

Abstract
Tissue plasminogen activator (tPA) is a serine protease involved in the degradation of blood clots through the activation of plasminogen to plasmin. Here we report on the identification of tPA as a specific protease able to activate platelet-derived growth factor C (PDGF-C). The newly identified PDGF-C is secreted as a latent dimeric factor (PDGF-CC) that upon proteolytic removal of the N-terminal CUB domains becomes a PDGF receptor α agonist. The CUB domains in PDGF-CC directly interact with tPA, and fibroblasts from tPA-deficient mice fail to activate latent PDGF-CC. We further demonstrate that growth of primary fibroblasts in culture is dependent on a tPA-mediated cleavage of latent PDGF-CC, generating a growth stimulatory loop. Immunohistochemical analysis showed similar expression patterns of PDGF-C and tPA in developing mouse embryos and in tumors, indicating both autocrine and paracrine modes of activation of PDGF receptor-mediated signaling pathways. The identification of tPA as an activator of PDGF signaling establishes a novel role for the protease in normal and pathological tissue growth and maintenance, distinct from its well-known role in plasminogen activation and fibrinolysis.
Keywords: activation, CUB, PDGF, proteolysis, tPA
Introduction
Platelet-derived growth factor (PDGF) signaling is critical for normal tissue growth and maintenance, and is mediated through two structurally related tyrosine kinase receptors, PDGFR-α and PDGFR-β. The PDGF family of growth factors consist of disulfide-bonded dimers involving four polypeptide chains: the classical PDGF-A and PDGF-B chains (Heldin and Westermark, 1999), and the novel PDGF-C (Li et al, 2000), and PDGF-D chains (Bergsten et al, 2001; LaRochelle et al, 2001). Unique for the novel PDGF chains are that they share a two-domain organization not found within the classical PDGF chains, with an N-terminal CUB domain in front of the conserved growth factor domain. The role of the CUB domain is currently not fully understood; however, proteolytic removal of the CUB domain is required before PDGF-CC and PDGF-DD can bind to and activate their cognate PDGFRs. Activated PDGF-C, like PDGF-A, signals through PDGFR-α, and activated PDGF-D through PDGFR-β, whereas PDGF-B binds to and activates both PDGFRs (Heldin and Westermark, 1999; Li and Eriksson, 2003). Other groups have demonstrated that both PDGF-C and PDGF-D are able to activate PDGFR-α/β heterodimeric complexes as well (Gilbertson et al, 2001; LaRochelle et al, 2001; Cao et al, 2002).
The identity of the proteases responsible for activation of the novel PDGFs remains elusive, although there are data indicating the involvement of serum-derived factors (Gilbertson et al, 2001; LaRochelle et al, 2001). Here we report that tissue plasminogen activator (tPA) cleaves and activates latent PDGF-CC. This is a novel role for tPA, which is a secreted serine protease with highly restricted substrate specificity. tPA is best characterized for its role in releasing the broad-specificity protease plasmin from the inactive zymogen plasminogen (Plg), which then digests the fibrin network of blood clots to form soluble products. Since the activity of tPA is substantially accelerated in the presence of fibrin (Hoylaerts et al, 1982; Ranby, 1982) thereby facilitating a localized generation of plasmin, tPA has been investigated as a potential thrombolytic agent. In fact, tPA is currently the only treatment of acute ischemic stroke approved by the FDA (The National Institute of Neurological Disorders and Stroke rtPA Stroke Study Group, 1995). Recently, there have been several reports suggesting that tPA plays normal and pathological roles that do not require plasminogen (Wu et al, 2000; Nicole et al, 2001; Yepes et al, 2002, 2003), but so far only one other substrate, apart from plasminogen, has been reported for tPA, that is, the NR1 subunit of the NMDA receptor (Nicole et al, 2001). The identification of latent PDGF-CC as a bona fide substrate for tPA suggests an important role of the protease in PDGF-mediated signaling.
Results
Identification and cloning of a PDGF-CC processing protease
In order to identify enzymes capable of activating latent PDGF-CC, conditioned media from different in vitro-grown cell lines were screened for expression of endogenous PDGF-CC, and for the capacity to cleave and activate the secreted latent growth factor. The human fibroblastic cell line AG1523 efficiently secreted full-length PDGF-CC, and also displayed the capacity to cleave specifically full-length PDGF-C chains, and thus releasing a distinct 22 kDa species under reducing conditions (Figure 1A). This species migrated similarly to the recombinant active growth factor domain of PDGF-C expressed in insect cells (Li et al, 2000).
Figure 1.
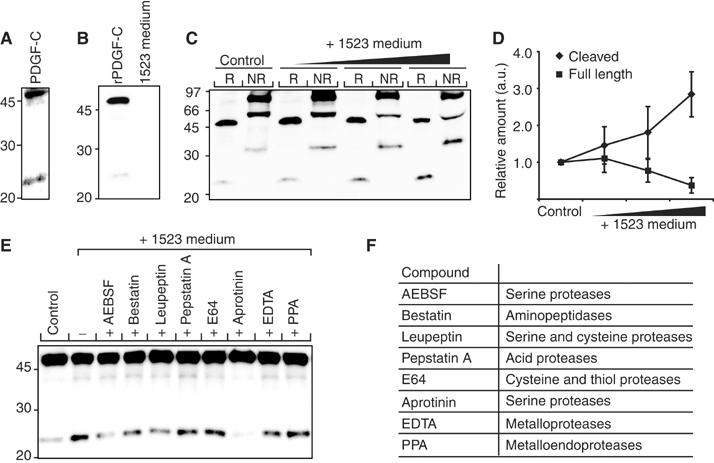
Characterization of a PDGF-CC processing activity. (A) Endogenous expression of PDGF-CC from AG1523 fibroblasts detected by a PDGF-C-specific antibody. Reduced latent PDGF-C migrated as a 48 kDa species, while the released core domain migrated as a 22 kDa species. (B) Using an anti-His6 antibody, immunoreactivity was detected only in recombinant latent PDGF-C expressed in baculovirus-infected cells and not in conditioned medium from AG1523 cells. (C) Increasing concentrations of conditioned medium from AG1523 cells were incubated with fixed amounts of recombinant latent PDGF-CC. The reduced (R) and nonreduced (NR) recombinant proteins were analyzed by immunoblotting using an anti-His6 antibody. Under reducing conditions, the 48 kDa latent PDGF-C and the released 22 kDa core domain of PDGF-C were visualized. Under nonreducing conditions, the 90 kDa latent homodimer of PDGF-CC, the 60 kDa hemidimer, and the 35 kDa homodimeric core domain of PDGF-CC were visualized. (D) Quantification of the amounts of reduced full-length 48 kDa (▪) and cleaved 22 kDa (⧫) PDGF-C species. The results are mean±s.d. of five independent experiments. (E) Different protease inhibitors were preincubated with AG1523 medium, and then incubated with recombinant full-length PDGF-CC. Recombinant PDGF-CC incubated with serum-free medium (control) or AG1523 medium only (−) were used as controls. All lanes with incubations pretreated with serine protease inhibitors displayed reduced PDGF-C processing activity. An anti-His6 antibody was used. (F) List of the protease inhibitors used and the specificity of the inhibitors.
In an in vitro assay, the properties of the enzyme(s) involved in cleavage and activation of PDGF-CC were studied by mixing serum-free conditioned media from AG1523 cells with His6-tagged recombinant full-length PDGF-CC. Control analysis demonstrated that immunoreactivity toward the His6 epitope was found only in recombinant PDGF-CC, and not in conditioned medium from AG1523 cells (Figure 1B). SDS–PAGE analysis under reducing and nonreducing conditions, and immunoblotting using an anti-His6 antibody, showed that increasing amounts of conditioned media from the AG1523 cells sequentially released the CUB domains of latent human PDGF-CC in a dose-dependent manner (Figure 1C and D). These data show that the enzymatic activity responsible for the cleavage of full-length PDGF-CC is derived from a secreted protease(s) present in the conditioned media from AG1523 cells.
The class of enzyme(s) responsible for cleavage and activation of latent PDGF-CC was established by generating an enzyme inhibitor profile of the enzymatic activity (Figure 1E). Eight different protease inhibitors (see Figure 1F) were separately preincubated with conditioned media from AG1523 cells, and then incubated with His6-tagged recombinant full-length PDGF-CC. Analysis of the incubation mixtures by SDS–PAGE and immunoblotting revealed that inhibitors of serine proteases (AEBSF, leupeptin, and aprotinin) inhibited the proteolytic cleavage of latent PDGF-CC (Figure 1E), while inhibitors of other protease classes, including matrix metalloproteinases, failed to inhibit efficiently the processing. These results suggest that a secreted trypsin-like serine protease is responsible for the proteolytic activation of latent PDGF-CC.
A coupled reverse transcription–polymerase chain reaction (RT–PCR) assay was employed to clone trypsin-like serine proteases expressed by AG1523 cells. Based on conserved amino-acid sequences around the catalytic triad in the serine protease domain, degenerate oligonucleotide mixtures were included in the RT–PCR reactions using single-stranded cDNA from the AG1523 cells as the template. Amplified products ranging from 500 to 650 bp were visualized by agarose gel electrophoresis (Figure 2A), subcloned, and inserts with the expected size range of approximately 550–600 bp were sequenced. The results revealed that the most abundant amplified cDNA was derived from tPA, while neurotrypsin (NT), coagulation factor X, and trypsinogen IV were other known serine proteases expressed by the AG1523 cells (Figure 2B).
Figure 2.

Cloning of candidate proteases from AG1523 fibroblastic cells. (A) Agarose gel electrophoresis of PCR products (arrowheads) amplified from AG1523 cDNA using degenerate oligonucleotide mixtures derived from trypsin-like serine protease domains. The amplified PCR fragments were cloned into the pCR2.1-TOPO vector and the nucleotide sequences of 18 clones were determined. (B) Histogram showing the identification of candidate proteases and distribution of the sequenced PCR-generated clones obtained from AG1523 cells.
tPA is a specific activator of latent PDGF-CC
A cotransfection assay was established to identify serine proteases able to cleave and activate latent PDGF-CC. Expression plasmids encoding the relevant enzymes and full-length PDGF-C were cotransfected into COS-1 cells, and aliquots of the conditioned media from the transfectants were subjected to SDS–PAGE and immunoblotting using antibodies to the growth factor domain of PDGF-C. The results showed that tPA released the growth factor domain of latent PDGF-CC, and the fragment migrated as a 22 kDa species under reducing conditions (Figure 3A). In contrast, NT lacked proteolytic activity toward latent PDGF-CC. As a specificity control, we analyzed the ability of tPA and NT to use full-length PDGF-DD as the substrate in the cotransfection assay. The results revealed that neither of the two enzymes was able to cleave and activate latent PDGF-DD (Figure 3B). Using purified tPA and recombinant latent PDGF-CC, or recombinant latent PDGF-DD, in an in vitro assay, we confirmed these observations showing that PDGF-CC, but not PDGF-DD, is a substrate for tPA (Figure 3C and D). One difference in the latter results, as compared with the results from the cotransfection assay, was that purified tPA generated a second intermediate species of 32 kDa using latent PDGF-CC as the substrate. It is possible that this intermediate is the result of digestion by plasmin contamination in the tPA preparation, since the size of the fragment is similar to that of plasmin-cleaved PDGF-CC previously reported (Li et al, 2000).
Figure 3.
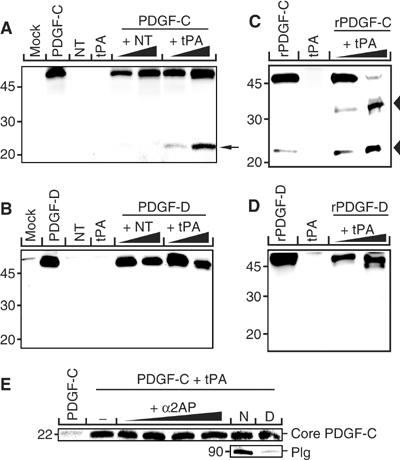
tPA specifically cleaves latent PDGF-CC. Coexpression and functional analysis of tPA and NT on the proteolysis of PDGF-CC and PDGF-DD. (A, B, E) COS-1 cells were transfected with combinations of expression vectors encoding for PDGF-C or PDGF-D and different concentrations encoding for tPA and NT, respectively. Empty vector (mock) and the expression vectors alone were used as negative control. When coexpressed with PDGF-C, tPA released a 22 kDa fragment of PDGF-C (A, arrow), while tPA did not release the corresponding part of PDGF-D (B). In transfected cells, coexpressing NT and PDGF-C or PDGF-D, or mock transfection, did not release the core domains of PDGF-CC nor PDGF-DD. (C, D) In vitro cleavage of recombinant PDGF-CC (C) and PDGF-DD (D) using purified tPA in two different concentrations. PDGF-CC but not PDGF-DD is readily cleaved by tPA generating a 22 kDa band under reducing conditions, corresponding to the released core domain (lower arrowhead in C). Note the intermediate 32 kDa PDGF-C species (C, upper arrowhead), possibly due to cleavage by plasmin contamination in the tPA preparation (see Results). (E) Addition of the specific plasmin inhibitor α2-anti-plasmin (α2AP) into the cotransfection medium had no effect on the release of core PDGF-C by tPA nor had removal of Plg from the culture medium. N, normal FCS medium; D, Plg-depleted FCS medium.
To ensure that the cleavage of PDGF-C observed in the cotransfection assay was a direct effect of tPA, and not an indirect effect due to cleavage by remnants of plasmin, we cultured the COS-1 cells in the absence or presence of the specific plasmin inhibitor α2-anti-plasmin or in Plg-depleted medium prior to transfection (Figure 3E). Neither α2-anti-plasmin treatment nor culturing in Plg-depleted medium had any effect on the processing of PDGF-C, showing that the cleavage of PDGF-C is performed by tPA directly.
To demonstrate that the proteolytic activity of tPA accounted for the major PDGF-CC processing activity produced by AG1523 cells, a well-characterized inhibitor of tPA, tPA-STOP™ (Sturzebecher et al, 1997), and the serine protease inhibitor aprotinin (see above) were added to the serum-free culture medium of growing AG1523 cells. Analysis of conditioned media showed that tPA-STOP™, in a dose-dependent way, prevented processing of full-length PDGF-CC (Figure 4A). Similarly, aprotinin efficiently inhibited processing of latent PDGF-CC in comparison with the untreated control. These results showed that tPA accounts for a majority of the PDGF-CC processing activity in conditioned media from AG1523 cells.
Figure 4.

tPA is the major PDGF-CC processing protease secreted from AG1523 cells and from primary mouse fibroblasts in culture. (A) Inhibition of cleavage of endogenous PDGF-CC produced by AG1523 cells using aprotinin and different concentrations of the specific tPA inhibitor tPA-STOP™. The inhibitors blocked processing of latent PDGF-CC showing that tPA accounts for the majority of the PDGF-C processing activity in conditioned media from AG1523 cells. (B) Serum-free media from wild-type and tPA-deficient fibroblasts were analyzed by immunoblotting. The results showed that both wild-type (+/+) and tPA-deficient (−/−) cells expressed latent PDGF-CC. However, tPA-deficient cells displayed a greatly reduced ability to process and activate the latent growth factor. tPA expression was analyzed by immunoblotting of conditioned media (middle panel). Agarose gel electrophoresis of PCR reactions from the genotyping of the animals used to establish the primary cultures of fibroblasts (lower panel). The immunoblot analyses were performed using protein-specific antibodies.
We also examined the ability of primary cultures of lung and kidney fibroblasts from wild-type and tPA-deficient mice to produce and activate latent PDGF-CC. SDS–PAGE and immunoblotting analyses of TCA-precipitated proteins from serum-free conditioned media showed that the primary fibroblasts secreted latent PDGF-CC migrating as a 48 kDa species in SDS–PAGE under reducing conditions (Figure 4B). In the medium from wild-type cells, processing of latent PDGF-CC into species migrating as 35 kDa species and as double bands of 22–25 kDa was seen. In contrast, in medium from tPA-deficient cells, the generation of double species migrating as 22–25 kDa was reduced to less than 10%, and the intensity of the 35 kDa species was also significantly reduced. These data demonstrate an essential role of tPA in activation of latent PDGF-CC in vivo.
tPA-mediated activation of PDGF-CC generates a PDGFR-α agonist
We verified that the growth factor domain in PDGF-CC released by tPA-mediated proteolysis is an efficient PDGFR-α ligand. Conditioned media from transfected COS-1 cells were applied onto porcine aortic endothelial (PAE) cells with stable expression of PDGFR-α (Figure 5A). Stimulation of the cells using conditioned medium from mock-transfected COS-1 cells, or media from transfected COS-1 cells separately expressing tPA, or latent PDGF-CC, failed to induce receptor activation measured as induction of receptor tyrosine phosphorylation. In contrast, stimulation using medium from COS-1 cells coexpressing tPA and full-length PDGF-CC induced strong PDGFR-α activation. This showed that the growth factor domain of full-length PDGF-CC released by tPA is a bona fide ligand and activator of PDGFR-α.
Figure 5.
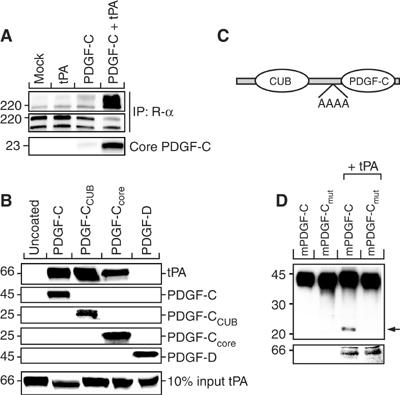
tPA-mediated proteolysis of latent PDGF-CC generates a PDGFR-α agonist. Conditioned serum-free media from transfected COS-1 cells were used to induce tyrosine phosphorylation of PDGFR-α expressed in PAE cells. (A) The 22 kDa fragment of PDGF-C, generated by tPA-mediated cleavage of latent PDGF-CC, induced efficient tyrosine phosphorylation of PDGFR-α as compared to mock, tPA, and PDGF-C controls as analyzed using antibodies against phosphotyrosine (PY99) (upper panel). The amount of precipitated PDGFR-α was monitored using antibodies to PDGFR-α (CED, middle panel). The amount of PDGF-C core domain in the media from the transfected cells was monitored by immunoblotting (lower panel). (B) Direct interaction of PDGF-CC with tPA. Ni-NTA beads coated with recombinant His6-tagged latent PDGF-CC, CUB domain, and core domains of PDGF-CC, or latent PDGF-DD, were incubated with purified tPA. Proteins eluted from the beads using a buffer containing 400 mM imidazole were analyzed by immunoblotting using specific antibodies. The results show that latent PDGF-CC interacts directly with tPA both via the CUB and the core domains. (C) Illustration of the cleavage site mutant. (D) Analysis of the cleavage site mutant of PDGF-CC using the cotransfection assay. Normal and mutant latent PDGF-CC forms were expressed in transfected COS-1 cells, without or with the coexpression of tPA. Analysis by immunoblotting showed that cleavage of latent PDGF-CC by tPA was abolished in the alanine cleavage site mutant (upper panel) suggesting that the tribasic site is the cleavage site for tPA. The expression of tPA was also monitored (lower panel).
The possibility of a direct protein–protein interaction between tPA and latent PDGF-CC was explored by developing a pull-down assay. Ni-NTA beads were allowed to bind recombinant His6-tagged latent PDGF-CC or PDGF-DD, and purified tPA was added and incubated. Following extensive washings, bound proteins were subsequently eluted with an imidazole-containing buffer, and the eluates were analyzed by immunoblotting using specific antibodies. The results showed that full-length PDGF-CC-coated beads specifically bound tPA, while uncoated Ni-NTA beads or PDGF-DD-coated beads failed to do so (Figure 5B). Similar experiments using Ni-NTA beads separately coated with recombinant ‘free' CUB domain or recombinant core domain of PDGF-CC showed that both domains were able to interact with tPA.
We have previously identified a conserved site of four amino acids containing three basic amino-acid residues (amino-acid residues -R-K-S-R-) as a potential site for proteolytic activation of latent PDGF-CC (Li et al, 2000). It is notable that the corresponding regions in PDGF-A and PDGF-B are the cleavage sites for furine-like proteases that act in the exocytic pathway during secretion of these PDGFs (Östman et al, 1992; Siegfried et al, 2003). To verify this, a mutant with the tribasic site replaced with alanine residues was created (schematically illustrated in Figure 5C). Analysis using the cotransfection assay verified that the mutant was resistant to tPA-mediated cleavage, while the wild-type PDGF-CC was readily cleaved (Figure 5D). These data suggest that tPA cleaves latent PDGF-CC in, or at least around, the conserved tribasic site.
tPA-dependent activation of latent PDGF-CC drives proliferation of primary fibroblasts
We observed that primary fibroblasts derived from tPA-deficient mice grew slower in culture than fibroblasts derived from wild-type animals, raising the possibility that activation of latent PDGF-CC by tPA generated autocrine and paracrine growth stimulatory loops for primary fibroblasts in culture. To analyze this effect, isolated wild-type and tPA-deficient fibroblasts were serum-starved overnight, and the growth of the cells during the next 24 h was monitored using an enzyme-based viability assay (see Materials and methods). The results confirmed our initial observation and showed that tPA-deficient cells displayed a reduced growth rate in serum-free medium as compared to wild-type cells (Figure 6A). Rescue of the tPA-deficient cells by the addition of 50 ng ml−1 of activated PDGF-CC or recombinant tPA to the serum-free culture medium allowed the cells to grow similar to the wild-type fibroblasts.
Figure 6.
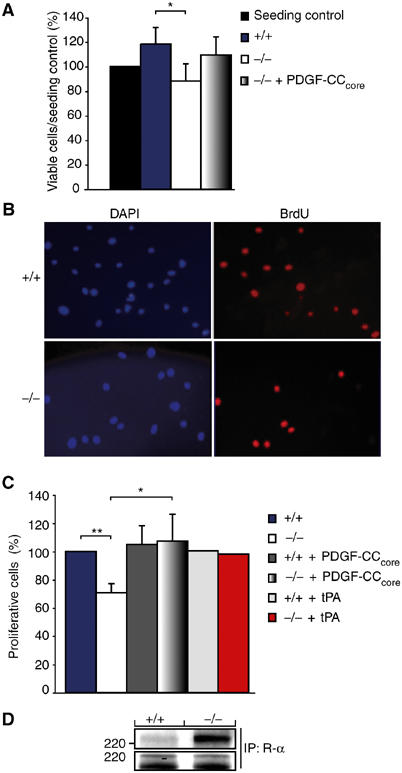
An autocrine tPA-dependent growth stimulatory loop involving activation of latent PDGF-CC drives proliferation of fibroblasts in primary culture. Primary cultures of fibroblasts were established from wild-type and tPA-deficient animals. (A) Total cell numbers of wild-type (+/+) and tPA-deficient cells (−/−) after 36 h of culture in serum-free conditions (mean±s.d., n=4). Significantly less tPA-deficient cells were observed after the culture period (P<0.05). The tPA-deficient cells were stimulated to grow by the addition of activated PDGF-CC (mean±s.d., n=3). The seeding control was set to 100%. (B) Microphotographs showing wild-type and tPA-deficient fibroblasts following labeling with BrdU. Cell nuclei were visualized using DAPI (blue), while BrdU-labeled nuclei were identified by immunofluorescence using a specific antibody (red). (C) Quantification showed that significantly less tPA-deficient cells incorporated BrdU as compared to wild-type cells. Stimulation of the tPA-deficient cells with activated PDGF-CC or tPA enhanced BrdU incorporation, while wild-type cells were not markedly stimulated by this treatment (mean±s.d., n=3; n=2 for tPA treatment). *P<0.05, **P<0.01. (D) Activated PDGF-CC protein induced more efficient tyrosine phosphorylation of PDGFR-α in the tPA-deficient cells as compared to wild-type cells as analyzed using antibodies against phosphotyrosine (PY99) (upper panel). The amount of precipitated PDGFR-α was monitored using antibodies to PDGFR-α (CED, lower panel). These results show that growth of primary fibroblasts in culture is dependent on a growth stimulatory loop involving a tPA-dependent activation of latent PDGF-CC.
To further demonstrate that growth of primary fibroblasts in culture was dependent on a tPA-mediated growth stimulatory loop, serum-starved fibroblast cultures were labeled with 5-bromo-2′-deoxyuridine (BrdU) for 24 h in order to identify dividing cells. Cell nuclei were visualized with 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI), and BrdU-labeled cells were determined by immunofluorescence using antibodies to BrdU (Figure 6B). Quantification of the results showed that the fraction of BrdU-labeled nuclei were significantly higher in wild-type fibroblasts as compared to the tPA-deficient cells (Figure 6C). Addition of 50 ng ml−1 of activated PDGF-CC or recombinant tPA strongly stimulated BrdU incorporation in the tPA-deficient cells but had less effect on wild-type cells. These data suggest that autocrine and paracrine growth stimulatory loops are present in primary fibroblasts, and that these loops are generated by a tPA-mediated activation of latent PDGF-CC.
It is known that constitutive activation of PDGFRs by PDGFs leads to receptor desensitization (Heldin and Westermark, 1999), and we therefore investigated whether the differences observed in growth between the wild-type and tPA-deficient fibroblasts upon PDGF-CC treatment were due to differential activation of PDGFR-α. Recombinant PDGF-CC protein was applied onto the primary fibroblasts and receptor activation was measured as induction of PDGFR-α tyrosine phosphorylation (Figure 6D, upper panel). Stimulation of PDGFR-α was more pronounced in the tPA-deficient cells as compared to wild type, which might explain the efficient stimulation of proliferation seen in these cells following PDGF-CC treatment.
To determine whether PDGF-C and tPA are coexpressed, or at least expressed in adjacent cells, both proteins were localized by immunohistochemistry using tissue sections from E14.5 mouse embryos and T241 tumor xenografts (Figure 7). Furthermore, the expressions of PDGF-C and tPA reported in previous publications were compiled and compared (Carroll et al, 1994; Ding et al, 2000; Aase et al, 2002). The results from these analyses suggest that PDGF-C and tPA are coexpressed in several locations in the developing embryo such as the kidney and the surface ectoderm of the skin (Figure 7A–D). In tumor tissue sections, PDGF-C expression was observed mostly in tumor cells located at the center of the tumor in close apposition to larger blood vessels, while tPA was mainly expressed in the endothelium of the tumor blood vessels (Figure 7E and F). Scattered PDGF-C-positive tumor cells were also seen at the edge of the tumor. These observations support our findings and suggest that PDGF-CC can be activated by tPA in vivo.
Figure 7.
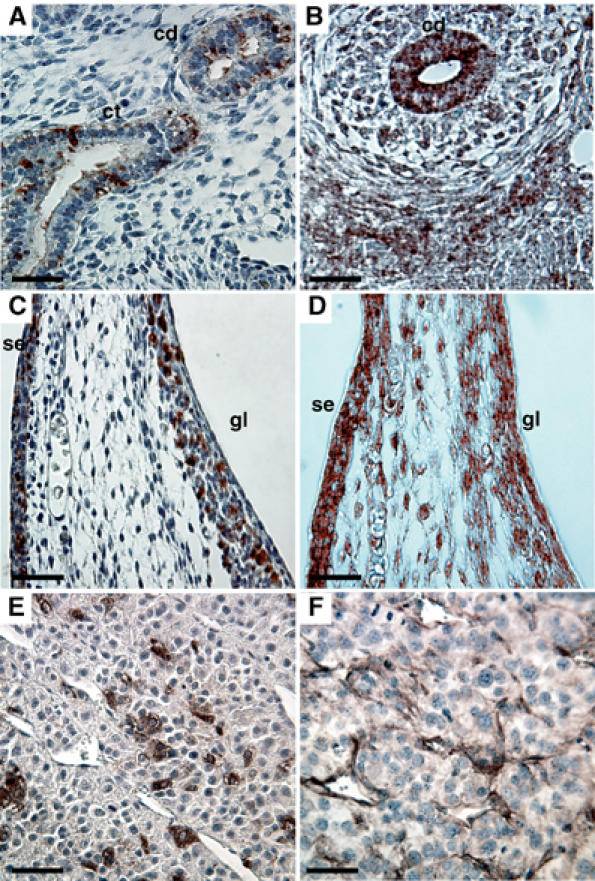
Colocalization of PDGF-CC and tPA. Immunohistochemical localization of PDGF-C (first column) and tPA (second column) in E14.5 mouse embryo and in T241 tumor xenografts. Tissue sections were stained using specific antibodies. (A, B) Developing kidney; overlapping staining for both PDGF-C and tPA was observed in the collecting ducts (cd). PDGF-C was also expressed in the collecting tubules (ct). (C, D) Skin of abdomen; colocalization of PDGF-C and tPA was seen in the germinal layer of the skin (gl) and in the surface ectoderm (se). (E, F) Expression of PDGF-C and tPA in T241 tumor xenografts. Scale bars, 50 μm.
Discussion
It is well established that PDGF-CC expression is widespread in both normal adult and embryonic tissues, as well as in several pathological conditions including tumors. In order to understand the physiological roles of PDGF-CC-mediated signal transduction in these processes, it is important to gain further insight into how latent PDGF-CC becomes proteolytically activated to generate a PDGF receptor agonist. We have previously shown that the relatively nonspecific protease plasmin can be used to activate both PDGF-CC and PDGF-DD from their latent precursors (Li et al, 2000; Bergsten et al, 2001); however, given the wide substrate specificity of plasmin, we considered this protease unlikely to be the physiologically relevant protease in activation of the novel PDGFs. Elucidating the identity, localization, and regulation of this protease(s) will greatly enhance our understanding of PDGF regulation in vivo.
To identify the enzyme responsible for activation of latent PDGF-CC, we developed an in vitro assay to monitor cleavage of latent PDGF-CC, and by using a combination of protease inhibitor profiling (so-called reverse biochemistry; Takeuchi et al, 1999), molecular cloning with RT–PCR using degenerate primers, and a functional assay, we identified tPA as a specific protease able to activate latent PDGF-CC. Despite the close structural similarities between PDGF-C and PDGF-D, the latter factor was not activated by tPA, demonstrating that distinct pathways are involved in activation of the two factors.
tPA is a multidomain trypsin-like serine protease best known for its role in fibrinolysis via proteolytic activation of plasminogen into plasmin (for reviews, see Vassalli et al, 1991; Collen, 2001). However, the expression pattern of tPA in the mouse embryo, especially in neuronal tissue and in areas undergoing extensive tissue remodeling, suggests that the protease may serve additional functions (Rickles and Strickland, 1988; Carroll et al, 1994). Also, several reports have suggested that tPA plays normal and pathological roles that do not require plasminogen activation (Strickland, 2001; Tsirka, 2002), but apart from plasminogen, only one additional substrate has been identified, that is, the NR1 subunit of the NMDA receptor (Nicole et al, 2001). Our identification of tPA as a specific activator of latent PDGF-CC is thus rather unexpected, but it provides additional evidence for roles of tPA in nonthrombolytic events, including fibrosis, angiogenesis, and tumor growth (see below).
The mechanisms underlying the specific cleavage and activation of latent PDGF-CC by tPA involve the formation of a stable substrate·protease complex. Our data show that tPA specifically interacts with both the CUB and the PDGF/VEGF-like growth factor domain in PDGF-CC. The specific binding of tPA to the CUB domain of PDGF-C, and not that of PDGF-D, is required for proteolytic activation of the factor (our unpublished observation). Thus, the role of the CUB domain in PDGF-CC appears two-fold: to prevent an agonistic role of the unprocessed growth factor (Li et al, 2000) and to bind specifically tPA to allow a site-specific cleavage of the factor. It is well documented that CUB domains in different proteins are involved in protein·protein interactions (e.g., see Thielens et al, 1999; Nakamura and Goshima, 2002), and it is possible that the released CUB domains might act as a competitive inhibitor in the activation of latent PDGF-CC. At present, the structural domains of tPA interacting with the CUB domain of PDGF-C are unknown. In Figure 8A, we have summarized our findings regarding the complex formation of full-length PDGF-CC and tPA, and the functional consequences of the growth factor when only one or both CUB domains have been removed by tPA-mediated proteolysis.
Figure 8.

Hypothetical mechanisms involved in the activation of PDGF-CC by tPA. (A) tPA binds to both the CUB domain and the growth factor domain of latent PDGF-CC. Released CUB domains might act as competitive inhibitors of the subsequent proteolytic activation of PDGF-CC. (B) A tPA-mediated activation of latent PDGF-CC drives proliferation of primary fibroblasts in culture.
The tight complex formation of tPA and PDGF-CC allows a precise cleavage of the substrate. Previously, we suggested that a conserved tribasic region (amino-acid residues -R231-K232-S233-R234- in human PDGF-C), 15 amino-acid residues N-terminal of the first cysteine in the PDGF/VEGF-like domain, represented a putative proteolytic cleavage site (Li et al, 2000). This suggestion was based on the location of this site in relation to the well-defined cleavage sites found in the intracellular proforms of PDGF-A and PDGF-B. Our data verify that the corresponding site in PDGF-C is the cleavage site for tPA.
Components of the fibrinolytic system, including tPA, urokinase-type plasminogen activator (uPA), the urokinase-type plasminogen activator receptor (uPAR), and the plasminogen activator inhibitors (PAIs), are often overexpressed in tumors (Kwaan, 1992 and references therein). So far, strong evidence suggests that overexpression of uPA, uPAR, and PAIs is linked to increased tumor growth, invasion, and metastatic spreading, whereas less is known about the role of tPA in these processes. In addition, many types of tumors overexpress PDGF-C (Uutela et al, 2001; Zwerner and May, 2001; Andrae et al, 2002; Dijkmans et al, 2002; Lokker et al, 2002; U Eriksson, unpublished observation), raising the possibility that in PDGF-C-expressing tumors, tPA would contribute to the activation of the growth factor. Several studies have shown that PDGF-C overexpression in tumor cells enhances tumor growth by promoting cellular transformation, and stimulates stromogenesis and tumor vascularization (Zwerner and May, 2001; Cao et al, 2002; Li et al, 2003). The source of tPA could either be PDGF-CC-expressing tumor cells themselves or as shown here for the T241 tumor the enzyme may be released by the invading endothelial cells of the tumor vasculature (Figure 7F).
tPA administration is the only FDA-approved thrombolytic therapy for acute ischemic stroke, and increasing evidence from studies in animal models of embolic stroke cautions against the use of tPA, as it might mediate neuronal damage (Tsirka, 2002). At least part of the neuronal damage might be caused by a tPA-dependent, plasminogen-independent opening of the blood–brain barrier mediated via the low-density lipoprotein receptor-related protein (LRP) and the cleavage of an as yet unidentified substrate (Yepes et al, 2003). Interestingly, LRP is a negative regulator of PDGF signaling (Boucher et al, 2003), raising the possibility that part of the plasminogen-independent action of tPA is indeed mediated via modulation of PDGF signaling.
The finding that the growth of fibroblasts is dependent on a tPA-mediated activation of latent PDGF-CC, thus generating autocrine and paracrine growth stimulatory loops, raises several possible roles of PDGF-CC in normal and pathological conditions involving fibroblast growth and recruitment. For example, such conditions may include tissue morphogenesis and regeneration, wound healing, and tumor growth (see Figure 8B). In part, this mechanism may also be the explanation for the long-standing observation that it is relatively easy to establish primary cultures of fibroblasts in comparison to most other cell types.
In summary, the identification of tPA as a potent activator of latent PDGF-CC has provided novel insights into PDGF-mediated signaling with broad implications in normal and pathological conditions, in particular in tumor biology and cardiovascular medicine.
Materials and methods
Cell culture
All cells used were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS), 2 mM glutamine, 100 U ml−1 penicillin, and 100 μg ml−1 streptomycin, except PAE cells that were kept in supplemented F12 medium. The cells were cultured at 37°C in a humidified 5% CO2 atmosphere. Kidney and lung primary fibroblast cultures were prepared essentially as described (Eghbali et al, 1991) from 5-week-old wild-type (+/+) and tPA-deficient (−/−) mice (Carmeliet et al, 1994) (kindly provided by Prof. P Carmeliet, Leuven). In short, kidneys and lungs were dissected, washed in ice-cold PBS, cut into smaller pieces, and incubated with trypsin/collagenase in PBS for 20 min at 37°C. Dissociated cells were pelleted and plated. Experiments were performed on cells at passages 4–7.
Protein expression and immunoblotting
To test the endogenous expression of PDGF-CC, subconfluent AG1523 cells and primary fibroblast cultures were cultured in serum-free DMEM overnight. Recombinant His6-tagged human PDGF-CC species and full-length PDGF-DD were expressed in serum-free medium from Sf9 insect cells using the baculovirus expression system as described previously (Li et al, 2000; Bergsten et al, 2001). To explore the extracellular proteolytic activities in conditioned serum-free AG1523 medium, the medium was coincubated with recombinant latent PDGF-CC-containing medium (ratios 1:2, 3:2, and 10:2) at 37°C overnight. To identify PDGF-CC activating serine proteases, the protease expression constructs were cotransfected with full-length PDGF-C (Li et al, 2000), full-length PDGF-D (Bergsten et al, 2001), or PDGF-C cleavage site mutant constructs into subconfluent COS-1 cells using LipofectaminePlus in serum-free DMEM (Life Technology). Transfection with empty vectors served as negative control. After 24 h, the transfection medium was replaced by DMEM only, with or without the addition of α2-anti-plasmin (10 ng–1 μg, #4030, American Diagnostica Inc.), for an extra 24 h. In addition, the COS-1 cells were grown in DMEM supplemented with 10% Plg-depleted FCS prior to transfection. Plg was removed from the FCS by affinity chromatography on lysine-Sepharose (Deutsch and Mertz, 1970) and the Plg-depleted FCS was tested by immunoblotting with rabbit anti-human Plg (A0081, DAKO). The conditioned serum-free medium was collected, and proteins were TCA precipitated as described previously (Li et al, 2000). In the case of the primary cultures, total protein concentration was measured and normalized (Bradford, 1976). All precipitates were subjected to SDS–PAGE under reducing conditions if not stated otherwise, immunoblotting, and visualization by chemiluminescence. PDGF-C and PDGF-D were detected by immunoblotting using affinity-purified polyclonal rabbit antibodies against PDGF-C (Li et al, 2000) and PDGF-D (Bergsten et al, 2001), respectively. The His6-tagged proteins were detected using an anti-His monoclonal antibody (C-terminal, Invitrogen). tPA was detected using sheep polyclonal antibodies against human tPA (ab9030, Abcam).
Reverse biochemistry
All protease inhibitors were purchased from Sigma and the concentrations used were as follows: AEBSF 1 mM, bestatin 100 μM, leupeptin 100 μM, pepstatin A 10 μM, E64 100 μM, aprotinin 100 μM (∼3 TIU), EDTA 50 mM, and phosphoramidon 100 μM. The protease inhibitors were preincubated with conditioned AG1523 medium at room temperature for 30 min, and then incubated with recombinant PDGF-CC (ratio 10:2) at 37°C overnight. Recombinant PDGF-CC species were analyzed by immunoblotting as above.
To determine whether tPA is the major proteolytic enzyme responsible for the PDGF-CC processing in AG1523 conditioned medium, AG1523 cells were cultured in serum-free medium, with or without the addition of a synthetic tPA inhibitor tPA-STOP™ (3.5–35 μM, #544, American Diagnostica Inc.) or 100 μM aprotinin as a positive control. The conditioned serum-free medium was collected, and proteins were precipitated before SDS–PAGE and immunoblotting using antibodies against PDGF-C (see above).
Cloning of serine proteases and plasmid construction
To clone trypsin-like serine proteases in AG1523 fibroblastic cells, total cellular RNA was prepared using the guanidinium thiocyanate/acid phenol method (Chomczynski and Sacchi, 1987). Single-stranded cDNA was synthesized using AMV Reverse Transcriptase (Amersham) and oligo-dT to prime the reaction. Degenerate oligonucleotide primers flanking the conserved histidine and serine residues in the catalytic triad were designed as follows: 5′-CAR TGG GTN YTN WCN GCN GCN CAY TG (corresponding to the amino-acid sequence Q W V L/F S/T A A H C, forward) and 5′-NCC NCC NGA RTC NCC YTG RCA NGC RTC (corresponding to the amino-acid sequence D A C Q G D S G G, reverse). The oligonucleotides were used to prime PCRs utilizing cDNA from the AG1523 cells as template. The PCR products were cloned into the pCR2.1-TOPO vector (TOPO TA Cloning kit, Invitrogen) and clones of the expected size of 500–600 bp were sequenced.
Full-length human tPA was amplified by PCR using cDNA from the AG1523 cells as template and the 1750-bp product was subcloned into the pCR2.1-TOPO vector. The primers used, including a BamH1 site (underlined), were as follows: 5′-CGGGATCCGCCGTGAATTTAAGGGAC (forward) and 5′-CGGGATCCTTGCTTTTGAGGAGTCGG (reverse). The BamH1 fragment was excised and cloned into the eukaryotic expression vector pSG5.
The fully sequenced MGC clone containing the 5′ part of human NT in the pOTB7 vector was purchased from Research Genetics whereas the 3′ part was amplified by PCR using AG1523 cDNAs as template. The primers used were as follows: 5′-GAGCTGAATACATACGTG (forward) and 5′-GCAGATCTGCTGCTTTGAAGTTTCCA (reverse, including a BglII site, underlined). The resulting 1400-bp 3′ fragment was subcloned into the pCR2.1-TOPO vector and then excised with NdeI–BglII. A full-length cDNA for hNT was constructed by fusing the excised 3′ fragment with NdeI–BglII-digested 5′-hNT/pOTB7. The full-length cDNA for hNT was excised and directionally cloned into the EcoRI–BglII sites of the eukaryotic expression vector pSG5.
To generate the cleavage site mutant, mouse PDGF-C cDNA was used as template. The predicted processing site in murine PDGF-C, amino-acid residues -K-K-S-K-, was replaced by four alanines. The N-terminal fragment of PDGF-C, containing an EcoRI and a NotI site (underlined), and the C-terminal fragment, containing a NotI and an XbaI site (underlined), were amplified using the following primers: 5′-GGAATTCAGCCAAATGCTCCTCCTCGGCCTC (forward, N-terminal) and 5′-TGCCGCGGCCGCCCCATACAGGAAAGCCTT (reverse, N-terminal, alanine replacement in bold), 5′-GCGGCCGCGGCAGTGGTGAATCTGAATCTCCTC (forward, C-terminal, alanine replacement in bold), and 5′-GCTCTAGACTGCAGTTACCCTCCTGCGTT (reverse, C-terminal). The amplified fragments were ligated and cloned in-frame into pcDNA3.1 (+) expression vector.
To produce recombinant CUB domain of human PDGF-C using the baculovirus system, the sequence encoding amino-acid residues 23–163 of PDGF-C was amplified by PCR. Primers used were as follows: 5′-CGGGATCCCGAATCCAACCTGAGTAG (forward, including a BamHI site for in-frame cloning) and 5′-CCGGAATTCCTAATGGTGATGGTGATGATGTTTGT CATCGTCGTCGACAATGTTGTAGTG (reverse, including an EcoRI site and sequences encoding a C-terminal His6 tag). The amplified product was cloned into the baculovirus expression vector pAcGP67A.
All primers used were purchased from Invitrogen and all the constructs were verified by nucleotide sequencing. The nucleotide and amino-acid sequences of human tPA can be found in the GenBank under accession number NM_000930 and of hNT under accession number NM_003619. The MGC clone containing the 5′ part of hNT has GenBank accession ID BC007761.
In vitro cleavage and protein–protein interaction studies
Recombinant latent PDGF-CC and PDGF-DD were digested with human tPA in 100 mM Tris–HCl pH 7.5, 0.1% Tween 20, and 0.1 mg ml−1 CNBr activated fibrinogen (Sigma) for 4 h at 37°C using 0.2–20 μg ml−1 tPA purified from human melanoma cells (T7776, Sigma). The digestions were analyzed by SDS–PAGE under reducing conditions and immunoblotted using affinity-purified antibodies against PDGF-C and PDGF-D, respectively (see above).
To determine a direct protein–protein interaction between tPA and PDGF-CC, His6-tagged recombinant protein species were bound to Ni-NTA-agarose (Qiagen) and then incubated with 1 μg of purified tPA for 2 h at room temperature. Uncoated and PDGF-DD-coated Ni-NTA beads were used as controls. The beads were washed thoroughly, and His6-tagged proteins were specifically eluted with 400 mM imidazole. Eluted proteins were analyzed by SDS–PAGE under reducing conditions and immunoblotted with antibodies against human tPA (see above). The membranes were subsequently stripped and reprobed with specific antibodies.
Receptor activation and proliferation analysis
To monitor growth factor-induced tyrosine phosphorylation of PDGFR-α, serum-starved PAE cells stably expressing human PDGFR-α were incubated for 120 min on ice with conditioned medium from COS-1 cells transfected with full-length PDGF-C in the absence or presence of tPA. Alternatively, primary wild-type and tPA-deficient fibroblasts were stimulated with 100 ng ml−1 activated PDGF-CC protein. The cells were lysed as described previously (Li et al, 2000) and PDGFR-α was immunoprecipitated using a specific antiserum (Eriksson et al, 1992). Precipitated proteins were separated by SDS–PAGE under reducing conditions. Tyrosine-phosphorylated receptors were detected by immunoblotting using an antiphosphotyrosine antibody (PY99, Santa Cruz). The membranes were stripped and reprobed using a polyclonal antibody against the C-terminal of the PDGFRs (CED) to detect receptor expression levels.
To monitor cell growth, both the cell proliferation reagent WST-1 (Roche) and BrdU (Sigma) were used. A total of 0.4 × 104 (WST-1) or 1 × 104 (BrdU) wild-type and tPA-deficient fibroblasts were seeded in triplicate–hexaplicate, and after attachment they were serum-starved overnight. Serum-starved cells were counted (WST-1 seeding control) and alternatively incubated for 24 h in serum-free medium supplemented with 1 mg ml−1 BSA, and 50 μM BrdU in the BrdU experiment, in the absence or presence of 50 ng ml−1 activated PDGF-CC or tPA protein (#116, American Diagnostica Inc.). Upon counting, WST-1 reagent was added and measured according to the manufacturer's protocol using an ELISA reader. In the BrdU experiment, the cells were fixed in 4% paraformaldehyde in PBS for 30 min at room temperature and the DNA was denatured in 2 M HCl for 20 min at room temperature and then blocked in 0.5% BSA, 0.5% Tween, and 10% goat serum in PBS. BrdU was localized by a monoclonal anti-BrdU antibody (DAKO), and proliferating cells were visualized by an Alexa594-conjugated mouse secondary antibody (Molecular Probe). To visualize all nuclei, DAPI (1 μg ml−1, Roche) was included in the secondary antibody solution. Quantification of the BrdU-positive cells was performed by counting all cells along the vertical and horizontal diameters of all wells.
Immunohistochemical analysis of PDGF-C and tPA expression
Expression analysis of PDGF-C and tPA was performed by immunohistochemistry using tissue sections from E14.5 mouse embryos and T241 tumor xenografts generated from syngenic mice essentially as described previously (Aase et al, 2002). The primary antibodies used were affinity-purified rabbit antibodies directed against human PDGF-C and rabbit anti-mouse tPA IgG (#387, American Diagnostica Inc.). As negative controls, the sections were incubated only with secondary Ig or preimmune rabbit IgG, and in all cases only background staining was observed.
Acknowledgments
We thank Erika Folestad for the kind gifts of recombinant PDGF-D and antibodies to PDGF-D, and Peter Carmeliet for the gift of tPA-deficient mice. This work was supported by grants from The Novo Nordisk Foundation, The Swedish Research Council, and Karolinska Insitutet.
References
- Aase K, Abramsson A, Karlsson L, Betsholtz C, Eriksson U (2002) Expression analysis of PDGF-C in adult and developing mouse tissues. Mech Dev 110: 187–191 [DOI] [PubMed] [Google Scholar]
- Andrae J, Molander C, Smits A, Funa K, Nister M (2002) Platelet-derived growth factor-B and -C and active α-receptors in medulloblastoma cells. Biochem Biophys Res Commun 296: 604–611 [DOI] [PubMed] [Google Scholar]
- Bergsten E, Uutela M, Li X, Pietras K, Östman A, Heldin CH, Alitalo K, Eriksson U (2001) PDGF-D is a specific, protease-activated ligand for the PDGF β-receptor. Nat Cell Biol 3: 512–516 [DOI] [PubMed] [Google Scholar]
- Boucher P, Gotthardt M, Li WP, Anderson RG, Herz J (2003) LRP: role in vascular wall integrity and protection from atherosclerosis. Science 300: 329–332 [DOI] [PubMed] [Google Scholar]
- Bradford M (1976) A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem 72: 248–254 [DOI] [PubMed] [Google Scholar]
- Cao R, Bråkenhielm E, Li X, Pietras K, Widenfalk J, Östman A, Eriksson U, Cao Y (2002) Angiogenesis stimulated by PDGF-CC, a novel member in the PDGF family, involves activation of PDGFR-αα and -αβ receptors. FASEB J 16: 1575–1583 [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Schoonjans L, Kieckens L, Ream B, Degen J, Bronson R, De Vos R, van den Oord JJ, Collen D, Mulligan RC (1994) Physiological consequences of loss of plasminogen activator gene function in mice. Nature 368: 419–424 [DOI] [PubMed] [Google Scholar]
- Carroll PM, Tsirka SE, Richards WG, Frohman MA, Strickland S (1994) The mouse tissue plasminogen activator gene 5′ flanking region directs appropriate expression in development and a seizure-enhanced response in the CNS. Development 120: 3173–3183 [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction. Anal Biochem 162: 156–159 [DOI] [PubMed] [Google Scholar]
- Collen D (2001) Ham-Wasserman lecture: role of the plasminogen system in fibrin-homeostasis and tissue remodeling. Hematology (Am Soc Hematol Educ Program) 1–9 [DOI] [PubMed] [Google Scholar]
- Deutsch DG, Mertz ET (1970) Plasminogen: purification from human plasma by affinity chromatography. Science 170: 1095–1096 [DOI] [PubMed] [Google Scholar]
- Dijkmans J, Xu J, Masure S, Dhanaraj S, Gosiewska A, Geesin J, Sprengel J, Harris S, Verhasselt P, Gordon R, Yon J (2002) Characterization of platelet-derived growth factor-C (PDGF-C): expression in normal and tumor cells, biological activity and chromosomal localization. Int J Biochem Cell Biol 34: 414–426 [DOI] [PubMed] [Google Scholar]
- Ding H, Wu X, Kim I, Tam PP, Koh GY, Nagy A (2000) The mouse Pdgfc gene: dynamic expression in embryonic tissues during organogenesis. Mech Dev 96: 209–213 [DOI] [PubMed] [Google Scholar]
- Eghbali M, Tomek R, Woods C, Bhambi B (1991) Cardiac fibroblasts are predisposed to convert into myocyte phenotype: specific effect of transforming growth factor beta. Proc Natl Acad Sci USA 88: 795–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson A, Siegbahn A, Westermark B, Heldin C-H, Claesson-Welsh L (1992) PDGF α- and β-receptors activate unique and common signal transduction pathways. EMBO J 11: 543–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbertson DG, Duff ME, West JW, Kelly JD, Sheppard PO, Hofstrand PD, Gao Z, Shoemaker K, Bukowski TR, Moore M, Feldhaus AL, Humes JM, Palmer TE, Hart CE (2001) Platelet-derived growth factor C (PDGF-C) a novel growth factor that binds to PDGF α and β receptor. J Biol Chem 276: 27406–27414 [DOI] [PubMed] [Google Scholar]
- Heldin C-H, Westermark B (1999) Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev 79: 1283–1316 [DOI] [PubMed] [Google Scholar]
- Hoylaerts M, Rijken DC, Lijnen HR, Collen D (1982) Kinetics of the activation of plasminogen by human tissue plasminogen activator—role of fibrin. J Biol Chem 257: 2912–2919 [PubMed] [Google Scholar]
- Kwaan HC (1992) The plasminogen–plasmin system in malignancy. Cancer Metast Rev 11: 291–311 [DOI] [PubMed] [Google Scholar]
- LaRochelle WJ, Jeffers M, McDonald WF, Chillakuru RA, Giese NA, Lokker NA, Sullivan C, Boldog FL, Yang M, Vernet C, Burgess CE, Fernandes E, Deegler LL, Rittman B, Shimkets J, Shimkets RA, Rothberg JM, Lichenstein HS (2001) PDGF-D, a new protease-activated growth factor. Nat Cell Biol 3: 517–521 [DOI] [PubMed] [Google Scholar]
- Li H, Fredriksson L, Li X, Eriksson U (2003) PDGF-D is a potent transforming and angiogenic growth factor. Oncogene 22: 1501–1510 [DOI] [PubMed] [Google Scholar]
- Li X, Eriksson U (2003) Novel PDGF family members: PDGF-C and PDGF-D. Cytokine Growth Factor Rev 14: 91–98 [DOI] [PubMed] [Google Scholar]
- Li X, Pontén A, Aase K, Karlsson L, Abramsson A, Uutela M, Bäckström G, Hellström M, Boström H, Li H, Soriano P, Betsholtz C, Heldin C-H, Alitalo K, Östman A, Eriksson U (2000) PDGF-C is a new protease-activated ligand for the PDGF-α receptor. Nat Cell Biol 2: 302–309 [DOI] [PubMed] [Google Scholar]
- Lokker NA, Sullivan CM, Hollenbach SJ, Israel MA, Giese NA (2002) Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer Res 62: 3729–3735 [PubMed] [Google Scholar]
- Nakamura F, Goshima Y (2002) Structural and functional relation of neuropilins. Adv Exp Med Biol 515: 55–69 [DOI] [PubMed] [Google Scholar]
- Nicole O, Docagne F, Ali C, Margaill I, Carmeliet P, MacKenzie ET, Vivien D, Buisson A (2001) The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat Med 7: 59–64 [DOI] [PubMed] [Google Scholar]
- Östman A, Thyberg J, Westermark B, Heldin C-H (1992) PDGF-AA and PDGF-BB biosynthesis: proprotein processing in the Golgi complex and lysosomal degradation of PDGF-BB retained intracellularly. J Cell Biol 118: 509–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranby M (1982) Studies on the kinetics of plasminogen activation by tissue plasminogen activator. Biochim Biophys Acta 704: 461–469 [DOI] [PubMed] [Google Scholar]
- Rickles RJ, Strickland S (1988) Tissue plasminogen activator mRNA in murine tissues. FEBS Lett 229: 100–106 [DOI] [PubMed] [Google Scholar]
- Siegfried G, Khatib AM, Benjannet S, Chretien M, Seidah NG (2003) The proteolytic processing of pro-platelet-derived growth factor-A at RRKR(86) by members of the proprotein convertase family is functionally correlated to platelet-derived growth factor-A-induced functions and tumorigenicity. Cancer Res 63: 1458–1463 [PubMed] [Google Scholar]
- Strickland S (2001) Tissue plasminogen activator in nervous system function and dysfunction. Thromb Haemost 86: 138–143 [PubMed] [Google Scholar]
- Sturzebecher J, Prasa D, Hauptmann J, Vieweg H, Wikstrom P (1997) Synthesis and structure–activity relationships of potent thrombin inhibitors: piperazides of 3-amidinophenylalanine. J Med Chem 40: 3091–3099 [DOI] [PubMed] [Google Scholar]
- Takeuchi T, Shuman MA, Craik CS (1999) Reverse biochemistry: use of macromolecular protease inhibitors to dissect complex biological processes and identify a membrane-type serine protease in epithelial cancer and normal tissue. Proc Natl Acad Sci USA 96: 11054–11061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The National Institute of Neurological Disorders and Stroke rtPA Stroke Study Group (1995) Tissue plasminogen activator for acute ischemic stroke. N Engl J Med 333: 1581–1587 [DOI] [PubMed] [Google Scholar]
- Thielens NM, Bersch B, Hernandez JF, Arlaud GJ (1999) Structure and functions of the interaction domains of C1r and C1s: keystones of the architecture of the C1 complex. Immunopharmacology 42: 3–13 [DOI] [PubMed] [Google Scholar]
- Tsirka SE (2002) Tissue plasminogen activator as a modulator of neuronal survival and function. Biochem Soc Trans 30: 222–225 [DOI] [PubMed] [Google Scholar]
- Uutela M, Lauren J, Bergsten E, Li X, Horelli-Kuitunen N, Eriksson U, Alitalo K (2001) Chromosomal location, exon structure, and vascular expression patterns of the human PDGFC and PDGFD genes. Circulation 103: 2242–2247 [DOI] [PubMed] [Google Scholar]
- Vassalli JD, Sappino AP, Belin D (1991) The plasminogen activator/plasmin system. J Clin Invest 88: 1067–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YP, Siao CJ, Lu W, Sung TC, Frohman MA, Milev P, Bugge TH, Degen JL, Levine JM, Margolis RU, Tsirka SE (2000) The tissue plasminogen activator (tPA)/plasmin extracellular proteolytic system regulates seizure-induced hippocampal mossy fiber outgrowth through a proteoglycan substrate. J Cell Biol 148: 1295–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yepes M, Sandkvist M, Coleman TA, Moore E, Wu JY, Mitola D, Bugge TH, Lawrence DA (2002) Regulation of seizure spreading by neuroserpin and tissue-type plasminogen activator is plasminogen-independent. J Clin Invest 109: 1571–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yepes M, Sandkvist M, Moore EG, Bugge TH, Strickland DK, Lawrence DA (2003) Tissue-type plasminogen activator induces opening of the blood–brain barrier via the LDL receptor-related protein. J Clin Invest 112: 1533–1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwerner JP, May WA (2001) PDGF-C is an EWS/FLI induced transforming growth factor in Ewing family tumors. Oncogene 20: 626–633 [DOI] [PMC free article] [PubMed] [Google Scholar]