

Abstract
The growth factor/receptor pair HGF/c-Met exerts control on proliferation, morphogenesis and motility, and through overexpression and mutation is implicated in cancer. Here we have investigated the relationship between receptor signalling and traffic, and its control by specific PKC isotypes. It is shown that c-Met signalling to the ERK cascade occurs within endosomal compartments and that it is in this compartment that PKCɛ specifically exerts its control on the pathway with the consequent accumulation of ERK in focal complexes. These events are clearly separated from the subsequent microtubule-dependent sorting of c-Met to its perinuclear destination, which is shown to be under the control of PKCα. Thus while it is shown that traffic to endosomes is essential for HGF/c-Met to trigger an ERK response, the subsequent traffic and signalling of c-Met controlled by these two PKC isotypes are unconnected events. The dynamic properties conferred by the PKCɛ control are shown to be essential for a normal HGF-dependent migratory response. Thus PKCs are shown to control both receptor traffic and signal traffic to relay HGF/c-Met responses.
Keywords: c-Met, ERK, PKC, signalling, traffic
Introduction
Growth factor receptors play major regulatory roles in normal cellular processes and their deregulation can have profound effects. Overexpression of c-Met, the receptor for hepatocyte growth factor (HGF), has been observed in a large number of human tumours, correlating closely with metastatic tendency and poor prognosis (Di Renzo et al, 1995; Ghoussoub et al, 1998; Tsarfaty et al, 1999). Furthermore, germline missense mutations of c-Met, which lead to increased tyrosine kinase activity, have been reported in childhood hepatocellular carcinoma (Park et al, 1999). A molecular understanding of how this receptor is switched on and off will provide the basis for developing rational interventions in such situations.
HGF is a mesenchymal-derived cytokine, which controls a wide spectrum of biological events including proliferation, scattering, invasion, branching morphogenesis, transformation and angiogenesis on various epithelial target cells. It is thus a mitogenic, motogenic and morphogenic factor. HGF receptor, c-Met, encoded by the c-met proto-oncogene is a disulphide-linked α/β heterodimer. The β-chain p145met is composed of an extracellular domain, which binds the ligand, a transmembrane domain and an intracellular domain including a tyrosine kinase domain and a multifunctional docking site. The binding of HGF to c-Met triggers phosphorylation of two tyrosine residues (Y1234 and Y1235) in the kinase domain and consequently the phosphorylation of two other tyrosine residues (Y1349 and Y 1356) in the docking site. Several signalling molecules are then directly recruited (and activated) to the docking site including PI3Kinase, src. The recruitment of the adaptors Grb-2 and Gab-1 brings a number of further effectors to the docking site including Ras, PLC-γ, Shc, SHP2, CRKL and PI3Kinase (reviewed in Zhang and Vande Woude, 2003). The PI3Kinase and Ras/MAPK cascades are two major pathways required for HGF-induced invasion and branching morphogenesis.
Although ligand-induced endocytosis was originally thought to be a mechanism of receptor inactivation, recent studies suggest that receptors remain active within endosomes (Daaka et al, 1998; Burke et al, 2001) and that signalling can occur from endosomes (McPherson et al, 2001; Jiang and Sorkin, 2002). Moreover, certain signalling events, such as the activation of ERK, appear to require endocytosis (Vieira et al, 1996; Ceresa et al, 1998; Roy et al, 2002). In view of the critical role played by c-Met in cancer, it is important to know how its internalisation and trafficking are regulated and how these are related to signal output. The internalisation of several membrane receptors is activated by PKC, including the γ-aminobutyric type A receptor and the sst2A somatostatin receptor (Chapell et al, 1998; Hipkin et al, 2000). By contrast, we showed recently that PKC does not influence c-Met internalisation but positively controls c-Met traffic along microtubules towards a perinuclear compartment (Kermorgant et al, 2003). Prior evidence indicates that PKC plays a negative role in controlling c-Met function (Sipeki et al, 2000); however, it is not known how this relates to the traffic of c-Met.
In this study we show that in HeLa cells, two distinct PKC isotypes control post-early endosomal c-Met traffic and endosomal c-Met signalling to MAPK pathways. PKCα positively regulates c-Met traffic from endosomes to a perinuclear compartment and this operates independently of c-Met signalling to MAPK. In a distinct pathway, the c-Met control of ERK is found to be controlled in endosomes by the action of PKCɛ. This input serves to control both the level of the sustained ERK response and its subcellular localisation, with consequent effects on migratory responses. The data thus define dual pathways impinging on c-Met responses, controlling localisation of the receptor and the strength and location of signal output.
Results
PKC inhibition sustains dynamic c-Met-dependent activation of ERK1/2 via MEK1
It was reported previously that broad inhibition of PKC with bisindolylmaleimide 1 (BIM-I) in HepG2 cells leads to a more sustained ERK activation in response to HGF (Sipeki et al, 2000). This behaviour was confirmed here in HeLa cells with both an enhanced acute response and a higher sustained ERK1/2 phosphorylation following HGF treatment in the presence of BIM-I (Figure 1A). For example, after 120 min of HGF stimulation, ERK2 phosphorylation was increased 21.8-fold in the presence of BIM-I as compared to 3.4-fold with HGF alone. This was not due to an aberrant PKC–ERK pathway, since as in other cell types, direct activation of PKCs by phorbol 12-myristate 13-acetate (TPA) stimulates ERK1/2 phosphorylation and this response is inhibited by BIM-I (Figure 1A).
Figure 1.
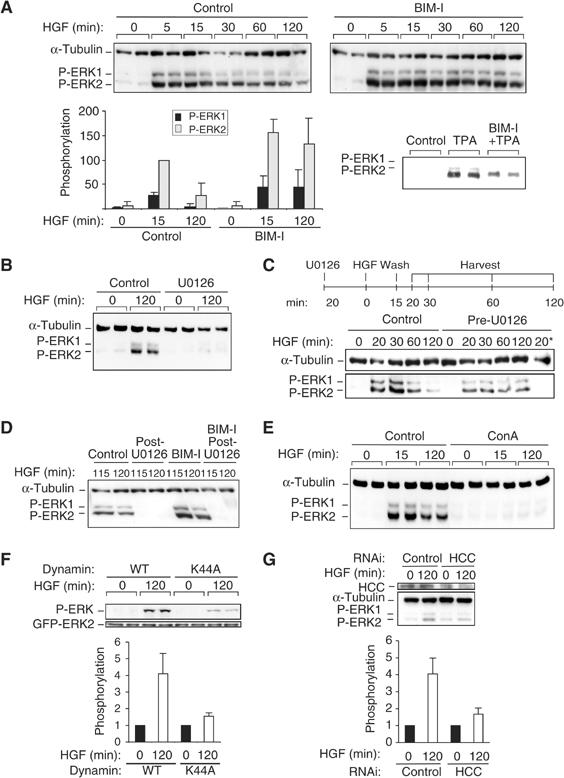
BIM-I sustains dynamic activation of ERK1/2. (A–E) Representative P-ERK1/2 and α-tubulin Western blots. (A) HeLa cells were stimulated with HGF in the absence (control) or presence of BIM-I (1 μM) or TPA (400 nM) or BIM-I plus TPA. Densitometric results are in arbitrary units. (B, C) Cells were stimulated with HGF in the absence (control) or presence of U0126 (10 μM) added 20 min before HGF. In (C) only, after 15 min of HGF exposure, the cells were washed, some fresh medium was replaced and cells were harvested at the indicated time. The last lane, ‘20*', is a control: cells pretreated with U0126 were harvested after 20 min of HGF treatment without washing. (D) Cells pretreated or not (control) for 20 min with BIM-I (1 μM) were treated with HGF. After 110 min, U0126 was added. Cells were harvested 5 or 10 min later (at 115 or 120 min). (E) Cells were pretreated or not (control) with concanavalin A (ConA) (250 μg/ml) before HGF stimulation. (F) Cells cotransfected with GFP-ERK2 and dynamin WT or dominant-negative (K44A) were stimulated with HGF for 0 or 120 min. Representative P-ERK and GFP Western blots are shown. (G) Cells transfected with an RNAi specific to clathrin heavy chain (HCC) were stimulated with HGF for 0 or 120 min. Representative HCC, α-tubulin and P-ERK Western blots are shown. (F, G) Densitometric results are in arbitrary units.
Inhibition of MEK activity with the drug UO126 was sufficient to block HGF-induced ERK1/2 phosphorylation after 15 min (data not shown) and after 120 min (Figure 1B). To assess the dynamics of this HGF signal and its BIM-I sensitivity, cells were pulsed with HGF (15 min) in the presence or absence of UO126 followed by wash-out of HGF and the inhibitor (at 15 min) (Figure 1C). In control cells, HGF-induced ERK1/2 phosphorylation attenuated over the following 1–2 h. The MEK inhibitor UO126 does not inhibit c-Met internalisation and on removal of U0126, post-HGF-induced internalisation, ERK1/2 activation still occurs, that is, via the internalised c-Met compartment. To ensure the effectiveness of HGF wash-out, the medium in contact with cells after wash-out was transferred to fresh cells and no c-Met internalisation was observed in these cells, indicative of effective HGF removal. Similarly by immunostaining for HGF, no extracellular matrix-associated HGF was observed following the pulsed treatment (data not shown). Therefore, the pattern of ERK1/2 phosphorylation in response to pulsed HGF was not due to residual HGF continuing to signal from the cell surface. This indicates that HGF/c-Met retains the capacity to signal to MEK1/ERK following endocytosis; this is effectively complete within 15 min of HGF treatment (see Kermorgant et al, 2003).
To determine whether ongoing phosphorylation of ERK1/2 was maintained during the late, sustained phase of c-Met-induced phosphorylation, UO126 was added 110 min post-HGF. As shown in Figure 1D, this delayed addition of UO126 was sufficient to lead to the complete dephosphorylation of ERK1/2 within 5 min. This indicates that maintenance of the active upstream kinase MEK is essential for maintaining sustained ERK1/2 phosphorylation. Furthermore, inhibition of PKC with BIM-I had no effect on this rapid UO126-induced ERK1/2 dephosphorylation, indicating that the dominant influence of BIM-I is on the upstream c-Met/MEK pathway rather than through inactivation of ERK1/2 phosphatase(s).
The evidence above indicates that HGF can induce ERK1/2 phosphorylation within an endosomal compartment. To assess the requirement for endocytosis to trigger this response, the inhibitor concanavalin A was employed. Blocking endocytosis with this inhibitor (see Supplementary Figure 2D) completely blocked HGF-dependent ERK1/2 phosphorylation (Figure 1E), suggestive of a requirement for endocytosis and entirely consistent with the data above. Consistent with this, expression in HeLa cells of the K44A dominant-negative form of dynamin with GFP-ERK2 significantly reduced the phosphorylation of coexpressed GFP-ERK2 by 2.6-fold (P<0.02) compared to cells coexpressing wild-type (WT) dynamin (Figure 1F). Furthermore, knocking down the heavy chain of clathrin with a specific RNAi reduced HGF-induced endogenous ERK2 phosphorylation by 2.4-fold compared to an RNAi control (Figure 1G).
The phosphorylation of ERK1/2 by HGF correlated with phosphorylation of MEK1 with little change in the constitutive basal MEK2 phosphorylation. This link was also reflected in the BIM-I response, since PKC inhibition was found to enhance selectively the acute HGF-induced phosphorylation of MEK1 but not MEK2 (Supplementary Figure 1A). Consistent with the requirement for endocytosis defined above, no HGF-induced MEK1 phosphorylation is detected in cells pretreated with concanavalin A (Supplementary Figure 1B).
PKC inhibition alters HGF-induced ERK localisation at focal complexes
We observed by immunofluorescence that HGF stimulated ERK1/2 or phospho-ERK1/2 accumulation at plasma membrane structures. The colocalisation with actin (Figure 2Af), vinculin (Figure 2Bf) and paxillin (data not shown) indicated that this ERK localisation corresponded to focal complexes. As previously shown, BIM-I blocks the perinuclear accumulation of c-Met after 120 min of HGF stimulation, leading to a sustained endosomal localisation (Kermorgant et al, 2003) (Figure 2Ag and k). BIM-I also strongly reduced the c-Met-dependent ERK localisation at focal complexes (Figure 2Ai, j and Bk, l), maintaining ERK in the cytoplasm where it partially colocalises with endosomal c-Met (Figure 2Al). These data suggest that BIM-I sustains c-Met-dependent ERK activation by maintaining the ERK cascade in proximity to c-Met and upstream activators in the endosome.
Figure 2.
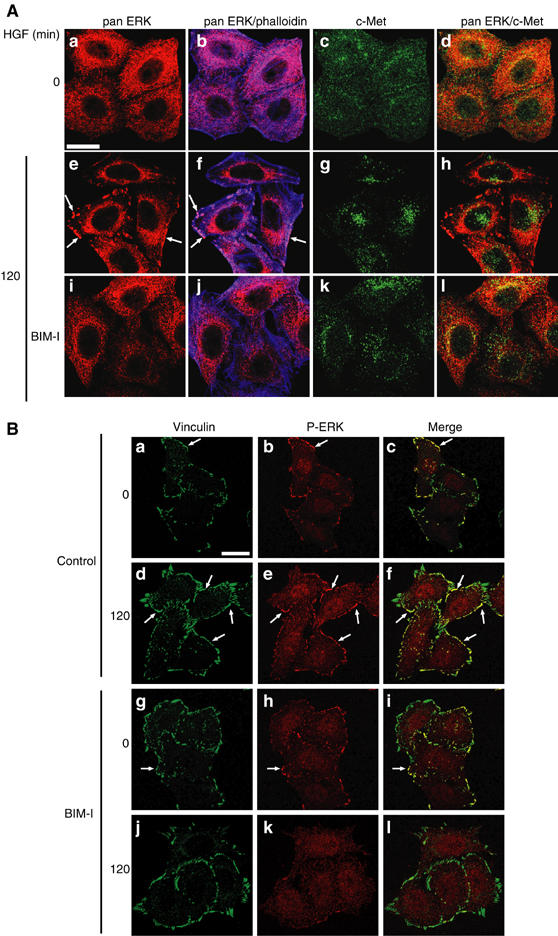
BIM-I inhibits c-Met-dependent ERK translocation to focal complexes. Representative confocal images for (A) pan ERK (red), phalloidin (blue) and c-Met (green), and (B) vinculin (green) and P-ERK (red). The arrows show examples of colocalisation. Bar, 20 μm. HeLa cells were stimulated for 0 or 120 min with HGF alone or with BIM-I.
PKC inhibition has selective effects on c-Met signals
To investigate if BIM-I action was specific to c-Met–ERK signalling or whether it could influence other c-Met signals, we investigated the effects of BIM-I on other downstream signals of c-Met. BIM-I was found not to increase c-Met-dependent STAT-3 phosphorylation (Figure 3A); however, it did enhance and sustain the phosphorylation of the MAPK family protein JNK. For example, following 120 min of HGF treatment, JNK phosphorylation was increased six-fold in the presence of BIM-I as compared to 1.8-fold without BIM-I (Figure 3B). c-Met internalisation seems to be required for c-Met-dependent JNK activation as treatment of cells with concanavalin A inhibited c-Met-dependent JNK phosphorylation (Figure 3C). Interestingly, MEK inhibition by U0126 also blocked c-Met-dependent JNK phosphorylation, indicating that HGF activates JNK through a MEK–ERK pathway (Figure 3D). In parallel to the JNK response, BIM-I also sustained the c-Met-dependent phosphorylation of the JNK downstream effector c-Jun. The activation of c-Jun by HGF was transient, and reached a peak at 15 min of stimulation (data not shown) and was back close to the basal level at 120 min. The number of phospho-c-Jun-positive cells at 120 min of HGF stimulation increased from 13.6 to 62.3% in the presence of BIM-I (Figure 3E).
Figure 3.
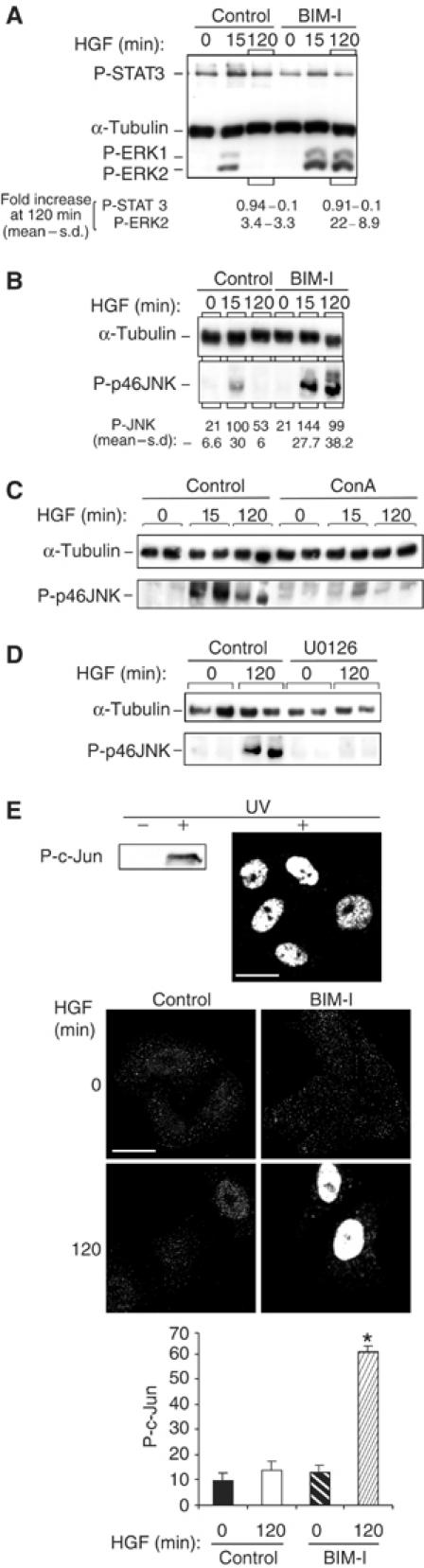
BIM-I does not modify STAT3 phosphorylation but sustains dynamic activation of JNK and c-Jun. (A) Representative P-STAT3 (PY705), α-tubulin and P-ERK1/2 Western blots from cells stimulated for 0 or 120 min with HGF alone or with BIM-I. Densitometric results are in fold increase. (B–D) Representative P-JNK and α-tubulin Western blots. (B) HeLa cells were stimulated with HGF in the absence (control) or presence of BIM-I (1 μM). The densitometric results are in arbitrary units. (C, D) Cells were pretreated or not (control) with concanavalin A (ConA) (250 μg/ml) (C) or U0126 (10 μM) (D) before HGF stimulation. (E) Representative Western blot and confocal medial sections for P-c-Jun. Bar, 20 μm. HeLa cells were UV treated (100 μJ) (positive control) or stimulated with HGF alone (control) or in the presence of BIM-I (1 μM). For each experiment, the ratio of P-c-Jun-positive nuclei was counted in 10 random fields of 10 cells (*P<0.05). The graph is in arbitrary units and is the mean of three independent experiments.
PKC inhibition sustains signalling of internalised c-Met independently of trans-cytosolic traffic
By the use of the PKC inhibitor BIM-1, the PKC activator TPA and the microtubule-disrupting agent vinblastine, we showed previously that PKC positively controls the trans-cytosolic movement of c-Met along microtubules from an early endosomal compartment to a perinuclear compartment (Kermorgant et al, 2003). These BIM-I and vinblastine effects are visible here where after 120 min of HGF stimulation, c-Met vesicles are dispersed in the cell instead of accumulating around the nucleus (Figure 4A (top panels), see also Figure 2, and see Supplementary Figure 2A and B (left panels) for a detailed analysis).
Figure 4.
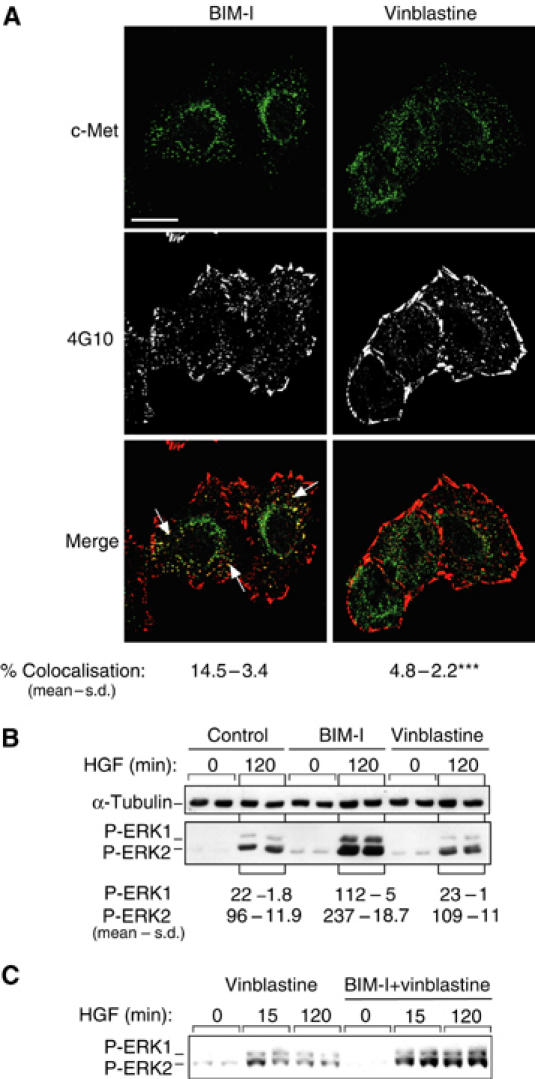
BIM-I sustains endocytic signalling from c-Met independent of trans-cytosolic traffic. (A, B) HeLa cells were stimulated by HGF in the presence of BIM-I (1 μM) or vinblastine (1 μM). (A) Representative confocal projections of five Z-sections for c-Met (green) and 4G10 (white or red). Bar, 20 μm. The numbers represent the statistical analysis of the colocalisation between c-Met and 4G10; *P<0.0001 (see Materials and methods). (B, C) Representative P-ERK1/2 and α-tubulin Western blots. Densitometric results are in arbitrary units (*P<0.01). Cells were stimulated by HGF in the presence or not (control) of BIM-1 (1 μM) or vinblastine (1 μM) alone or vinblastine and BIM-I (1 μM).
To investigate the basis of the sustained signal output from c-Met and its response to PKC inhibition, HGF-induced tyrosine phosphorylation in parallel to the endocytosis/traffic of c-Met was monitored, employing immunofluorescence with the antiphosphotyrosine antibody 4G10, frequently used to analyse the tyrosine phosphorylation of c-Met and/or of c-Met downstream effectors. Within 5 min of treatment, HGF induced the accumulation of phosphotyrosine in cytoplasmic vesicles, which overlapped with endocytosed c-Met (Supplementary Figure 2Ad–f). Both the abundance of cytoplasmic phosphotyrosine and its coincidence with the increasingly perinuclear c-Met were diminished at 30 and 120 min post-HGF treatment (Supplementary Figure 2Ag–l). By contrast, cells pretreated with BIM-I showed sustained vesicular phosphotyrosine (Figure 4 (left middle panel) and Supplementary Figure 2Ah, k and 2Bb, e), which substantially overlapped with the maintained vesicular c-Met (Figure 4 (left bottom panel) and Supplementary Figure 2Bc, f). In the presence of concanavalin A, which blocks c-Met internalisation (Supplementary Figure 2C), 15 min of HGF stimulation did not induce the accumulation of phosphotyrosine in cytoplasmic vesicles.
Pretreatment with BIM-I and concanavalin A neither induced cytoplasmic phosphotyrosine nor modified the phosphotyrosine detected at the plasma membrane (Supplementary Figure 2Ch). Similarly, BIM-I did not induce ERK1/2 phosphorylation on cells pretreated with concanavalin A and stimulated with HGF (data not shown). Thus the effects of BIM-I are restricted to signals from the endocytosed c-Met. When the heavy chain of clathrin was knocked down by transient transfection with a specific RNAi oligonucleotide, HGF was still able to stimulate the phosphorylation of the kinase domain of c-Met (Supplementary Figure 2D). Nevertheless, the phosphorylation was two-fold less at 15 min and four-fold less at 120 min as compared to cells transfected with an RNAi control. These results suggest that c-Met internalisation contributes to the maintenance of its full activation and to sustained ERK phosphorylation.
The sustained signalling to MEK1–ERK1/2 and JNK–c-Jun through c-Met suggests that PKC might control this response by controlling c-Met traffic as opposed to effector uncoupling per se. PKC could effectively attenuate c-Met signalling by promoting its exit from endosomal compartments to further compartments where its activity would be reduced. To monitor the relationship between the effects on traffic and those on signalling and to relate these to ERK1/2 phosphorylation, the effects of BIM-I and vinblastine on c-Met/phosphotyrosine were compared. In opposition to BIM-I, vinblastine caused only the accumulation of c-Met-positive, 4G10-negative vesicles. The quantitation of c-Met (green) and 4G10 (red) colocalisation in the presence of BIM-I or vinblastine indicates that the difference is highly significant (P<0.0001; Figure 4A). In keeping with this dissociation of traffic and signal attenuation, vinblastine had no effect on HGF-induced ERK1/2 phosphorylation (Figure 4B). Furthermore, BIM-I still increased HGF-induced ERK phosphorylation even in the presence of vinblastine (Figure 4C). These results indicate that blocking c-Met traffic between the early endosome and the perinuclear compartment is not sufficient to modify its signalling. A dissociation must exist between the PKC influence on trans-cytosolic traffic of c-Met and its effect on signalling to the ERK1/2 pathway, which must therefore be independent.
PKCα and PKCɛ control traffic of c-Met at two different steps
The dissociation of these two BIM-I-sensitive properties (traffic and signalling) suggested that distinct PKC species might be involved. They could belong to either the classical and/or the novel class of PKC since BIM-I can inhibit their activities. We therefore aimed to identify first which PKC controls c-Met traffic. We determined that HeLa cells express the PKCα, ɛ and δ isotypes (see Figure 6A) and observed by immunofluorescence that PKCα and PKCɛ partially colocalise with endosomal c-Met (see Supplementary Figure 3 for PKCɛ). As shown previously (Kermorgant et al, 2003), HGF treatment of HeLa cells induces acute endocytosis and a delayed perinuclear accumulation of c-Met (Figure 5A, left panels). We show here that, like BIM-I, another PKC inhibitor, Gö6976, selective for classical PKC isotypes α and β1 (Martiny-Baron et al, 1993), has no effect on the acute HGF-induced endocytosis of c-Met, but blocks the traffic of c-Met to the perinuclear compartment (Figure 5A, right-hand panels). The quantitation of this effect, as indicated in Materials and methods and as previously described (Kermorgant et al, 2003), showed that it is significant (see Table I). Although a classical PKC appears to control c-Met traffic, it could not attenuate c-Met signalling responses through controlling c-Met degradation, because Gö6976 does not affect HGF-induced c-Met degradation (Figure 5B). Some colocalisation was detected between vesicular c-Met and transfected WT GFP-PKCα (data not shown).
Figure 6.
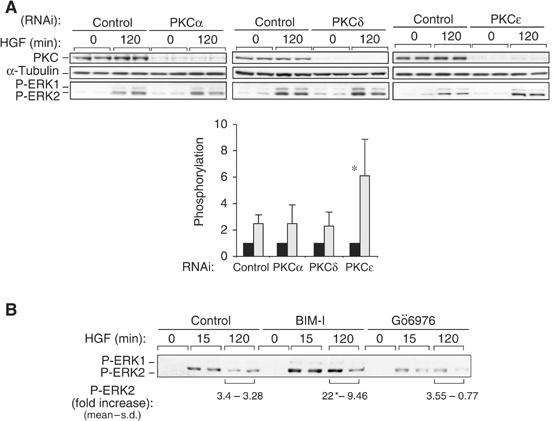
PKCɛ but not PKCα controls c-Met-dependent ERK activation. (A) HeLa cells transfected with RNAi control or RNAis specific to PKCα, δ or ɛ were stimulated with HGF for 0 or 120 min. Western bots were performed for each PKC, α-tubulin and P-ERK. The densitometric results for P-ERK2 are in arbitrary units (*P<0.05). (B) HeLa cells were pretreated or not (control) with BIM-I (1 μM) or Gö6976 (1 μM) for 10 min and then stimulated with HGF for 120 min. Representative P-ERK1/2 Western blots and the densitometric results in fold increase are shown.
Figure 5.
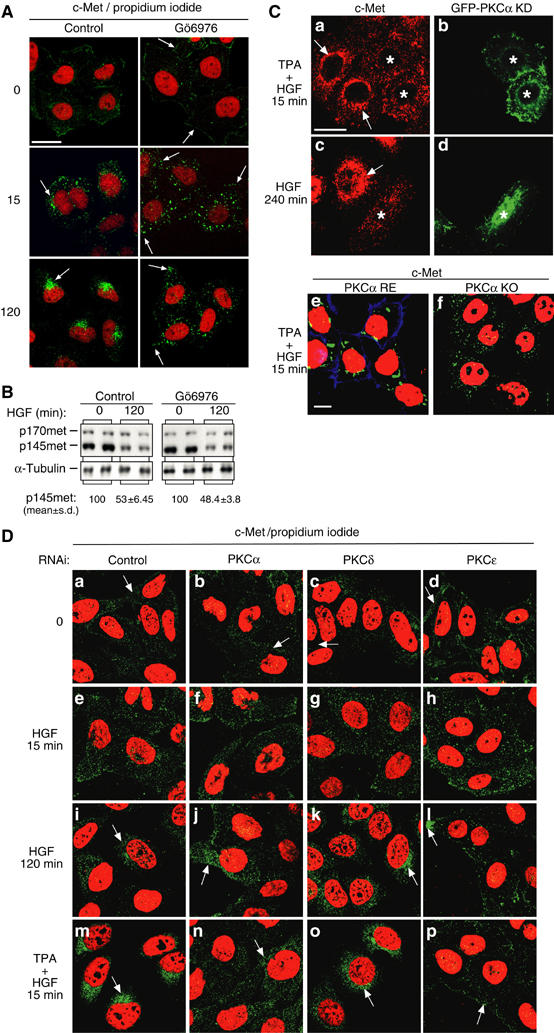
PKCα controls traffic of c-Met. (A–C) HeLa cells were pretreated or not (control) with Gö6976 (1 μM) for 10 min and stimulated with HGF. (A) Representative basomedial confocal images for c-Met (green) and propidium iodide (red). Bar, 20 μm. (B) Representative c-Met and α-tubulin Western blots. Densitometric results are in arbitrary units. (C) Representative medial confocal pictures for c-Met (red for (a, c); green for (e, f)). Bar, 20 μm. HeLa cells transfected with a kinase-inactive GFP-PKCα mutant (GFP-PKCα KD) were treated for 10 min with TPA plus 15 min with HGF (a, b) or with HGF alone for 240 min (c, d). * Indicates transfected cells. (e, f) Mouse embryonic fibroblasts knocked out for PKCα (PKCα KO) or re-expressing PKCα were treated for 10 min with TPA plus 15 min with HGF. (D) Representative confocal images for c-Met (green) and propidium iodide (red). Bar, 20 μm. HeLa cells transfected with RNAi control (a, e, i, m) or RNAis specific to PKCα (b, f, j, n), PKCδ (c, g, k, o) or PKCɛ (d, h, l, p) were stimulated with HGF alone for 0, 15 or 120 min or for 10 min with TPA plus 15 min with HGF.
Table 1.
Effect of Gö6976 on relative distances of c-Met-positive vesicles from the cell boundary to the nucleus boundary
| Vesicle movement | ||||
|---|---|---|---|---|
| Treatment | Time (min) | Relative distance (mean±s.d.) | Number of cells | P-Value (ANOVA) |
| HGF | 15 | 0.65±0.04 | 29 | |
| HGF | 120 | 0.76±0.10 | 31 | + |
| Gö6976 | 15 | 0.54±0.05 | 99 | ** |
| Gö6976 | 120 | 0.61±0.06 | 84 | * |
| Treated cell cultures were preincubated for 10 min with Gö6976 before addition of HGF. Analysis of variance (ANOVA) was used for comparison of the cells incubated with HGF only, for 15 and 120 min. P<0.05 (* or +), P<0.001 (**). + indicates a significant difference compared to HGF, 5 min. | ||||
We previously showed that the perinuclear accumulation of c-Met is promoted by either a short TPA pretreatment in combination with an acute (15 min) HGF stimulation or a long-term (120–240 min) HGF stimulation alone (Kermorgant et al, 2003). However, cells transfected with a kinase-inactive GFP-cPKCα (GFP-PKCα KD) failed to display either acute TPA+HGF- (Figure 5Ca and b) or long-term HGF (Figure 5Cc and d) stimulation-induced perinuclear accumulation of endocytosed c-Met. To confirm the selective nature of the effect of cPKCα on c-Met traffic, we monitored c-Met localisation in PKCα knockout or re-expressing mouse embryo fibroblasts (Srivastava et al, 2002), which express c-Met mRNA and protein (data not shown). TPA+HGF did not induce a perinuclear accumulation of c-Met in the knockout cells as compared to the re-expressing cells (Figure 5Ce and f). These results show that PKCα is responsible for the positive control of c-Met trans-cytosolic traffic.
In order to confirm these results and to investigate the potential modification of c-Met traffic by other PKCs, we then analysed the influence of knock-down of PKCα, δ or ɛ on c-Met traffic by RNAi. These PKCs were not detectable by Western blot in cells transfected with the specific RNAi oligonucleotides as compared to cells transfected with a control RNAi (Figure 6A). In cells knocked down for PKCs as in cells transfected with the RNAi control, c-Met was predominantly expressed at the plasma membrane and HGF triggered its internalisation as shown here after 15 min (Figure 5Da–h). We first confirmed that PKCα promotes the HGF-dependent perinuclear accumulation of c-Met. This is the case for long-term HGF stimulation or short-term HGF+TPA stimulation (Figure 5Dj and n). Second, the knock-down of PKCδ does not modify c-Met traffic (Figure 5Dk and o). Third, in cells knocked down for PKCɛ, the HGF-induced (long term or short term plus TPA) perinuclear accumulation of c-Met is blocked (Figure 5Dl and p). It is of interest to note that in cells knocked down for PKCɛ, internalised c-Met vesicles remain very close to the plasma membrane unlike in cells knocked down for PKCα. In addition, under basal conditions, PKCɛ knock-down cells displayed a stronger c-Met localisation at the plasma membrane and a weaker localisation in the cytoplasm than cells transfected by the RNAi control or knocked down for PKCα or PKCδ RNAis. These observations indicate that PKCɛ may negatively control c-Met recycling to the plasma membrane.
Altogether, these results indicate that PKCα controls c-Met traffic from the early endosome to a perinuclear compartment and that PKCɛ would regulate c-Met traffic upstream of PKCα by controlling the recycling of internalised c-Met to the plasma membrane.
PKCɛ but not PKCα controls c-Met signalling
In order to investigate which PKC controls internalised c-Met signalling, we analysed the effect of knocking down PKCα, δ or ɛ on HGF-dependent ERK1 and 2 phosphorylation. The loss of PKCɛ led to a significantly enhanced HGF-dependent ERK phosphorylation (by 6.1-fold for ERK2, P<0.05) with no change on loss of the other PKCs (2.3- to 2.5-fold of stimulation for RNAi control, PKCα and δ) (Figure 6A). This indicates that PKCɛ selectively controls c-Met endosomal signalling. Further evidence that PKC-controlled traffic to the perinuclear compartment and signalling to ERK were unrelated events came from the observation that Gö6976 did not affect HGF-induced ERK1/2 phosphorylation (Figure 6B). Consistent with this, the phosphotyrosine association with c-Met was not enhanced with Gö6976 (data not shown) as seen for vinblastine (see above). Thus despite effects on trans-cytosolic traffic, PKCα inhibition does not influence the coupling of c-Met to MAPK.
PKCɛ knockout mouse embryo fibroblasts displayed an elevated basal ERK1/2 phosphorylation compared to PKCɛ-expressing cells, confounding an unequivocal analysis of HGF responses (data not shown). Notwithstanding this limitation, treatment of PKCɛ−/− cells with BIM-I had no effect on the HGF induction of ERK phosphorylation. By contrast, a positive effect of BIM-I similar to that in HeLa cells was detected in PKCɛ+/+ cells (data not shown). This is consistent with PKCɛ dominating the PKC control of c-Met>ERK signalling, which occurs in the endosome. Indeed, as mentioned above, PKCɛ, endogenous or expressed as a GFP fusion protein, partially colocalises with internalised c-Met (Supplementary Figure 3).
Similar results on HGF-dependent c-Jun activation were observed. PKCɛ knock-down led to significantly enhanced HGF-dependent phosphorylation of c-Jun (by 2.7-fold, P<0.0005; Figure 7A). Knock-down of PKCα (Figure 7A) or PKCδ (data not shown) had no effect; Gö6976 inhibition did not affect c-Jun phosphorylation (Figure 7B). We also employed single cell assays to compare the HGF-dependent activation of c-Jun in HeLa cells transiently transfected with WT or kinase-inactive mutants (KD) of GFP-PKCα, δ or ɛ constructs. In each case, in cells stimulated by HGF or not (control), the proportion of GFP-PKC-transfected cells positive for phospho-c-Jun has been determined as a function of the empty EGFP vector control. HGF stimulates c-Jun activation by 4.4-fold (P<0.001) in cells expressing GFP-PKCɛ KD; expressions of GFP-PKCɛ WT, GFP-PKCα or δ, WT or KD, do not induce significant differences between HGF stimulation and control (Figure 7C). Thus PKCɛ and not PKCα negatively controls c-Met-dependent c-Jun phosphorylation.
Figure 7.
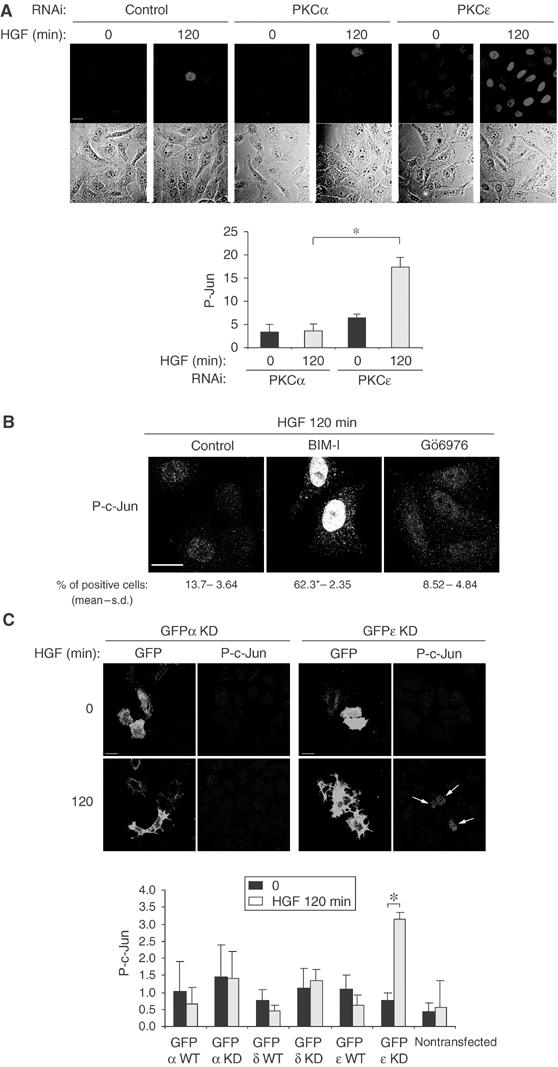
PKCɛ controls c-Met-dependent activation of c-Jun. (A) Representative confocal images for the same fields show P-Jun and phase. Bar, 20 μm. HeLa cells transfected with RNAi control or RNAis specific to PKCα or ɛ were stimulated with HGF alone for 0 or 120 min, fixed and stained for P-Jun. The graph represents the intensity of P-Jun in arbitrary units measured on 100 cells for each condition. (B) Representative basomedial confocal images for P-c-Jun. Bar, 20 μm. HeLa cells were pretreated or not (control) with BIM-I (1 μM) or Gö6976 (1 μM) for 10 min and stimulated with HGF for 120 min. The number of positive nuclei was determined as in Figure 3. (C) HeLa cells were transfected with GFP-PKCα, δ or ɛ constructs, WT or kinase-inactive (KD) or an empty EGFP vector. After 24 h in 0.1% FBS medium, the cells were stimulated or not with HGF, fixed and stained for P-c-Jun. A total of 10 confocal medial sections (10 cells/section) were randomly acquired. The ratio of transfected cells positive for P-c-Jun was obtained. The value obtained for each PKC construct was divided by the value obtained with empty GFP vector. Representative confocal sections for GFP-PKCɛ KD and GFP-PKCα KD are shown. Bar, 20 μm. The graph represents the P-Jun activation for each GFP-PKC at 0- and 120-min HGF stimulation; data are derived from three independent experiments (*P<0.001). For each coverslip, the proportions of nontransfected cells positive for P-c-Jun have been counted and represent an internal control. The last column represents this control for GFP-PKCɛ KD.
PKCɛ controls c-Met-dependent ERK translocation to focal complexes
Gö6976 does not inhibit the HGF-dependent accumulation of phosphorylated ERK at focal complexes (Figure 8A). To distinguish more specifically PKC isotype functions, transient transfections with the different PKC constructs were employed to determine whether effects on ERK1/2 activation were associated with altered HGF-dependent ERK1/2 localisation at the plasma membrane. HGF was found not to induce ERK1/2 translocation to focal complexes in cells expressing GFP-PKCɛ KD. Cells expressing GFP-PKCɛ WT, GFP-PKCα or δ KD (Figure 8B) or GFP-PKCα or δ WT (data not shown) showed an unchanged translocation of ERK as compared to surrounding untransfected cells. This altered ERK location precisely mirrors the behaviour observed with BIM-I, indicative of the key role played by PKCɛ in this response. In addition, no role of PKCα was detected. Figure 8C shows that HGF induces ERK translocation to focal complexes when PKCα is knocked down but not in the case where PKCɛ is lost. Thus, the c-Met–ERK pathway control and the c-Met-dependent ERK localisation at focal complexes appear to be related since they are both promoted by PKCɛ (see Discussion).
Figure 8.
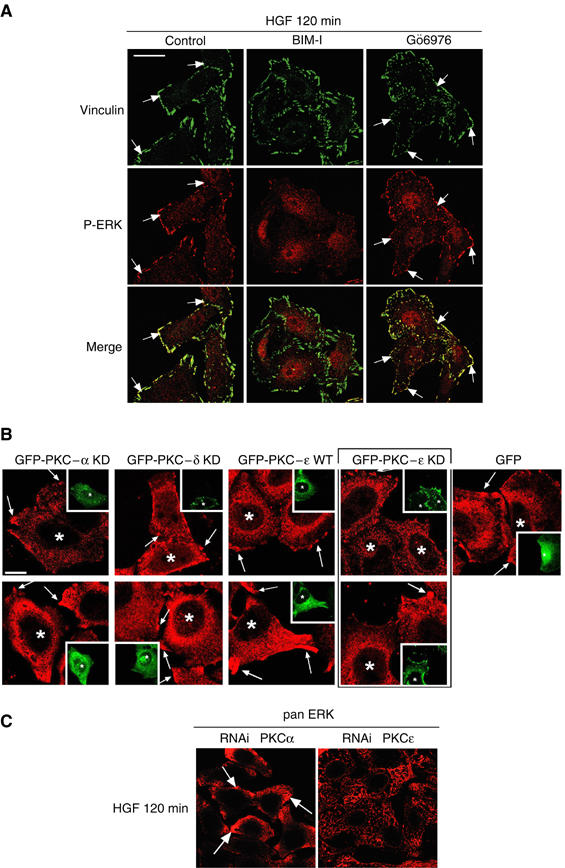
PKCɛ controls c-Met-dependent ERK translocation to focal complexes. (A) Representative confocal images for vinculin (green) and P-ERK (red). The arrows indicate examples of colocalisation. Bar, 20 μm. HeLa cells were pretreated or not (control) with BIM-I (1 μM) or Gö6976 (1 μM) for 10 min and stimulated with HGF for 120 min. (B) HeLa cells were transfected and HGF treated as indicated in Figure 7C and stained for pan ERK (red). Representative medial confocal sections are shown for PKCα and δ KD, PKɛ WT and KD and GFP alone. Bar, 10 μm. The arrows indicate examples of pan ERK localisation at the plasma membrane. * Indicates transfected cells. (C) Confocal images for pan ERK of HeLa cells transfected with RNAis control or specific to PKCɛ and stimulated with HGF for 120 min. Bar, 20 μm.
HGF-dependent cell migration is perturbed in cells defective in PKCɛ
Despite the reduced steady-state level of HGF-induced ERK1/2 phosphorylation associated with a functional PKCɛ, the focal complex association of ERK1/2 observed under these conditions suggested that this plays a role in migratory responses to HGF. To determine such a role, wounding assays were performed in PKCɛ knockout mouse embryo fibroblasts (PKCɛ KO) or the same cells in which PKCɛ has been reintroduced (PKCɛ RE clone 5) (Ivaska et al, 2002). ERK1/2 phosphorylation was found to be essential for HGF-induced migration since both cell types were sensitive to U0216 (data not shown). Evidence that the dynamic shift in ERK1/2 phosphorylation and/or localisation is critical in the migratory response was provided by the observation that the wound healing stimulated by HGF was substantially delayed in PKCɛ KO cells as compared to the PKCɛ replete cells as illustrated in Figure 9A. The wound was closed at 30 h of HGF treatment for PKCɛ-expressing cells and was still not fully closed after 72 h for PKCɛ KO cells (data not shown). The specificity of the HGF effect was confirmed by the preincubation of both cell types with an inhibitory anti-HGF antibody, which strongly inhibited migration (data not shown). Similar results were obtained in HeLa cells (although these cells migrate slower than fibroblasts) knocked down for PKCɛ versus transfected with the control RNAi. The loss of PKCɛ resulted in a slower HGF-dependent cell migration (Figure 9B). Thus ERK1/2 phosphorylation is required for HGF-induced migration and this is positively regulated by PKCɛ influencing its localisation.
Figure 9.
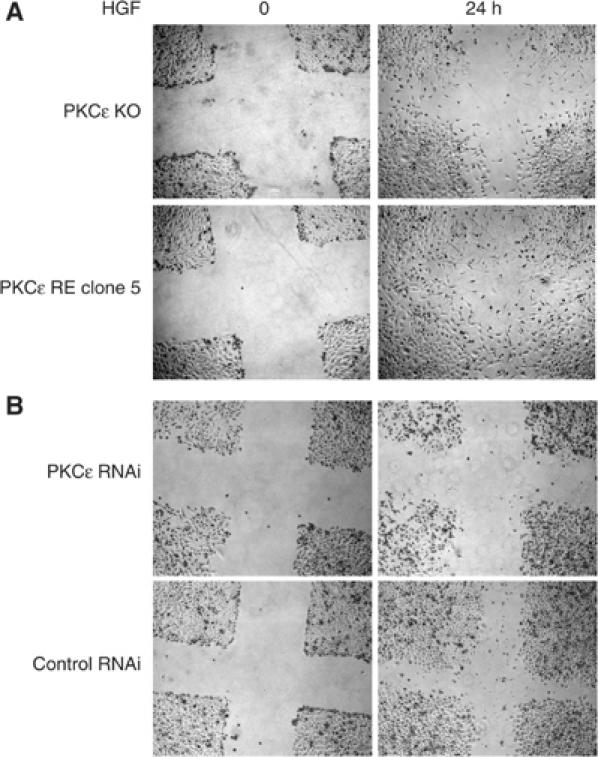
Confluent cells in 0.5% FBS medium for 24 h were wounded with a tip, washed and replaced with 0.5% FBS medium containing HGF. The same wound was photographed at 0 and 24 h. (A) Mouse embryo PKCɛ knockout (PKCɛ KO) or re-expressing (PKCɛ RE, clone 5). (B) HeLa cells transfected with RNAis specific to PKCɛ or control.
Discussion
The endocytosis of ligand-activated receptors has been considered to be the initiating step in their desensitisation. Recent studies, however, have provided evidence that this removal from the plasma membrane may not of itself cause signal attenuation; on the contrary, there is growing evidence that receptors remain competent to signal in endosomal compartments (Xue and Lucocq, 1998) and that the nature of signal output is distinct in these compartments (Daaka et al, 1998; Alves dos Santos et al, 2001). Moreover, the idea is emerging that receptor signalling is regulated by internalisation and intracellular trafficking. Thus, the blocking of receptor internalisation decreases receptor-dependent ERK1/2 activation. This has been shown for tyrosine kinase receptors such as EGFR (Vieira et al, 1996), insulin receptor (Ceresa et al, 1998), IGF-1 receptor (Chow et al, 1998) and TrKA receptor (Howe et al, 2001). The TGFβ response may also require internalisation of the TGFβ receptor in endosomes (Hayes et al, 2002).
Here it is shown that c-Met-dependent ERK1/2 phosphorylation occurs and is sustained in endosomes. Direct blockade of internalisation suppresses ERK1/2 phosphorylation. Furthermore, inhibition of MEK, an hour or more after HGF has induced c-Met internalisation, also blocks ERK1/2 phosphorylation. High and sustained phosphorylation of c-Met itself seems to require internalisation. In combination with the evidence for sustained phosphotyrosine in c-Met-positive endosomes in association with sustained ERK1/2 phosphorylation, it is concluded that the coupling of signals downstream of HGF/c-Met remains intact in the endosome, is required for sustained signal output and as evidenced here is the target for the control of c-Met signals by PKCɛ (Figure 10). The role of PKCɛ is defined by manipulation of its expression/function and is clearly distinguished from the demonstrated effects of PKCα on the microtubule-based movement of c-Met from an early endosomal compartment to a perinuclear compartment. The use of PKC isotype class-selective inhibitors and of RNAis specific to each PKC isotype independently confirms this conclusion. The endosomal location of the PKCɛ regulatory event is indicated by the finding that BIM-I does not influence (the lack of) HGF-dependent ERK1/2 phosphorylation or endosomal phosphotyrosine when c-Met internalisation is blocked by concanavalin A. In addition, PKCɛ/BIM-I can exert its influence under conditions where c-Met is retained in endosomal compartments by prevention of its traffic to the perinuclear compartment (i.e. in the presence of vinblastine). Finally, GFP-PKCɛ itself is partially colocalised to the c-Met-positive endosomal compartment, indicative of its action in this location.
Figure 10.
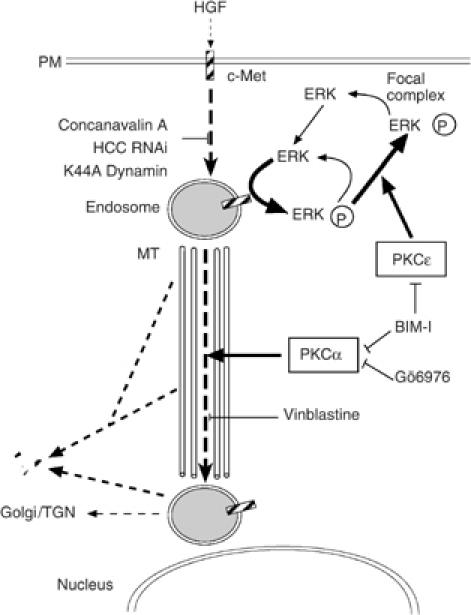
Scheme of PKC, c-Met controls. The figure illustrates the elements in the HGF-induced c-Met pathway under the control of PKC (see Discussion).
Associated with increased steady-state ERK activation under conditions of PKCɛ inhibition or loss, there is a loss of immunoreactive phospho-ERK1/2 at focal complexes. This indicates that one element of PKCɛ action is to facilitate the accumulation of active ERK1/2 at focal complexes. It is entirely possible that PKCɛ control of ERK1/2 location is sufficient to confer the observed altered steady-state phosphorylation of the protein. The finding that the dephosphorylation of ERK1/2 following addition of U0216 is not inhibited by BIM-I indicates that the process of uncoupling is directed at the upstream pathway. Hence if location per se dominates the maintenance of active ERK1/2, it is a function of MEK (or upstream) accessibility rather than ERK phosphatase exposure.
Activation of the JNK pathway by HGF/c-Met has been reported to be essential for transformation by the met oncogene (Rodrigues et al, 1997). It is notable that in HeLa cells, in response to HGF, the activation of JNK is sensitive to U0216. This is not a general property of the MEK inhibitor U0216 and there appears to be an as yet undefined link between the pathways in these cells. It is also apparent that PKC family proteins are linked in very different ways to the triggering of ERK1/2 activation in response to different stimuli. Thus while BIM-I enhances c-Met>ERK coupling through the control exerted by PKCɛ, the characteristic phorbol ester-induced phosphorylation of ERK1/2 is inhibited by BIM-I. This is consistent with the reports that HGF activates PKC and ERK1/2 through parallel pathways and that the activation of ERK in response to HGF does not depend on PKC (Awasthi and King, 2000; Sipeki et al, 2000).
We identify PKCα as the PKC isotype responsible for promoting post-early endosome c-Met traffic. We showed previously that upon ligand stimulation, c-Met is rapidly internalised and traffics towards a perinuclear compartment. This trans-cytosolic movement is microtubule dependent and is promoted by TPA and inhibited by BIM-I. Gö6976, an inhibitor more selective for the classical isotypes α and β1 or the knock-down of PKCα, is shown here to block this traffic. Moreover, in HeLa cells overexpressing a kinase-inactive GFP-PKCα and in PKCα knockout mouse fibroblasts, neither acute TPA+HGF nor long-term HGF stimulated perinuclear accumulation of endocytosed c-Met. PKCα itself has been shown to traffic to a perinuclear compartment in response to its activation at the plasma membrane by phorbol ester (Prevostel et al, 2000). In response to HGF, there is a partial colocalisation of GFP-PKCα with c-Met in endosomes, indicating that these proteins may traffic to the perinuclear compartment in a similar PKCα activity-dependent fashion.
The initial hypothesis tested here was that the enhancement of c-Met signalling by the PKC inhibitor BIM-I was a direct consequence of the blocking of post-early endosomal receptor traffic: the retention of the receptor in the endosome leading to more sustained signals. Unexpectedly, the evidence is that the control by PKC of c-Met signalling is not a direct consequence of the control of post-early endosomal c-Met traffic itself. This is evident when comparing the effects of the microtubule-disrupting agent vinblastine and those of BIM-I on c-Met: both block c-Met trans-cytosolic traffic but unlike BIM-I, vinblastine does not modify the HGF-dependent c-Met signalling to ERK1/2. Similarly, Gö6976 inhibits c-Met traffic, but affects neither HGF-dependent c-Met signals nor c-Met degradation. Finally, the expression of a kinase-inactive GFP-PKCα does not modify HGF-dependent c-Met signals.
The localisation of active ERK at focal complexes has been reported previously and has implications in the regulation of adhesion, the cytoskeletal network and cell motility (Fincham et al, 2000). We show here that HGF stimulates the translocation of ERK1/2 to membrane ruffles corresponding to focal complexes since they colocalise with actin, vinculin and paxillin (see also Ishibe et al, 2003). PKCɛ positively controls HGF-dependent translocation of ERK to focal complexes, perhaps in a process related to its control of recycling β-integrins (Ivaska et al, 2002). The consequent colocalisation with paxillin may well underlie the ERK1/2 localisation requirement since phosphorylation events between paxillin and ERK are implicated in HGF-dependent cell spreading, adhesion (Liu et al, 2002) and epithelial morphogenesis (Ishibe et al, 2003). A likely mechanism is that PKCɛ progressively dissociates the c-Met>ERK cascade in the endosome, and the consequent release of ERK and its accumulation at focal complexes would contribute to the motogenic responses to HGF. The consequent dissociation from the activating input and accumulation at focal complexes appear sufficient to shift the steady-state level of ERK1/2 phosphorylation. This relationship between the apparent c-Met attenuation of signal and the stimulation of a cell function may appear contradictory, especially since sustained signals from c-Met are known to be involved in cell functions; sustained c-Met-dependent ERK signalling is essential for promoting DNA proliferation in mammary myoepithelial cells (Sergeant et al, 2000) and for disassembly of adherens junctions, one of the early steps in cell dissociation (Potempa and Ridley, 1998). It is also notable that HGF induces a long-term activation of ERK1/2 in the course of cell scattering in contrast to EGF, which fails to induce scattering and stimulates the activation of the ERK cascade for only a short time (Sipeki et al, 1999). Nevertheless, the results here demonstrate that while PKCɛ exerts an apparently negative effect upon ERK1/2 activation, it appears that it is the control of localisation that is critical to the PKCɛ input affecting the ability of HGF to induce efficient migration, the effect on steady-state phosphorylation simply being a consequence of this altered location.
In conclusion, it is established that two distinct PKC isotypes control two properties of c-Met, namely post-early endosomal traffic and signal output. These are shown to operate selectively and not to be interdependent. The consequence of these regulatory events is that the PKCɛ input to the c-Met>ERK pathway has a dominant effect on the localisation of this signalling output conferring HGF-induced cell migration.
Materials and methods
Growth factor, antibodies, inhibitors and plasmid constructs
HGF (100 ng/ml), BIM-I, vinblastine sulphate, propidium iodide, TPA, antibodies against c-Met, EEA1, tubulin, secondary fluorescent and peroxidase-labelled antibodies were obtained as described previously (Kermorgant et al, 2003). The following antibodies were used: mouse monoclonal anti-4G10 (Upstate Biotechnology), -pan ERK (BD Sciences), -vinculin (Sigma); rabbit polyclonal anti-phospho-ERK1/2, -MEK1/2, -Jnk, c-Jun, -c-Met (Tyr1234/1235) (Cell Signalling), anti-heavy-chain clathrin (Santa Cruz). Gö6976 was from Calbiochem, phalloidin from Molecular Probes and concanavalin A from Sigma. GFP-PKCα KD was derived from PKCα by cloning into the eGFP vector from Clontech (S Parkinson, unpublished). The GFP-ERK2 construct was kindly provided by Dr C Marshall (Institute for Cancer Research, London).
Cell culture, transfections, Western blots and densitometric analysis
They were performed as described previously (Kermorgant et al, 2003). Each value of the densitometric results corresponds to the mean of at least three independent experiments performed in duplicate.
Immunofluorescence, measure of colocalisation and intensity
Immunofluorescence was performed as described (Kermorgant et al, 2003). For analysis of colocalisation, images were acquired using a confocal laser scanning microscope (LSM510, Carl Zeiss Inc.). High-quality (12 bits, 2048) images were acquired, interactively thresholded and the colocalised (red and green) area was evaluated as a fraction of the red or green area using specially developed software in Mathematica (Wolfram Research). Comparisons were analysed by one-way ANOVA followed by the t-test. Each value corresponds to the mean of five independent experiments where 3–5 images containing 4–5 cells each have been analysed. The analysis of the intensity of P-Jun was performed on confocal images with the Acquisition Manager (Kinetic Imaging) program. For each condition, 100 cells were analysed and statistics (t-test) calculated.
Semiautomatic assessment of vesicle distribution in the cytosol
It was performed as described (Kermorgant et al, 2003).
RNAi knock-down
The following 21-mer oligoribonucleotide pairs were obtained from Qiagen: control, 5′-UUCUCCGAACGUGUCACGUTT-3′ and 5′-ACGUGACACGUUCGGAGAATT-3′; HCC, 5′-GAAGGCUCGAGAGUCCUAUTT-3′ and 5′-AUAGGACUCUCGAGCCUUCTT-3′; PKCα, 5′-GGCUUCCAGUGCCAAGUUUTT-3′ and 5′-AAACUUGGCACUGGAAGCCTT-3′; PKCδ, 5′-GGCUACAAAUGCAGGCAAUTT-3′ and 5′-AUUGCCUGCAUUUGUAGCCTT-3′; PKCɛ, 5′-GAUCGAGCUGGCUGUCUUUTT-3′ and 5′-AAAGACAGCCAGCUCGAUCTT-3′. The uniqueness of each RNAi recognition sequence was confirmed by blasting against the GenBank database. The RNAi control does not match to any known sequence. Cells were plated at 105/well in six-well plates and transfected the next day in medium without serum using oligofectamine (Invitrogen) with 10 μl of 20 μM RNAi and 5 μl of transfection reagent/well. Serum was added 4 h later. The cells were stimulated and harvested after 72 h.
Wounding assays
Confluent cells, deprived for 24 h in 0.5% FBS were wounded with a pipette tip in order to obtain two perpendicular wounds in each well. They were rinsed one time and fresh medium with or without HGF was added. A picture was acquired on an Axiovert TM 135 microscope (Carl Zeiss) equipped with a × 5 objective lens and an Orca ER CCD camera (Hamamatsu) using Acquisition Manager (Kinetic Imaging) in the same area of each wound at time 0 and 24 h. Experiments were performed in duplicate with two wounds per duplicate; knockout cells were tested on five independent occasions.
Supplementary Material
Supplementary Figures
Acknowledgments
We thank Sharon Tooze and Clive Dickson for their comments, J Ivaska, S Parkinson and J Strivastava for the GFP-PKC constructs, and also thank Amir Faisal, Angus Cameron, Richard Whelan and Alastair Nicoll.
References
- Alves dos Santos CM, van Kerkhof P, Strous GJ (2001) The signal transduction of the growth hormone receptor is regulated ubiquitin/proteasome system and continues after endocytosis. J Biol Chem 276: 10839–10846 [DOI] [PubMed] [Google Scholar]
- Awasthi V, King RJ (2000) PKC, p42/p44 MAPK, and p38 MAPK are required for HGF-induced of H441 cells. Am J Physiol Lung Cell Mol Physiol 279: L942–L949 [DOI] [PubMed] [Google Scholar]
- Burke P, Schooler K, Wiley HS (2001) Regulation of epidermal growth factor receptor signalling by endocytosis and intracellular trafficking. Mol Biol Cell 12: 1897–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceresa BP, Kao AW, Santeler SR, Pessin JE (1998) Inhibition of clathrin-mediated endocytosis selectively attenuates specific insulin receptor signal transduction pathways. Mol Cell Biol 18: 3862–3870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapell R, Bueno OF, Alvarez-Hernandez X, Robinson LC, Leidenheimer NJ (1998) Activation of protein kinase C induces gamma-aminobutyric acid type A receptor internalization in Xenopus oocytes. J Biol Chem 273: 32595–32601 [DOI] [PubMed] [Google Scholar]
- Chow JC, Condorelli G, Smith RJ (1998) Insulin-like growth factor-I receptor internalization regulates signalling via the Shc/mitogen-activated protein kinase pathway, but not the insulin receptor substrate-1 pathway. J Biol Chem 273: 4672–4680 [DOI] [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM, Ahn S, Rocca GJD, Ferguson SSG, Caron MG, Lefkowitz RJ (1998) Essential role for G protein-coupled receptor endocytosis in the activation of mitogen-activated protein kinase. J Biol Chem 273: 685–688 [DOI] [PubMed] [Google Scholar]
- Di Renzo MF, Olivero M, Giacomini A, Porte H, Chastre E, Mirossay L, Nordlinger B, Bretti S, Bottardi S, Giordano S, Plebani M, Gespach C, Comoglio PM (1995) Overexpression and amplification of the met/HGF receptor gene during the progression of colorectal cancer. Clin Cancer Res 1: 147–154 [PubMed] [Google Scholar]
- Fincham VJ, James M, Frame MC, Winder SJ (2000) Active ERK/MAP kinase is targeted to newly forming cell–matrix adhesions by integrin engagement and v-Src. EMBO J 19: 2911–2923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoussoub RA, Dillon DA, D'Aquila T, Rimm EB, Fearon ER, Rimm DL (1998) Expression of c-met is a strong independent prognostic factor in breast carcinoma. Cancer 82: 1513–1520 [DOI] [PubMed] [Google Scholar]
- Hayes S, Chawla A, Corvera S (2002) TGF beta receptor internalization into EEA1-enriched early endosomes: role in signalling to Smad2. J Cell Biol 158: 1239–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipkin RW, Wang Y, Schonbrunn A (2000) Protein kinase C activation stimulates the phosphorylation and internalization of the sst2A somatostatin receptor. J Biol Chem 275: 5591–5599 [DOI] [PubMed] [Google Scholar]
- Howe CL, Valletta JS, Rusnak AS, Mobley WC (2001) NGF signalling from clathrin-coated vesicles: evidence that signalling endosomes serve as a platform for the Ras–MAPK pathway. Neuron 32: 801–814 [DOI] [PubMed] [Google Scholar]
- Ishibe S, Joly D, Zhu X, Cantley LG (2003) Phosphorylation-dependent paxillin–ERK association mediates hepatocyte growth factor-stimulated epithelial morphogenesis. Mol Cell 12: 1275–1285 [DOI] [PubMed] [Google Scholar]
- Ivaska J, Whelan RD, Watson R, Parker PJ (2002) PKCepsilon controls the traffic of beta1 integrins in motile cells. EMBO J 21: 3608–3619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Sorkin A (2002) Coordinated traffic of Grb2 and Ras during epidermal growth factor receptor endocytosis visualized in living cells. Mol Biol Cell 13: 1522–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kermorgant S, Zicha D, Parker PJ (2003) Protein kinase C controls microtubule-based traffic but not proteasomal degradation of c-Met. J Biol Chem 278: 28921–28929 [DOI] [PubMed] [Google Scholar]
- Liu ZX, Yu CF, Nickel C, Thomas S, Cantley LG (2002) Hepatocyte growth factor induces ERK-dependent paxillin phosphorylation and regulates paxillin–focal adhesion kinase association. J Biol Chem 277: 10452–10458 [DOI] [PubMed] [Google Scholar]
- Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marme D, Schachtele C (1993) Selective inhibition of protein kinase C isozymes by the indolocarbazole Go 6976. J Biol Chem 268: 9194–9197 [PubMed] [Google Scholar]
- McPherson PS, Kay BK, Hussain NK (2001) Signalling on the endocytic pathway. Traffic 2: 375–384 [DOI] [PubMed] [Google Scholar]
- Park WS, Dong SM, Kim SY, Na EY, Shin MS, Pi JH, Kim BJ, Bae JH, Hong YK, Lee KS, Lee SH, Yoo NJ, Jang JJ, Pack S, Zhuang Z, Schmidt L, Zbar B, Lee JY (1999) Somatic mutations in the kinase domain of the Met/hepatocyte growth receptor gene in childhood hepatocellular carcinomas. Cancer Res 59: 307–310 [PubMed] [Google Scholar]
- Potempa S, Ridley AJ (1998) Activation of both MAP kinase and phosphatidylinositide 3-kinase by required for hepatocyte growth factor/scatter factor-induced adherens junction disassembly. Mol Biol Cell 9: 2185–2200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevostel C, Joubert D, Alice V, Parker PJ (2000) Protein kinase Cα actively downregulates through caveolae-dependent traffic to an endosomal compartment. J Cell Sci 113: 2575–2584 [DOI] [PubMed] [Google Scholar]
- Rodrigues GA, Park M, Schlessinger J (1997) Activation of the JNK pathway is essential for transformation by the Met oncogene. EMBO J 16: 2634–2645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Wyse B, Hancock JF (2002) H-Ras signalling and K-Ras signalling are differentially dependent on endocytosis. Mol Cell Biol 22: 5128–5140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeant N, Lyon M, Rudland PS, Fernig DG, Delehedde M (2000) Stimulation of DNA synthesis and cell proliferation of human mammary myoepithelial-like cells by hepatocyte growth factor/scatter factor depends on heparan sulfate proteoglycans and sustained phosphorylation of mitogen-activated protein kinases p42/44. J Biol Chem 275: 17094–17099 [DOI] [PubMed] [Google Scholar]
- Sipeki S, Bander E, Buday L, Farkas G, Bacsy E, Ways DK, Farago A (1999) Phosphatidylinositol 3-kinase contributes to Erk1/Erk2 MAP kinase activation associated with hepatocyte growth factor-induced cell scattering. Cell Signal 11: 885–890 [DOI] [PubMed] [Google Scholar]
- Sipeki S, Bander E, Farkas G, Gujdar A, Ways DK, Farago A (2000) Protein kinase C decreases the hepatocyte growth factor-induced of Erk1/Erk2 MAP kinases. Cell Signal 12: 549–555 [DOI] [PubMed] [Google Scholar]
- Srivastava J, Procyk KJ, Iturrioz X, Parker PJ (2002) Phosphorylation is required for PMA- and cell-cycle-induced degradation of protein kinase Cdelta. Biochem J 368: 349–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsarfaty I, Alvord WG, Resau JH, Altstock RT, Lidereau R, Bieche I, Bertrand F, Horev J, Klabansky RL, Keydar I, Vande Woude GF (1999) Alteration of Met protooncogene product expression and prognosis in carcinomas. Anal Quant Cytol Histol 21: 397–408 [PubMed] [Google Scholar]
- Vieira AV, Lamaze C, Schmid SL (1996) Control of EGF receptor signalling by clathrin-mediated endocytosis. Science 274: 2086–2089 [DOI] [PubMed] [Google Scholar]
- Xue L, Lucocq J (1998) ERK2 signalling from internalised epidermal growth factor receptor in broken A431 cells. Cell Signal 10: 339–348 [DOI] [PubMed] [Google Scholar]
- Zhang YW, Vande Woude GF (2003) HGF/SF-met signalling in the control of branching morphogenesis and invasion. J Cell Biochem 88: 408–417 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures