, Rad51OFF cells are not blocked in S phase or mitosis and do not become polyploid due to a failure in cytokinesis. During 24 h of transgene shutoff, the number of cells in S phase decreases from ≈65 to ≈20% (Figure 2). Rad51 deficiency does not induce polyploidy, as determined by FACS analysis (Figure 2A), and fewer than 1% of cells show multi- or micronucleation 24 h after Rad51 repression, as in control cells (data not shown). Furthermore, karyotype analysis of Rad51-deficient cells showed no evidence of polyploidy (Sonoda et al, 1998).
In order to determine the cell cycle stage at which the cells were duplicating their centrosomes, we carried out a BrdU pulse-chase experiment as diagrammed in Figure 4A. Cultures were pulsed with BrdU, in order to label cells in which DNA synthesis was occurring. After 15 min, BrdU was washed out and the cells were cultured in the presence or absence of doxycycline. Cells were chased for varying lengths of time through the cell cycle and all cells were analysed at the same final Rad51 repression time point of 24 h. Cells were then fixed in methanol, stained for both γ-tubulin and BrdU, and the number of centrosomes in BrdU-positive cells was scored (Figure 4B). Representative images of BrdU-positive cells with multiple centrosomes are shown in Supplementary Figure S2C.
Figure 4.
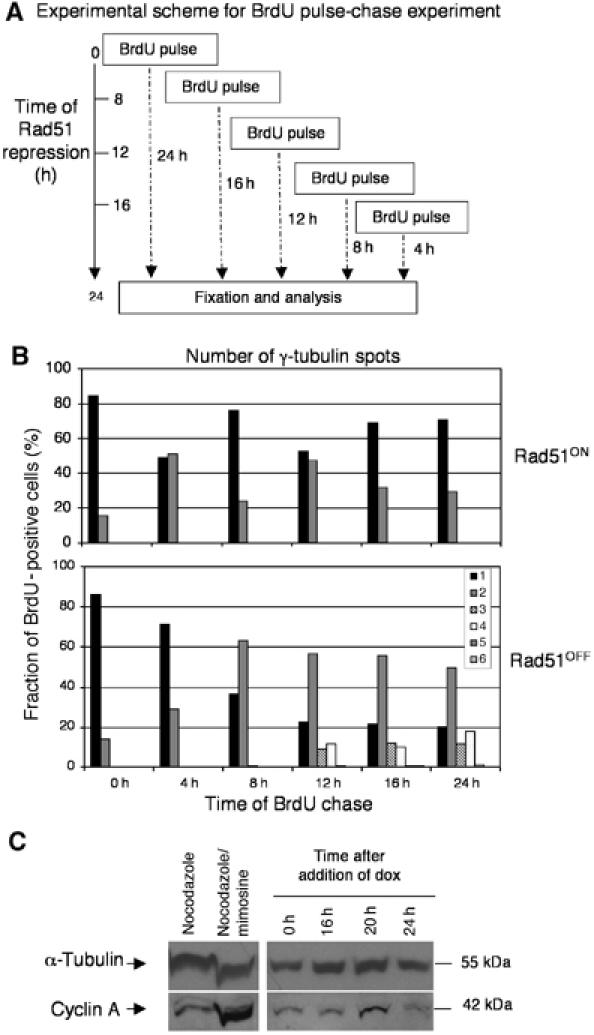
G2 arrest and centrosome duplication in Rad51OFF cells. (A) Experimental scheme of BrdU pulse-chase experiment. Rad51OFF cells were treated with a 15 min pulse of BrdU (20 μm), after which time they were washed three times in fresh medium and recultured in the presence or absence of doxycycline (10 ng/ml). Cells were all analysed at the same final time postrepression of 24 h. (B) Analysis of BrdU pulse-chase experiment. The number of centrosomes was counted in 200 BrdU-positive cells per data point; top, control cells (Rad51ON); bottom, Rad51OFF cells after Rad51 repression for the times indicated. A total of 200 cells were counted at each time point. The percentage of the total cells determined as BrdU positive by FACS analysis over the repression time course shown was 67% (8 h after Rad51 repression), 62% (12 h), 39% (16 h), 34% (20 h) and 22% (24 h). (C) Immunoblot analysis of cyclin A levels in mitotically arrested (nocodazole treated), early S phase (nocodazole/mimosine arrested; Sonoda et al, 2001) wild-type DT40 cells and from cells where the RAD51 transgene was repressed with doxycycline for 0, 16, 20 or 24 h as indicated. Immunoblot for α-tubulin was used as a loading control.
The pattern of centrosome staining can be used to monitor the progression of the control BrdU-positive Rad51ON cells through the ≈8-h DT40 cell cycle (Figure 4B, upper). Cultures in G1 or early S will have a much larger percentage of cells with a single centrosome (this could also be two closely associated centrosomes) than cells with separated centrosomes (0, 8, 16 and 24 h in Figure 4B, upper). Cultures in G2/M will have a much larger percentage of cells with separated centrosomes (4 and 12 h in Figure 4B, upper). Cultures treated with doxycycline progressed more slowly through the cell cycle and 8 h after being labeled with BrdU in S phase, approximately 60% of the cells had two separated γ-tubulin spots, consistent with the cultures being in either G2 or M phase (Figure 4B, lower). Strikingly, these cells did not continue to cycle since by 12 h, the number of cells in G1 or early S phase (as represented by the number of cells with only one γ-tubulin spot) actually decreased, and cells began to acquire multiple centrosomes.
This result suggests that Rad51-deficient cells may accumulate supernumerary centrosomes in a prolonged G2 phase. Since these cells are defective in homologous recombinational repair, they presumably activate the G2 DNA damage checkpoint (Sonoda et al, 1998). To confirm that these cells were in G2 phase, we quantified the number of cyclin B2-positive cells within the culture after the addition of doxycycline. The number of cyclin B2-positive cells was higher than that seen in control cultures (Supplementary Figure S3A), reaching a peak of 70% at 20 h after the addition of doxycycline. Representative images of cells stained for cyclin B2 and γ-tubulin are shown in Supplementary Figure S3B. To examine the cell cycle controls on centrosome amplification after DNA damage, we investigated the levels of cyclin A, the activity of which is required for normal centrosome duplication (Lacey et al, 1999; Meraldi et al, 1999). As shown in Figure 4C, cyclin A was not elevated in the absence of Rad51 to the level observed in the early S-phase population that we obtained by blocking the DT40 cells with nocodazole, releasing and then incubating with mimosine (Sonoda et al, 2001), showing that the centrosome amplification is not due to increased cyclin A levels.
Additive effects of induced S-phase arrest and Rad51 repression
Although we saw no evidence for an extended S phase in the Rad51-deficient population, we tested whether these cells undergo centrosome amplification during S-phase arrest, as has been described for mammalian cells (Balczon et al, 1995; Wong and Stearns, 2003). Treatment with hydroxyurea (HU) or aphidicolin for 12 h imposed a strong S-phase delay in wild-type DT40 cells, but resulted in only a minor induction of centrosome amplification (Figure 5A) as assessed using γ-tubulin as a centrosome marker. Longer treatments resulted in dramatic levels of centrosome amplification, consistent with previous data, although the cell cycle arrests were less robust (Figure 5B and C). Repression of Rad51 simultaneously with the induction of an S-phase delay caused moderate, additional levels of centrosome amplification (Figure 5C). These data are consistent with the idea that the extended DNA damage-induced G2 delay that would be imposed on the small number of cells exiting the HU/aphidicolin arrest is permissive for centrosome amplification.
Figure 5.
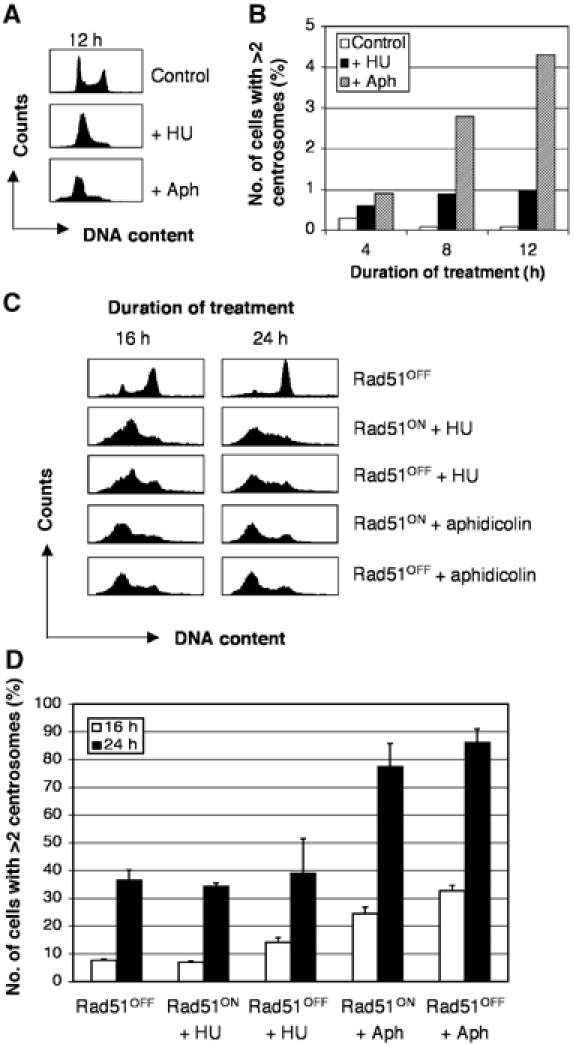
Centrosome amplification in Rad51OFF cells during extended S phase. (A) FACS profiles of wild-type DT40 cells treated as indicated. Aph, aphidicolin. (B) Histogram showing quantitation of cells with multiple centrosomes by immunofluorescence microscopy of γ-tubulin spots in 1000 cells per sample. (C) FACS analysis of the DNA content of Rad51-deficient or control cells after treatment with HU or aphidicolin as indicated. Sub-G1 cells have been gated from the analysis. (D) Histogram showing quantitation of cells with multiple centrosomes as for (A) in Rad51-deficient or control cells after treatment with HU or aphidicolin as indicated for the times shown in the key. Data shown are the mean+standard deviation of results from three separate experiments, each counting 1000 cells.
G2-to-M checkpoint over-ride suppresses centrosome amplification
To test directly whether the G2-to-M checkpoint that is induced by DNA damage contributes to centrosome amplification, we treated Rad51OFF cells with caffeine and wortmannin, known inhibitors of this checkpoint, and then monitored centrosome numbers. Over-ride by caffeine of the DNA damage-induced G2-phase checkpoint response has been known for decades (Lau and Pardee, 1982). Recent evidence has suggested that it may affect this checkpoint by targeting the ATM/ATR kinases, of which it is an in vitro inhibitor (Blasina et al, 1999; Zhou et al, 2000), although data exist showing that it does not necessarily inhibit these kinases in vivo (Cortez, 2003; Kaufmann et al, 2003). Wortmannin has been described as an inhibitor of PI3KK activity in vitro (Sarkaria et al, 1998), inhibiting ATM and DNA-PK more strongly than ATR. As shown in Figure 6A, both caffeine and wortmannin suppressed the ATM-dependent G2-phase cell cycle checkpoint arrest normally observed in DT40 cells after γ-irradiation (Takao et al, 1999). Abrogation of this checkpoint by the drug treatments had a striking effect on the extent to which multiple centrosomes were induced by Rad51 deficiency in both mitotic cells and in the entire population (Figure 6B). Treatment with 2 mM caffeine resulted in near-normal numbers of centrosomes in Rad51OFF cells, while 1 μM wortmannin effected almost as dramatic a normalisation of centrosome status. These data demonstrate that the G2-to-M checkpoint is required for centrosome amplification resulting from Rad51 deficiency-induced DNA damage.
Suppression of DNA damage-induced centrosome amplification by ATM disruption
While the repression of the G2-to-M checkpoint is a clear result of the caffeine and wortmannin treatment, the molecular target(s) of these drugs remain somewhat controversial. Since the G2-to-M checkpoint is known to be sustained at least in part by ATM signalling and since ATM is a putative target of caffeine and wortmannin, we next examined ATM's role in centrosome amplification. To test the role of the ATM-dependent checkpoint in centrosome amplification in the Rad51-deficient model, we disrupted ATM by gene targeting (Figure 7A). In order to achieve successful targeting, we were obliged to use a clone (#104) that expressed higher levels of Rad51 than the previously published Rad51 conditional clone (#110) (Sonoda et al, 1998). As shown in Figure 7B, efficient Rad51 repression occurs in this cell line after addition of the tetracycline analogue doxycycline. Extensive overexposure of the Rad51 immunoblot revealed residual Rad51 in both the ATM+/− and ATM−/− #104 derivatives; while not identical, the residual Rad51 levels do not vary more than two-fold. Rad51 repression causes centrosome amplification (Figure 7C), albeit with slightly different kinetics from Clone #110, which we used for all other experiments in this study. ATM targeting greatly diminishes the extent of centrosome amplification after Rad51 repression (Figure 7C), in both mitotic cells and in the entire population, implicating ATM as a major target of caffeine and wortmannin in repressing the G2-to-M checkpoint and allowing centrosome amplification in cells with extensive DNA damage.
Figure 7.
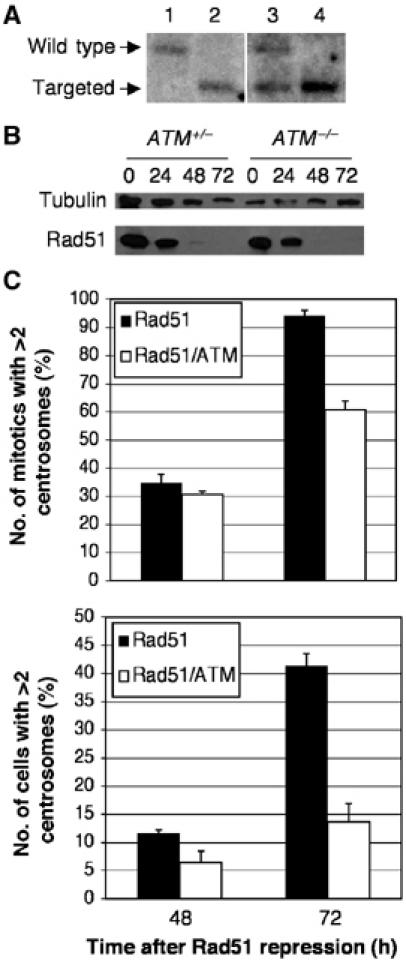
Suppression of centrosome amplification in Rad51OFF cells by ATM disruption. (A) Southern blot analysis of the ATM locus in (1) wild-type, (2) ATM−/−, (3) Clone #104 Rad51−/−/ATM+/− and (4) Clone #104 Rad51−/−/ATM−/− DT40 cells. (B) Immunoblot analysis of Rad51 repression kinetics in the Clone #104-derived Rad51−/−/ATM+/− and Rad51−/−/ATM−/− DT40 cells used for centrosome analysis below. Cells were treated with doxycycline for the times indicated prior to harvest. Immunoblot for α-tubulin was used as a loading control. (C) Quantitation of cells with multiple centrosomes in the mitotic (upper) or entire cell population (lower) of the indicated genotypes after the indicated length of time of Rad51 repression. Quantitation and histogram data are as for Figure 6B.
Next, to extend our findings regarding centrosome amplification to other models, we tested the induction of centrosomal numerical aberrations by γ-irradiation of DT40 cells. As shown in Supplementary Figure S4, 137Cs irradiation of DT40 cells induced a time- and dose-dependent increase in centrosome number in mitotic cells. While we have not yet visualised such spindle poles by electron microscopy and thus they may not be equivalent to the Rad51 deficiency-induced structures, they frequently contain two spots of centrin, suggesting they may contain centrioles (Supplementary Figure S5). We next investigated the impact that the loss of the recombinational repair protein Rad54 has on centrosome number. Gene targeted Rad54-deficient cells (Bezzubova et al, 1997) showed a slight elevation in the number of multipolar mitotics, which was potentiated greatly by 2 Gy γ-irradiation (Figure 8A). Over-ride of the G2-to-M checkpoint by wortmannin treatment reduced centrosome amplification in irradiated Rad54−/− cells, while caffeine treatment led to wild-type levels of multipolar spindles after irradiation of the Rad54 nulls (Figure 8A). As shown in Figure 8B, ATM loss notably reduced centrosome amplification in irradiated Rad54-deficient cells. The ATM knockout data in the Rad51- and Rad54-deficient models clearly reveal a significant role for ATM in allowing centrosome amplification. It is noteworthy that neither ATM deficiency nor wortmannin treatment completely abrogates DNA damage-induced multipolarity in the manner that caffeine treatment does. This suggests that there is a caffeine-sensitive, wortmannin-insensitive, ATM-independent activity also involved in the G2-to-M checkpoint that permits centrosome amplification.
Figure 8.
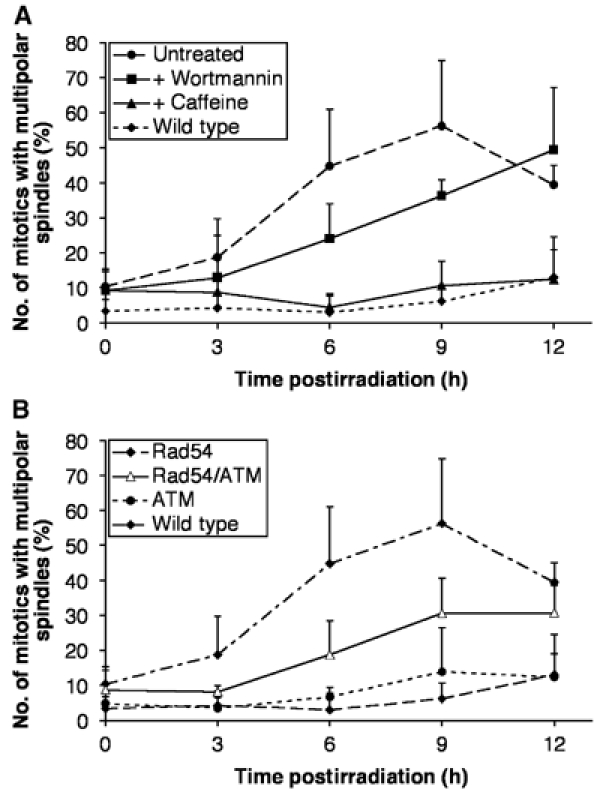
Suppression by G2-to-M checkpoint disruption of centrosome amplification in irradiated DNA repair mutant cells. Data points show the mean+standard deviation of results from three separate experiments in which at least 30 mitotic cells were counted. (A) Quantitation of the mitotic Rad54-deficient and wild-type DT40 cells with multiple spindle poles at the times indicated after 2 Gy γ-irradiation. Where shown, cells were treated with 1 μM wortmannin or 2 mM caffeine 1 h prior to irradiation. (B) Quantitation of the mitotic ATM−/−, Rad54−/− and Rad54−/−/ATM−/− DT40 cells with multiple spindle poles at the times indicated after 2 Gy γ-irradiation. Data from wild-type cells are as shown in (A) for comparison.
Discussion
Centrosome duplication can occur during a prolonged G2 delay
Present models for centrosome amplification fall into three classes. First, it has been suggested that under certain circumstances, centrosome duplication can become uncoupled from the normal cycles of DNA synthesis and mitosis. Uncoupling of centrosome duplication from cell cycle progression has been observed in specialised cells, such as early embryos (Sluder and Lewis, 1987; Raff and Glover, 1988; Gard et al, 1990; Sluder et al, 1990) and in certain cultured cells when their cycle is arrested at the G1/S-phase boundary using either HU or aphidicolin (Balczon et al, 1995; Wong and Stearns, 2003). Second, it has been proposed that the apparent centrosome multiplication seen in certain cells following DNA damage reflects fragmentation of the centrosome rather than bona fide duplication (Hut et al, 2003). Third, it has been suggested that numerical centrosome aberrations arise as a consequence of failures in cytokinesis, which can be induced, among other things, by abnormalities in the expression or regulation of several protein kinases (Meraldi et al, 2002). This model makes the clear prediction that cells with centrosomal abnormalities will be polyploid.
Centrosome amplification can happen repeatedly in cells with DNA damage induced by Rad51 deficiency or IR, as cells with up to eight centrosomes were observed during our studies. However, this amplification was not accompanied by DNA synthesis, as measured by BrdU incorporation, or polyploidisation, as detected by either FACS or microscopy analysis. These observations, together with the finding that cyclin B2 levels are elevated during the timeframe where the centrosome amplification is occurring, suggest that centrosome amplification in Rad51-deficient cells must occur during G2, presumably while the cells are blocked at the G2 DNA damage checkpoint. Although most of the Rad51-deficient cells are terminally arrested in G2 phase, a small number of cells do manage to escape into mitosis (because of the mitotic arrest, the precise number of cells that bypass this block cannot be determined; since there is no increase in mitotic index over time, this is clearly a relatively low, although significant number). Thus, our observations can explain the earlier finding that irradiation of tumour cells (which would also induce a prolonged G2 delay) can induce the formation of multipolar spindles (Sato et al, 1983). In testing this directly, by using caffeine and wortmannin to over-ride the G2-to-M checkpoint, we demonstrate that suppression of this checkpoint in Rad51OFF and in irradiated Rad54-deficient cells can reduce centrosome amplification dramatically, although the effects of the two drugs are not quite equivalent, as discussed below. These findings are consistent with centrosome amplification occurring during an extended G2 phase.
Our observations contrast with current models for centrosome amplification, which suggest that centrosome amplification is a consequence of either failed cytokinesis (Andreassen et al, 2001; Meraldi et al, 2002) or extended S-phase arrest (Balczon et al, 1995; Wong and Stearns, 2003). Consistent with previous data in mammalian cells, DT40 cells also undergo centrosome amplification after extended delays in S phase induced by HU or aphidicolin treatment. However, the imposition on any escapers of this arrest (G2 arrest) by Rad51 depletion causes additional amplification, consistent with our current hypothesis. Our data also argue against an alternative model, that centrosome ‘amplification' is actually centrosome fragmentation, as the supernumerary centrosomes detected here contain centrioles. This distinguishes them from the subcentrosomal fragments observed when Chinese hamster ovary (CHO) cells are forced into mitosis with incompletely replicated DNA (Hut et al, 2003) or after release from colcemid arrest (Keryer et al, 1984). In addition to centrioles, as shown by serial section electron microscopy, the amplified structures in Rad51OFF cells contain γ-tubulin and centrin, as visualised by light microscopy. We therefore conclude that they are ‘real' centrosomes.
Links between DNA damage and control of centrosome number
Several recent studies have implicated DNA damage in the generation of centrosome numerical aberrations. In Drosophila syncytial embryos, mitosis attempted in the presence of DNA damage causes a disruption of centrosome function and loss of the damaged nucleus by mitotic catastrophe (Takada et al, 2003). Intriguingly, induction of this catastrophe requires Chk2, which is a target of the ATM kinase in the G2 DNA damage checkpoint pathway, although another study of mitotic catastrophe reports that Chk2 inhibition can stabilise centrosomes (Castedo et al, 2004). p53-deficient mouse cells show a significant increase in centrosome number (Fukasawa et al, 1996), although there are multiple mechanisms by which deficiency in this key gene could cause such amplification (Tarapore and Fukasawa, 2002). The loss of p53 would be expected to impair the function of the G1–S checkpoints and might therefore allow damaged genomes to reach the G2–M transition, whereupon other signals might impose a cell cycle arrest in G2. Defects in a number of key genes involved in DNA repair have been shown to cause aberrations in centrosome number. Centrosome amplification is seen in cells deficient in Brca1 (Xu et al, 1999), Brca2, which binds Rad51 and plays a role in DNA repair (Tutt et al, 1999; Kraakman-van der Zwet et al, 2002), Mre11 (Yamaguchi-Iwai et al, 1999), the Rad51 paralogues Xrcc2 and Xrcc3 (Griffin et al, 2000) and Parp-1 (Kanai et al, 2003). Interestingly, the expression of a dominant-negative Rad51 in CHO cells resulted in increased centrosome numerical aberrations and aneuploidy (Bertrand et al, 2003). These cells gave rise to a higher frequency of tumors in nude mice than did CHO cells expressing wild-type Rad51, suggesting a role for Rad51 function in tumour suppression.
It is possible that the centrosome amplification during G2 phase that we describe here occurs because the centrosome cycle is controlled by an intrinsic timing mechanism, so that in cells arrested for an abnormally long time in G2, the centrosome cycle might begin again even though mitosis has not been traversed. Alternatively, a more active mechanism whereby specific signals or factors associated with DNA damage contribute to the centrosome overduplication could be hypothesised. Preliminary results suggest that induction of a G2 arrest by an alternative means, treatment with the topoisomerase II inhibitors ICRF-159 and ICRF-187, results in an ≈10-fold increase of multiple centrosomes over controls, from 0.1 to 1% of the total population. Clearly, the centrosome amplification following Rad51 depletion is much more significant, suggesting that a specific signal, rather than just a delay, may be required. Centrosome amplification in response to DNA damage is caffeine sensitive and comprises wortmannin-sensitive and -insensitive elements, along with ATM-dependent and -independent components. Although its in vivo role is controversial, caffeine inhibits ATM and ATR in vitro (Blasina et al, 1999; Sarkaria et al, 1999; Zhou et al, 2000; Cortez, 2003; Kaufmann et al, 2003). Wortmannin also inhibits ATM, DNA-PK and, to a lesser extent, ATR (Sarkaria et al, 1998; Chan et al, 2000). Taking these points together, ATR is a good candidate for a protein involved in mediating centrosome duplication after DNA damage, although DNA-PK might also be cited as a possibility. It will be important in future experiments to determine whether the centrosome amplification described here is actively triggered or whether it is a passive consequence of an extended cell cycle delay.
Centrosome amplification as a cellular defence mechanism?
Although Rad51-deficient and DNA-damaged DT40 cells frequently manage to enter mitosis, the majority of these cells accumulate in prometaphase and show strong kinetochore binding of the spindle assembly checkpoint protein BubR1. It remains to be explained how even the highly penetrant DNA damage level induced by loss of Rad51 could have such a strong impact on the kinetochore–microtubule interactions as to block the bulk of the cells at this point in mitosis. This prometaphase delay is not absolute, however, and a small number of Rad51-deficient cells do continue through mitosis, ultimately undergoing a highly aberrant mitotic exit followed by death early in the next cell cycle. Although it is possible that aneuploid offspring resulting from such multipolar divisions could somehow proliferate (Brinkley, 2001), this is likely to be an infrequent event and has never been observed in the absence of Rad51 (Sonoda et al, 1998 and unpublished observations).
Our results suggest the novel possibility that far from being an entirely negative and aberrant phenomenon, centrosome amplification occurring during a prolonged G2 arrest might offer a means to ensure the deletion of cells with DNA damage, which escape the G2- and M-phase checkpoints. Only cells with substantial levels of DNA damage would be expected to undergo a protracted G2 delay. If such a protracted arrest were to trigger centrosome amplification, this would ensure that any cells that did escape from the arrest would be unable to undergo a productive mitosis and produce viable progeny. In fact, it has been proposed that induction of multipolar spindles following DNA damage may be a cause of death in therapeutic irradiation (Sato et al, 2000). Clearly, the known activities of the PI3KK pathways in establishing a G2 delay in response to DNA damage implicate them in such a mitotic catastrophe response and might represent an additional mechanism by which they control genomic stability.
Materials and methods
Cell culture and sorting
The chicken lymphoma B-cell line DT40 was cultured and the human RAD51 and RAD54 transgenes were repressed by the addition of doxycycline (Sigma) to 10 ng/ml as described (Sonoda et al, 1998; Morrison et al, 2000). Phenotypes we describe here were similar in both the tetracycline-repressible Rad54 (Morrison et al, 2000) and constitutive Rad54 null (Bezzubova et al, 1997) lines. The high-expressing, conditionally Rad51-deficient cell line Clone #104 was derived in the same screen as the published Rad51 mutant line Clone #110 (Sonoda et al, 1998) and RAD51 was repressed using 25 ng/ml doxycycline. Targeting of ATM and Southern analysis were performed as described (Takao et al, 1999). Cells were treated with BrdU (Amersham) at a concentration of 20 μM. Wortmannin (Sigma) was dissolved in dimethylsulphoxide at 20 mM and stored at −20°C, and caffeine (Sigma) was dissolved in water at 200 mM and stored at 4°C. Caffeine or wortmannin treatment consisted of a 1 h preincubation of cells with the drug prior to further treatment. Aphidicolin was dissolved in ethanol and used at 1 μg/ml and HU, dissolved in water, was used at 1 mM, and both drugs were from Sigma. IR experiments were performed using a 137Cs source at 23.5 Gy/min (Mainance Engineering). For flow cytometry, cells were fixed with 70% ethanol, then resuspended in phosphate-buffered saline (PBS) and incubated with 200 μg/ml RNase A for 20 min. Propidium iodide was added to 40 μg/ml. Cell sorting was performed on a FACScalibur (BD Biosciences).
Giemsa staining of chromosomes
Cells were grown in the presence of 0.1 μg/ml colcemid (Sigma), then spun out of medium, hypotonically swollen with 0.9% sodium citrate for 15–30 min at room temperature, fixed with freshly prepared methanol:acetic acid (3:1) and dropped onto glass slides, then air-dried and stained. Giemsa stain (BDH) was diluted 1:5 with Gurr buffer, pH 6.8 (BDH), prior to use. Slides were washed with distilled water.
Immunofluorescence microscopy
Cells were adhered to poly-L-lysine slides or coverslips and were then fixed with either 4% paraformaldehyde in PBS for 10 min at room temperature or methanol/5 mM EGTA at −20°C for 10 min. Cells were then washed three times in PBS before blocking in PBS/1% bovine serum albumin. Primary antibodies were diluted in blocking solution and incubations were performed for 1 h at 37°C. The cells were then washed three times in PBS before incubation with appropriate secondary antibodies (fluorescein isothiocyanate (FITC) and Texas red-coupled anti-mouse and anti-rabbit antibodies from Jackson Laboratories and Alexa 488-coupled anti-rat antibodies from Molecular Probes) diluted 1:200 in blocking buffer. Samples were washed and counterstained with DAPI prior to mounting in Vectashield (Vector Laboratories). Imaging was performed using an Olympus IX-70 microscope driven by the SoftWorx package system (Applied Precision). Z-sections (0.15 μm) were collected and the images then deconvolved using SoftWorx and quick projections saved as Adobe Photoshop images. Cell counting and some imaging were performed using an Olympus BX51 microscope, × 100 objective, NA 1.35 using Openlab software (Improvision). Primary antibodies used are described in Supplementary Methods.
Electron microscopy
Cells adhered to poly-L-lysine coverslips were dipped briefly into prewarmed PHEM buffer (60 mM PIPES, 25 mM HEPES, 1 mM EGTA, 2 mM MgCl2, pH 6.9) containing 0.1% Triton X-100, and then immediately fixed in 1% glutaraldehyde for 10 min in PHEM buffer. After permeabilisation in PHEM buffer containing 0.1% Triton X-100 for 10 min, cells were rinsed with PBS, and excess aldehyde residues were reduced with freshly prepared 0.1% sodium borohydride in PBS, twice for 10 min. Immunofluorescence of tubulin was carried out with the monoclonal antibody DM1α (Sigma) at 1:500 dilution, followed by FITC-conjugated secondary antibody. Coverslips with cells were postfixed with 3.7% formaldehyde in PBS for 10 min, stained with DAPI and mounted onto glass slides in Vectashield, using droplets of nail polish as spacers. Phase contrast and fluorescence images of cells were captured, and the coordinates of these cells were recorded relative to a fixed point on the coverslip. Coverslips were then removed from the slides and rinsed in PBS, followed by 0.1 M sodium cacodylate for 5 min. After treatment with 0.5% tannic acid in 0.1 M cacodylate buffer for 45 min, cells were rinsed and postfixed with 1% osmium tetroxide in cacodylate buffer for 1 h. Cells were dehydrated in a graded series of ethanol, followed by propylene oxide and infiltration with a 1:1 mixture of propylene oxide and epon. Coverslips were flat embedded in epon, and after polymerisation of the resin, the glass was removed with hydrofluoric acid. Mitotic cells were relocated and serially sectioned. Ultrathin sections were contrasted with uranyl acetate followed by lead citrate, and viewed in a Philips CM120 Biotwin electron microscope.
Immunoblotting experiments
Polyclonal rabbit anti-Rad51 antibodies from Calbiochem (Ab-1) were used at 1:1000 in Western analyses, and the anti-α-tubulin monoclonal B512 was used at 1:10 000. Rabbit polyclonal anti-cyclin A antibodies from Transduction Laboratories were used at 1:125.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Methods
Acknowledgments
We thank E Nigg and JL Salisbury for provision of antibodies and T van Amstel for assistance with microscopy. Financial support to ES and ST was provided in part by CREST, JST and Center of Excellence (COE) grants from the Ministry of Education, Science and Culture of Japan. Work in WCE's laboratory is supported by the Wellcome Trust, of which he is a Principal Research Fellow. AM's laboratory is supported by a Wellcome Trust Senior Research Fellowship. Work in CM's laboratory is supported by a Science Foundation Ireland Investigator award.
References
- Abraham RT (2001) Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 15: 2177–2196 [DOI] [PubMed] [Google Scholar]
- Andreassen PR, Lohez OD, Lacroix FB, Margolis RL (2001) Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol Biol Cell 12: 1315–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balczon R, Bao L, Zimmer WE, Brown K, Zinkowski RP, Brinkley BR (1995) Dissociation of centrosome replication events from cycles of DNA synthesis and mitotic division in hydroxyurea-arrested Chinese hamster ovary cells. J Cell Biol 130: 105–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand P, Lambert S, Joubert C, Lopez BS (2003) Overexpression of mammalian Rad51 does not stimulate tumorigenesis while a dominant-negative Rad51 affects centrosome fragmentation, ploidy and stimulates tumorigenesis, in p53-defective CHO cells. Oncogene 22: 7587–7592 [DOI] [PubMed] [Google Scholar]
- Bezzubova O, Silbergleit A, Yamaguchi-Iwai Y, Takeda S, Buerstedde JM (1997) Reduced X-ray resistance and homologous recombination frequencies in a RAD54−/− mutant of the chicken DT40 cell line. Cell 89: 185–193 [DOI] [PubMed] [Google Scholar]
- Blasina A, Price BD, Turenne GA, McGowan CH (1999) Caffeine inhibits the checkpoint kinase ATM. Curr Biol 9: 1135–1138 [DOI] [PubMed] [Google Scholar]
- Boveri T (1914) Zur Frage der Enstehung maligner Tumoren. Jena: Gustav Fischer Verlag [Google Scholar]
- Brinkley BR (2001) Managing the centrosome numbers game: from chaos to stability in cancer cell division. Trends Cell Biol 11: 18–21 [DOI] [PubMed] [Google Scholar]
- Castedo M, Perfettini JL, Roumier T, Yakushijin K, Horne D, Medema R, Kroemer G (2004) The cell cycle checkpoint kinase Chk2 is a negative regulator of mitotic catastrophe. Oncogene 23: 4353–4361 [DOI] [PubMed] [Google Scholar]
- Chan DW, Son SC, Block W, Ye R, Khanna KK, Wold MS, Douglas P, Goodarzi AA, Pelley J, Taya Y, Lavin MF, Lees-Miller SP (2000) Purification and characterization of ATM from human placenta. A manganese-dependent, wortmannin-sensitive serine/threonine protein kinase. J Biol Chem 275: 7803–7810 [DOI] [PubMed] [Google Scholar]
- Cortez D (2003) Caffeine inhibits checkpoint responses without inhibiting the ataxia-telangiectasia-mutated (ATM) and ATM- and Rad3-related (ATR) protein kinases. J Biol Chem 278: 37139–37145 [DOI] [PubMed] [Google Scholar]
- D'Assoro AB, Lingle WL, Salisbury JL (2002) Centrosome amplification and the development of cancer. Oncogene 21: 6146–6153 [DOI] [PubMed] [Google Scholar]
- Doxsey S (2001) Re-evaluating centrosome function. Nat Rev Mol Cell Biol 2: 688–698 [DOI] [PubMed] [Google Scholar]
- Duensing S, Lee LY, Duensing A, Basile J, Piboonniyom S, Gonzalez S, Crum CP, Munger K (2000) The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc Natl Acad Sci USA 97: 10002–10007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukasawa K, Choi T, Kuriyama R, Rulong S, Vande Woude GF (1996) Abnormal centrosome amplification in the absence of p53. Science 271: 1744–1747 [DOI] [PubMed] [Google Scholar]
- Gard DL, Hafezi S, Zhang T, Doxsey SJ (1990) Centrosome duplication continues in cycloheximide-treated Xenopus blastulae in the absence of a detectable cell cycle. J Cell Biol 110: 2033–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin CS, Simpson PJ, Wilson CR, Thacker J (2000) Mammalian recombination-repair genes XRCC2 and XRCC3 promote correct chromosome segregation. Nat Cell Biol 2: 757–761 [DOI] [PubMed] [Google Scholar]
- Hinchcliffe EH, Li C, Thompson EA, Maller JL, Sluder G (1999) Requirement of Cdk2–cyclin E activity for repeated centrosome reproduction in Xenopus egg extracts. Science 283: 851–854 [DOI] [PubMed] [Google Scholar]
- Hinchcliffe EH, Sluder G (2001) Centrosome reproduction in Xenopus lysates. Methods Cell Biol 67: 269–287 [DOI] [PubMed] [Google Scholar]
- Hut HM, Lemstra W, Blaauw EH, van Cappellen GW, Kampinga HH, Sibon OC (2003) Centrosomes split in the presence of impaired DNA integrity during mitosis. Mol Biol Cell 14: 1993–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai M, Tong WM, Sugihara E, Wang ZQ, Fukasawa K, Miwa M (2003) Involvement of poly(ADP-ribose) polymerase 1 and poly(ADP-ribosyl)ation in regulation of centrosome function. Mol Cell Biol 23: 2451–2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsenti E (1999) Centrioles reveal their secrets. Nat Cell Biol 1: E62–E64 [DOI] [PubMed] [Google Scholar]
- Kaufmann WK, Heffernan TP, Beaulieu LM, Doherty S, Frank AR, Zhou Y, Bryant MF, Zhou T, Luche DD, Nikolaishvili-Feinberg N, Simpson DA, Cordeiro-Stone M (2003) Caffeine and human DNA metabolism: the magic and the mystery. Mutat Res 532: 85–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keryer G, Ris H, Borisy GG (1984) Centriole distribution during tripolar mitosis in Chinese hamster ovary cells. J Cell Biol 98: 2222–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraakman-van der Zwet M, Overkamp WJ, van Lange RE, Essers J, van Duijn-Goedhart A, Wiggers I, Swaminathan S, van Buul PP, Errami A, Tan RT, Jaspers NG, Sharan SK, Kanaar R, Zdzienicka MZ (2002) Brca2 (XRCC11) deficiency results in radioresistant DNA synthesis and a higher frequency of spontaneous deletions. Mol Cell Biol 22: 669–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey KR, Jackson PK, Stearns T (1999) Cyclin-dependent kinase control of centrosome duplication. Proc Natl Acad Sci USA 96: 2817–2822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange BM (2002) Integration of the centrosome in cell cycle control, stress response and signal transduction pathways. Curr Opin Cell Biol 14: 35–43 [DOI] [PubMed] [Google Scholar]
- Lau CC, Pardee AB (1982) Mechanism by which caffeine potentiates lethality of nitrogen mustard. Proc Natl Acad Sci USA 79: 2942–2946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle WL, Lutz WH, Ingle JN, Maihle NJ, Salisbury JL (1998) Centrosome hypertrophy in human breast tumors: implications for genomic stability and cell polarity. Proc Natl Acad Sci USA 95: 2950–2955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraldi P, Honda R, Nigg EA (2002) Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53−/− cells. EMBO J 21: 483–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraldi P, Lukas J, Fry AM, Bartek J, Nigg EA (1999) Centrosome duplication in mammalian somatic cells requires E2F and Cdk2–cyclin A. Nat Cell Biol 1: 88–93 [DOI] [PubMed] [Google Scholar]
- Morrison C, Sonoda E, Takao N, Shinohara A, Yamamoto K, Takeda S (2000) The controlling role of ATM in homologous recombinational repair of DNA damage. EMBO J 19: 463–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti A, Moudjou M, Paintrand M, Salisbury JL, Bornens M (1996) Most of centrin in animal cells is not centrosome-associated and centrosomal centrin is confined to the distal lumen of centrioles. J Cell Sci 109 (Part 13): 3089–3102 [DOI] [PubMed] [Google Scholar]
- Piel M, Meyer P, Khodjakov A, Rieder CL, Bornens M (2000) The respective contributions of the mother and daughter centrioles to centrosome activity and behavior in vertebrate cells. J Cell Biol 149: 317–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihan GA, Purohit A, Wallace J, Malhotra R, Liotta L, Doxsey SJ (2001) Centrosome defects can account for cellular and genetic changes that characterize prostate cancer progression. Cancer Res 61: 2212–2219 [PubMed] [Google Scholar]
- Raff JW, Glover DM (1988) Nuclear and cytoplasmic mitotic cycles continue in Drosophila embryos in which DNA synthesis is inhibited with aphidicolin. J Cell Biol 107: 2009–2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder CL, Borisy GG (1982) The centrosome cycle in PtK2 cells: Asymmetric distribution and structural changes in the pericentriolar material. Biol Cell 44: 117–132 [Google Scholar]
- Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, Abraham RT (1999) Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res 59: 4375–4382 [PubMed] [Google Scholar]
- Sarkaria JN, Tibbetts RS, Busby EC, Kennedy AP, Hill DE, Abraham RT (1998) Inhibition of phosphoinositide 3-kinase related kinases by the radiosensitizing agent wortmannin. Cancer Res 58: 4375–4382 [PubMed] [Google Scholar]
- Sato C, Kuriyama R, Nishizawa K (1983) Microtubule-organizing centers abnormal in number, structure, and nucleating activity in x-irradiated mammalian cells. J Cell Biol 96: 776–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N, Mizumoto K, Nakamura M, Ueno H, Minamishima YA, Farber JL, Tanaka M (2000) A possible role for centrosome overduplication in radiation-induced cell death. Oncogene 19: 5281–5290 [DOI] [PubMed] [Google Scholar]
- Shiloh Y (2003) ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 3: 155–168 [DOI] [PubMed] [Google Scholar]
- Sluder G, Begg DA (1983) Control mechanisms of the cell cycle: role of the spatial arrangement of spindle components in the timing of mitotic events. J Cell Biol 97: 877–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluder G, Lewis K (1987) Relationship between nuclear DNA synthesis and centrosome reproduction in sea urchin eggs. J Exp Zool 244: 89–100 [DOI] [PubMed] [Google Scholar]
- Sluder G, Miller FJ, Cole R, Rieder CL (1990) Protein synthesis and the cell cycle: centrosome reproduction in sea urchin eggs is not under translational control. J Cell Biol 110: 2025–2032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluder G, Nordberg JJ (2004) The good, the bad and the ugly: the practical consequences of centrosome amplification. Curr Opin Cell Biol 16: 49–54 [DOI] [PubMed] [Google Scholar]
- Sonoda E, Matsusaka T, Morrison C, Vagnarelli P, Hoshi O, Ushiki T, Nojima K, Fukagawa T, Waizenegger IC, Peters JM, Earnshaw WC, Takeda S (2001) Scc1/Rad21/Mcd1 is required for sister chromatid cohesion and kinetochore function in vertebrate cells. Dev Cell 1: 759–770 [DOI] [PubMed] [Google Scholar]
- Sonoda E, Sasaki MS, Buerstedde JM, Bezzubova O, Shinohara A, Ogawa H, Takata M, Yamaguchi-Iwai Y, Takeda S (1998) Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J 17: 598–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada S, Kelkar A, Theurkauf WE (2003) Drosophila checkpoint kinase 2 couples centrosome function and spindle assembly to genomic integrity. Cell 113: 87–99 [DOI] [PubMed] [Google Scholar]
- Takao N, Kato H, Mori R, Morrison C, Sonada E, Sun X, Shimizu H, Yoshioka K, Takeda S, Yamamoto K (1999) Disruption of ATM in p53-null cells causes multiple functional abnormalities in cellular response to ionizing radiation. Oncogene 18: 7002–7009 [DOI] [PubMed] [Google Scholar]
- Tarapore P, Fukasawa K (2002) Loss of p53 and centrosome hyperamplification. Oncogene 21: 6234–6240 [DOI] [PubMed] [Google Scholar]
- Tutt A, Gabriel A, Bertwistle D, Connor F, Paterson H, Peacock J, Ross G, Ashworth A (1999) Absence of Brca2 causes genome instability by chromosome breakage and loss associated with centrosome amplification. Curr Biol 9: 1107–1110 [DOI] [PubMed] [Google Scholar]
- Vorobjev IA, Chentsov Yu S (1982) Centrioles in the cell cycle. I. Epithelial cells. J Cell Biol 93: 938–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RA, Pan Z, Salisbury JL (2000) GFP-centrin as a marker for centriole dynamics in living cells. Microsc Res Tech 49: 451–457 [DOI] [PubMed] [Google Scholar]
- Wong C, Stearns T (2003) Centrosome number is controlled by a centrosome-intrinsic block to reduplication. Nat Cell Biol 5: 539–544 [DOI] [PubMed] [Google Scholar]
- Xu X, Weaver Z, Linke SP, Li C, Gotay J, Wang XW, Harris CC, Ried T, Deng CX (1999) Centrosome amplification and a defective G2–M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol Cell 3: 389–395 [DOI] [PubMed] [Google Scholar]
- Yamaguchi-Iwai Y, Sonoda E, Sasaki MS, Morrison C, Haraguchi T, Hiraoka Y, Yamashita YM, Yagi T, Takata M, Price C, Kakazu N, Takeda S (1999) Mre11 is essential for the maintenance of chromosomal DNA in vertebrate cells. EMBO J 18: 6619–6629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BB, Chaturvedi P, Spring K, Scott SP, Johanson RA, Mishra R, Mattern MR, Winkler JD, Khanna KK (2000) Caffeine abolishes the mammalian G(2)/M DNA damage checkpoint by inhibiting ataxia-telangiectasia-mutated kinase activity. J Biol Chem 275: 10342–10348 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Methods