

Abstract
The generation of complex neuronal structures, such as the neocortex, requires accurate positioning of neurons and glia within the structure, followed by differentiation, formation of neuronal connections, and myelination. To understand the importance of TrkB signaling during these events, we have used conditional and knockin mutagenesis of the TrkB neurotrophin receptor, and we now show that this tyrosine kinase receptor, through docking sites for the Shc/FRS2 adaptors and phospholipase Cγ (PLCγ), coordinates these events in the cerebral cortex by (1) controlling cortical stratification through the timing of neuronal migration during cortex formation, and (2) regulating both neuronal and oligodendrocyte differentiation. These results provide genetic evidence that TrkB regulates important functions throughout the formation of the cerebral cortex via recruitment of the Shc/FRS2 adaptors and PLCγ.
Keywords: cell differentiation, cell migration, neocortex development, TrkB signaling
Introduction
During development of the central nervous system (CNS) of higher vertebrates, a complex pattern of neuronal connections is established. In the cerebral cortex, the generation of this intricate pattern is initially dependent on the proper migration of newly generated neurons toward their final position in a specific cortical layer. The birthday of a neuron seems to define its eventual position and properties, such that neurons born at early stages of cortical development migrate to the deepest cortical layers, while those born at later times end up in progressively more superficial layers (Gilmore and Herrup, 1997). A delay in neuronal migration could therefore affect subsequent events such as neuronal stratification. The neurotrophin brain-derived neurotrophic factor (BDNF) and its high-affinity receptor tyrosine kinase, TrkB, have been implicated in, among numerous other biological functions in the nervous system (Bibel and Barde, 2000), the stimulation of migration of cerebellar granule neurons. In that study, the authors showed that BDNF/TrkB signaling initiates the movement of granule cells from the external granule cell layer (EGL) to their final position in the internal granule cell layer (IGL) (Borghesani et al, 2002). In the present study, we have asked whether TrkB signaling is required for the formation of a more complex structure, the cerebral cortex, by regulating the timing of migration of newly generated cortical neurons to their final positions. Moreover, the signaling pathways downstream of the TrkB receptor, which are responsible for neurotrophin-induced cell migration, are not well defined. TrkB activation leads to the recruitment of adaptor proteins that trigger well-characterized signaling cascades. The binding of Shc/FRS2 to the phosphotyrosine (pTyr) 515 primarily leads to the activation of the Ras/MAPK and the PI3K pathways. Phospholipase Cγ (PLCγ), by binding pTyr 816, instead results in an increase in intracellular Ca2+ and the activation of the calcium/calmodulin kinase pathway.
The premature death of trkB null mutant mice has necessitated the use of conditional mutagenesis to study the role of TrkB signaling in the CNS. We have previously shown that specific deletion of trkB from the postnatal forebrain results in compromised learning and impaired hippocampal CA3–CA1 long-term potentiation (LTP) (Minichiello et al, 1999). By analysing mice carrying point mutations at either the PLCγ or the Shc site, we have further shown that the PLCγ site is necessary for TrkB-mediated hippocampal LTP, but not the Shc site (Minichiello et al, 2002).
In the present study, we have conditionally deleted trkB from the embryonic CNS in order to examine its role in neocortical development. In addition, to understand the signaling mechanisms by which TrkB regulates cortical development, we have generated mice with a targeted mutation in both the Shc- and PLCγ-docking sites of TrkB (trkBD/D mice), and compared these to existing strains carrying a single point mutation in either the Shc or the PLCγ site of the receptor (Minichiello et al, 2002). Our results indicate that the absence of TrkB signaling during embryogenesis affects the timing of migration of newly born cortical neurons, resulting in their altered positioning in the newly forming cortical layers. In addition, we observed differentiation defects in diverse populations of neurons and glia in the neocortex, hypomyelination, and altered brain physiology. Moreover, we find that together, but not singly, the Shc and PLCγ sites of TrkB are necessary for normal neuronal migration and differentiation of neurons and glia during cortical development. Biochemical analysis of primary cortical neurons derived from the different trkB point mutant and control mice indicate that the Ras/MAPK and PI3K/AKT pathways are likely responsible for mediating these functions of TrkB during cortical development.
Results
Removal of TrkB in developing cortex
To investigate the role of TrkB signaling during neocortex development, we have used two mouse lines, the trkB floxed and the transgenic NestinCre (NesCre), both of which have been described previously (Minichiello et al, 1999; Tronche et al, 1999). As the expression of TrkB receptors is widespread in the peripheral nervous system (PNS) and the CNS (Klein et al, 1990), and the NesCre line has previously been examined mainly for Cre activity in the CNS, we assayed for Cre activity in the PNS by crossing NesCre mice to a lacZ reporter mouse line (Akagi et al, 1997). Whole-mount X-gal staining at embryonic day 11.5 (E11.5) (Figure 1A) revealed that Cre recombination occurred throughout the neural tube in agreement with previous studies (Graus-Porta et al, 2001). In the PNS, we found very little X-gal staining in the V ganglia (trigeminal), and no staining was observed in either the VIII ganglia (vestibulocochlear) or the IX and X ganglia (glossopharingeal and vagus, respectively) (Figure 1B). We then crossed heterozygous trkB floxed mice bearing the NesCre transgene with homozygous trkBlox/lox mice to generate trkBlox/lox; NesCre+/− mice (or simply ‘trkBNesCre' mice). trkBNesCre were viable, and overall did not differ significantly from their control littermates in their growth from birth until postnatal day 21 (P21). Beyond this age, however, their growth was retarded, as shown in Supplementary Figure 1A. Moreover, trkBNesCre mice were hyperactive, and in general lived less than their heterozygous or wild-type (+/+) littermates (dying between 2 and 8 months). TrkB protein levels in forebrain lysates of trkBNesCre mice were greatly reduced by E12, barely detectable at P1, and undetectable at later stages such as P45 (Figure 1C and D). As TrkB expression has been reported in glial progenitor cells (Climent et al, 2000), we cultured glial cells from P4 cortices of trkBNesCre and control mice, enriching for astrocytes following the McCarthy and De Vellis method, and found undetectable levels of TrkB in mutant cells compared to controls (Figure 1E). The control mice used in this study include +/+, trkBlx/+, and trkBlx/+; Nescre+/− (or simply ‘trkBlx/+;NesCre' mice).
Figure 1.
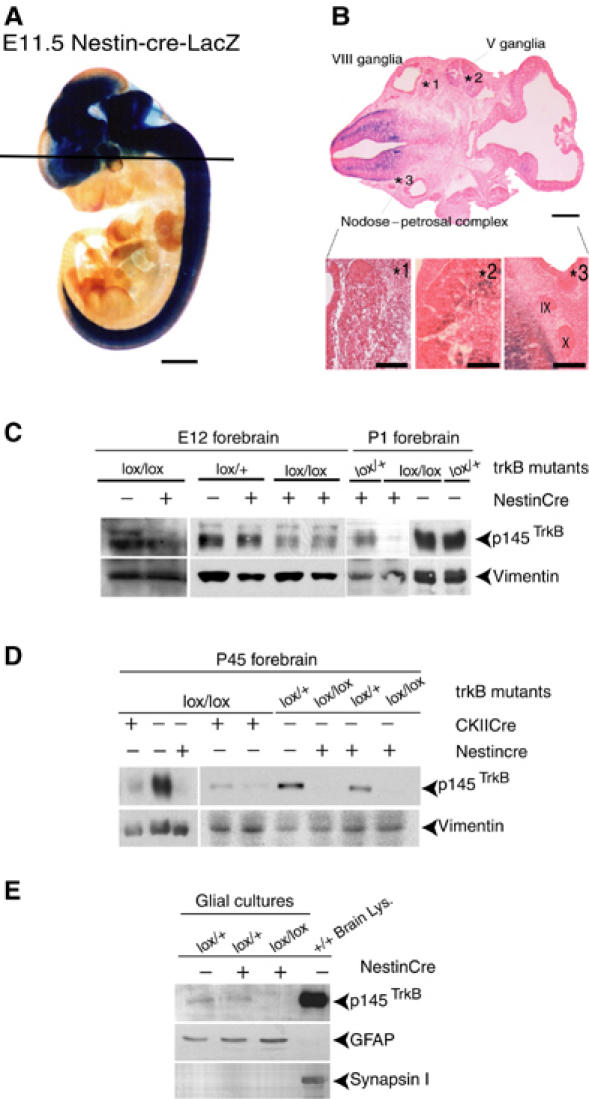
Analysis of NesCre transgenic line and gp145TrkB protein removal. (A, B) Spatial pattern of Cre activity. (A) E11.5 embryo double transgenic: lox-lacZ; NesCre mice stained for β-galactosidase activity. (B) Horizontal sections (anterior right) showing the VIII (*1), the V (*2), and the (*3; IX and X, respectively) peripheral ganglia. (C–E) Western blot analysis of gp145TrkB removal in forebrains of E12, and P1 trkBlox/+ and trkBlox/lox mice, either in the absence (−) or presence (+) of the NesCre transgene (C), in forebrains of P45 controls, trkBNesCre, or trkBlox/lox mice expressing the CKIICre transgene (D), blots were reprobed with anti-vimentin Ab to control for the protein levels, and in astrocyte cultures prepared from brains of P4 control or trkBNesCre mice. These cultures were positive for the astrocyte-specific marker, GFAP, and negative for the neuronal-specific marker, Synapsin I. Also note that, in panel (D), gp145TrkB protein is undetectable in trkBNesCre, whereas residual signal is observed in trkBCaMKIICre mice, as described previously (Minichiello et al, 1999). Scale bars: (A), 1 mm; (B), 400 μm; inset, 150 μm.
TrkB signaling regulates timing of cortical neuron migration
Since most cerebral cortical neurons are generated between E11 and E17 in the mouse, to detect potential defects in neuronal migration in the absence of TrkB signaling, we labeled proliferating neurons of control and conditional mutant mice with bromodeoxyuridine (BrdU) at two stages (E12.5 and E14.5), and analyzed the extent of their migration at E18.5. The body weight of trkBNesCre E18.5 embryos and their control littermates was found to be equivalent (Supplementary Figure 1B), as was their general anatomy (data not shown), suggesting normal embryonic development. As shown in Figure 2A–D, BrdU-labeled cells at E12.5, and analysed at E18.5, were distributed more diffusely in the central part of the cortical plate (CP) in trkBNesCre mice (Figure 2B, arrows) compared with control brains, which showed a more defined band of labeled cells in the inner layers (Figure 2A, arrows). In E18.5 control mice exposed to BrdU at E14.5, a well-defined band of labeled cells was found in the outer layers of the CP close to the marginal zone, while only a few labeled cells were seen in the inner layers (Figure 2C). Conversely, in the trkBNesCre, labeled cells were observed scattered throughout the CP (Figure 2D, arrows), indicating a delay in neuronal migration. Counts of cells BrdU labeled at E12.5, and analysed at E18.5, revealed a significantly decreased number in the deeper (Deep) layer of the cerebral cortex of trkBNesCre mice compared with their littermate controls, and an increased number of BrdU-positive cells in the middle (Mid) layer (Figure 2E and Table I). Similarly, quantitative analysis of cells BrdU labeled at E14.5 revealed a significant reduction in the number of BrdU-positive cells in the superficial (Sup) layer of the cerebral cortex in trkBNesCre mice at E18.5, and a significantly higher number of labeled cells in the Mid and Deep layers compared with their littermate controls (Figure 2F and Table I). The total number of BrdU-labeled cells in E18.5 trkBNesCre and control mice injected at either E12.5 or E14.5 was similar, indicating a comparable degree of neuronal proliferation (Figure 2, insets E and F and Table I). These results suggest that TrkB signaling is important for the timing of directed migration of newly generated neurons rather than for their proliferation. The expression of reelin, which is necessary for the formation of the inside-out layer organization of the cortex (Gilmore and Herrup, 1997), was not altered in calretinin-positive Cajal-Retzius cells of trkBNesCre (Figure 2G and H), suggesting that reelin is not involved in the delayed migration phenotype observed.
Figure 2.
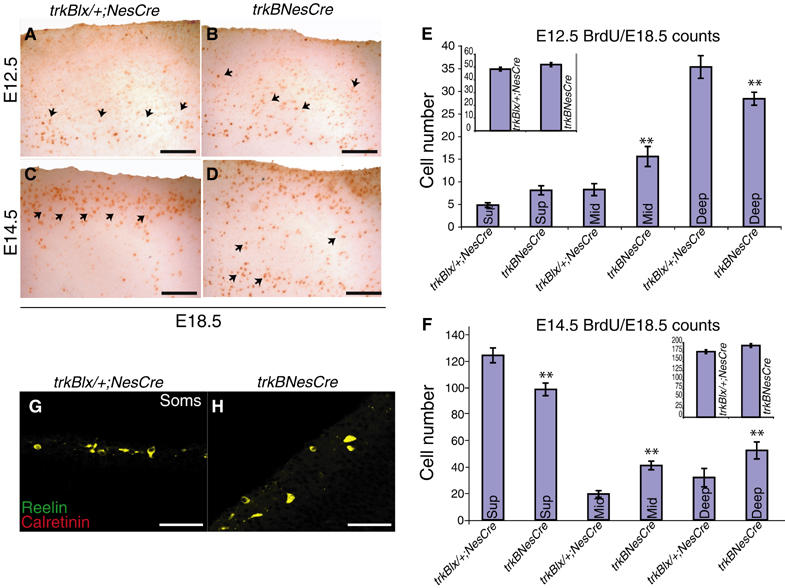
Cortical cell migration in trkBNesCre mice. (A–D) Comparable sagittal sections from the Soms cortex stained with anti-BrdU Ab. Cells labeled either at E12.5 (A,B) or at E14.5 (C, D) with BrdU were analyzed at E18.5. (E, F) Quantitative analysis of cortical BrdU-positive cells in trkBNesCre mice and littermate controls labeled either at E12.5 (E) or E14.5 (F) and analyzed at E18.5. (G, H) Cajal-Retzius cells in the Soms cortices of control (G) and trkBNesCre newborn mice (H) identified by double IMF with Abs against calretinin and reelin. **Statistically significant. Scale bars: (A–D), 100 μm; (G, H), 50 μm.
Table 1.
Counts of cells BrdU-labeled at E12.5 and E14.5 and analysed at E18.5
| Genotype |
|
||
|---|---|---|---|
| Cortical region | trkBlx/+;NesCre | trkBNesCre | t-test |
| E12.5>E18.5 | |||
| Sup | 5±1 | 8±1 | P=0.1 |
| Mid | 8±1 | 16±2 | P=0.002 |
| Deep | 35±1 | 28±3 | P=0.003 |
| Total cell number | 48±1 | 52±1 | P=0.7 |
| E14.5>E18.5 | |||
| Sup | 124±6 | 99±5 | P=0.0007 |
| Mid | 19±3 | 41±3 | P=0.004 |
| Deep | 32±6 | 52±6 | P=0.006 |
| Total cell number | 175±4 | 192±6 | P=0.7 |
| Genotype |
|||
| |
trkB+/+ |
trkBD/D |
|
| E14.5>E18.5 | |||
| Sup | 120±4 | 90±3 | P<0.0001 |
| Mid | 35±2 | 45±4 | P=0.02 |
| Deep | 23±2 | 32±2 | P=0.04 |
| Total cell number | 178±6 | 167±4 | P=0.6 |
| Three animals were analyzed per genotype. BrdU-positive cells± s.e.m. were counted in an area of 400 × 500 μm2, see Materials and methods. | |||
TrkB mediates timing of cortical neuronal migration through the Shc and PLCγ sites
To dissect the signaling pathways responsible for the migration defect observed in the absence of TrkB signaling, we compared mice carrying point mutations in either the Shc or the PLCγ site (Minichiello et al, 2002), or both sites of the TrkB receptor. For the latter mice (designated as trkBD/D), a cDNA knockin strategy (Minichiello et al, 2002) was used to introduce point mutations at both the Shc and the PLCγ site of TrkB (Figure 3A), thereby disrupting the binding of adaptor proteins and the signaling events mediated by these two sites downstream of activated TrkB. At E18.5, the body weight of trkBD/D embryos and their control littermates did not differ (Supplementary Figure 1C), nor did their general anatomy (data not shown), suggesting that trkBD/D embryos develop normally. Although the postnatal development of trkBD/D mice was found to be similar to control littermates up to P7, after this time the growth of the trkBD/D mice was retarded, and the mice showed a typical vestibular organ defect (Sciarretta and Minichiello, unpublished data, 2004). Their lifespan was 2–4 weeks. Mice with point mutations at either the Shc or PLCγ site of TrkB showed normal CNS development (data not shown; Minichiello et al, 1998, 2002), suggesting that multiple pathways mediate TrkB function during cortical development. To test this hypothesis, we examined neuronal migration in the cortices of trkBD/D mutants, using the same methods as discussed above for the trkBNesCre mice, with either +/+ or heterozygous (trkBD/+) littermates serving as controls. Similar to what was described for the trkBNesCre, trkBD/D cells labeled with BrdU at E12.5 resided mainly in the central and partially in the deepest region of the CP at E18.5, compared to the control situation where labeled cells were found in the deepest part of the CP (data not shown). By E18.5, control mice injected with BrdU at E14.5 showed a well-defined band of labeled cells in the outer layers of the CP, and a few scattered cells in the most inner layers (Figure 3C, arrows). At this same stage, trkBD/D mutant mice had many more BrdU-positive cells scattered throughout the CP (Figure 3D, arrows). Quantitative analysis of trkBD/D cells BrdU labeled at E14.5, and analyzed at E18.5, revealed a significant reduction in the number of BrdU-positive cells in the Sup layers of the CP, and a significantly higher number of labeled cells in the Mid and Deep layers of the CP compared to littermate controls (Figure 3E and Table I). Figure 3 inset E and Table I show that a similar total number of BrdU-labeled cells were present in the different layers of E18.5 trkBD/D and control mice, indicating a comparable degree of neuronal proliferation. Together, these results reveal a delay in neuronal migration in trkBD/D embryos that is similar, although less pronounced, to that observed in the trkBNesCre. Therefore, mainly the Shc and the PLCγ sites are involved in TrkB-mediated neuronal migration.
Figure 3.
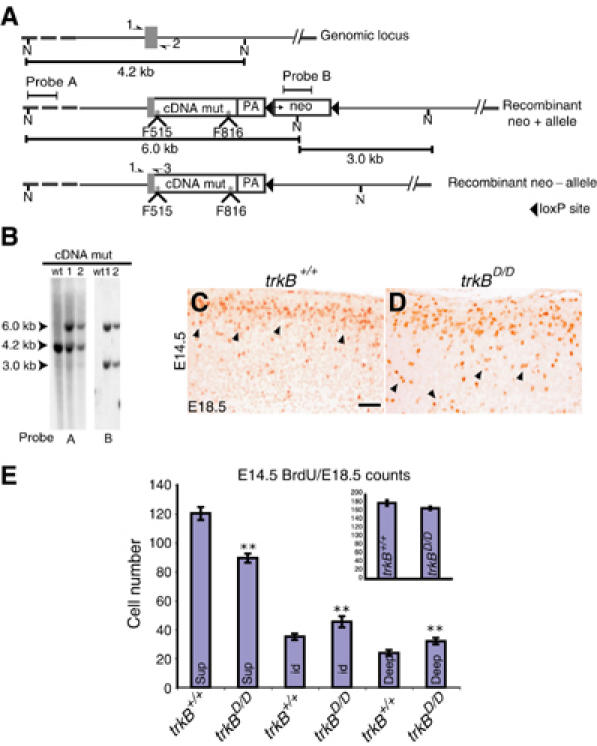
Cortical neuron migration is dependent on the Shc and pLCγ sites of TrkB. (A) Schematic diagram of +/+ trkB locus and mutant alleles. * indicates the point mutation introduced at the Shc and PLCγ sites (*F515, *F816). (B) Southern blot analysis of NcoI digests of genomic DNA prepared from neo-resistant (trkBD/+) and +/+ Es cell clones. (C, D) Delayed migration of neurons in the Soms cortex of E18.5 trkBD/D mutants compared to control littermate, as revealed by the BrdU staining of cells labeled at E14.5. (E) Quantitative analysis of BrdU-positive cells in trkBD/D and control mice labeled at E14.5 and analysed at E18.5. Scale bar (C, D), 50 μm.
Signaling pathways involved in TrkB-mediated neuronal migration
To investigate the signaling pathways that mediate neuronal migration downstream TrkB, we performed biochemical analyses on primary cortical neurons derived from E15.5 mouse embryos of the different point mutants and control mice. BDNF stimulation was used to induce rapid autophosphorylation of mutant and control TrkB receptors. As expected, the amount of autophosphorylated TrkBD receptor was highly diminished compared to the TrkBPLC and TrkBWT receptors, but not abolished (Figure 4A), whereas the expression levels of TrkBD versus TrkBPLC and TrkBWT receptors were not significantly different (P=0.3) (Figure 4B). To investigate if the mutant TrkBD receptor was still able to activate downstream effectors upon BDNF induction, we examined the phosphorylation of Gab1, a major adaptor protein recruited to activated TrkB. As shown in Figure 4C, mutation at the PLCγ site of TrkB did not affect the phosphorylation of Gab1 compared to controls (+/+, trkBWT/WT), whereas its phosphorylation was clearly decreased when the Shc site was mutated. A similar decrease was observed in the trkBD/D mutant neurons, suggesting first that TrkBD is catalytically active, and, second, that Gab1 activation downstream TrkB is mediated mainly through the Shc site. PLCγ is, however, able to recruit Gab1 to TrkB (Figure 4D), but, based on the results for the trkBD/D mutant, this recruitment does not contribute to Gab1 phosphorylation. We next examined the activation of two major pathways downstream TrkB receptors, the Ras/MAPK and the PI3K/AKT pathways. We have previously shown that, in dissociated cultures of primary trkBPLC/PLC cortical neurons stimulated with BDNF, ERK1/ERK2 is activated to the same extent as observed in +/+ or trkBWT/WT control cells, whereas BDNF-treated trkBSHC/SHC mutant cells show both a reduced and transient activation of MAPKs (Figure 4E; Minichiello et al, 2002). TrkBD-expressing cells showed a further reduced and more transient activation of ERK1/ERK2 in response to BDNF compared to TrkBSHC-expressing cells. Similar results were observed when we examined the activation of AKT, an effector of the PI3K pathway. Phosphorylation of AKT was not affected in the trkBPLC/PLC mutant cells compared to +/+ or trkBWT/WT controls, whereas mutation of the Shc site in TrkB significantly reduced AKT phosphorylation following BDNF treatment (Figure 4F; Postigo et al, 2002), and a further reduction was observed in trkBD/D mutant cells (Figure 4F). Together, these results indicate that both the Shc and PLCγ sites are necessary to fully activate MAPKs and AKT downstream TrkB, but additional uncharacterized pathways are also involved in their activation, although to a lesser extent. Furthermore, our data also suggest that threshold levels of Ras/MAPK and PI3K/AKT pathway activation are crucial for proper TrkB-mediated cortical neuronal migration.
Figure 4.
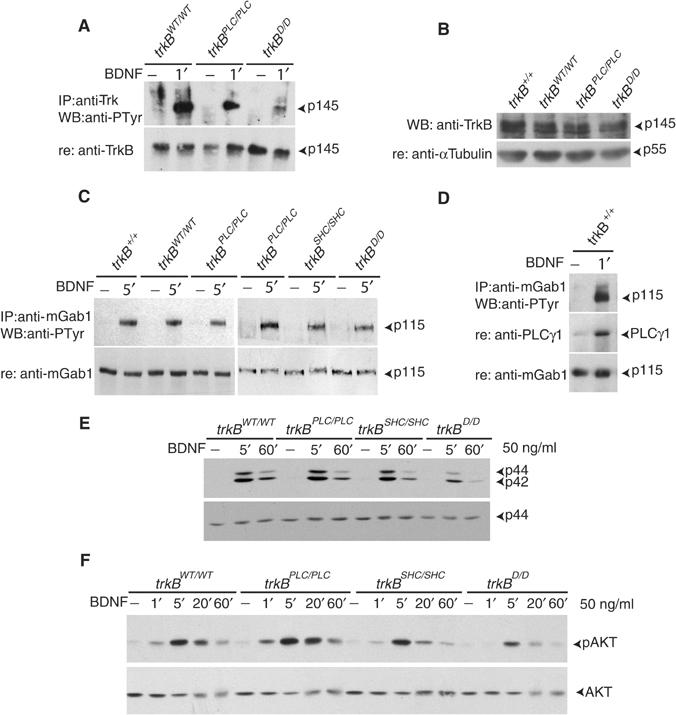
Signaling downstream of mutant TrkB receptors. (A, B) Autophosphorylation and expression levels of TrkB mutant receptors in primary neurons. Cell lysates of cortical neurons derived from either trkBWT/WT control or trkBPLC/PLC and trkBD/D mutants were either left untreated (−) or BDNF stimulated for 1′, immunoprecipitated with anti-Trk Abs followed by IB with anti-pTyr Abs. The blot was reprobed with anti-TrkB Abs to visualize TrkB levels (A). Untreated (−) cell lysates were subjected to SDS–PAGE, followed by IB with TrkB Abs to visualize TrkB levels (B). (C) Tyrosine phosphorylation of Gab1 in the different TrkB mutant and control cortical neurons upon BDNF stimulation. (D) Recruitment of Gab1 to the PLCγ site of TrkB upon BDNF stimulation. (E, F) Time course of MAPK and AKT phosphorylation upon BDNF stimulation of control and mutant primary neurons. Cell lysates were subjected to SDS–PAGE, followed by IB with Abs against the phosphorylated forms of p42 and p44 ERKs (E) or AKT (F). Blots were reprobed with Abs against p44 or AKT, respectively, to control for loading.
Loss of TrkB signaling affects neocortical development
Having observed a defect in migration due to the loss of TrkB signaling during neocortical development, we sought to determine if this was accompanied by defects in neuronal differentiation. Although little is known about the specific signals that neurons receive as they migrate in the CNS, it is apparent that their differentiation is directed by the changing cellular environment through which they travel (Purves et al, 2001). Thus, the position occupied by a cell early in development is crucial in determining its proper developmental program. To evaluate these effects, we initially examined the gross morphology of brains isolated from 28-week-old trkBNesCre. We observed severe caudal retraction of the cortical hemispheres, but normal olfactory bulbs and cerebellum (data not shown). The visual cortex was severely compressed compared to +/+ and trkBlx/+;NesCre controls (data not shown), and to trkBCaMKIICre mutants, where deletion of trkB occurs only after development (Minichiello et al, 1999). Similar results were observed in the somatosensory (Soms) cortex where, although the pattern of lamination was normal, the thickness of all layers (I–VI) was reduced (Figure 5A and B). Moreover, layer II/III of trkBNesCre was slightly compressed and neurons within the layer were tightly packed compared to the other layers and to that of control mice (Figure 5A and B). To further characterize the effects of loss of TrkB signaling during neocortical development, we examined specific populations of cortical neurons in brain sections of P35–P42 trkBNesCre and control mice by immunostaining (IMS). Analysis of calbindin-expressing neurons confirmed that neurons in layer II/III were tightly packed in trkBNesCre compared to controls (data not shown). Conversely, the distribution and expression of interneurons positive for either parvalbumin (PARV) or calretinin was normal in the trkBNesCre compared to control mice (data not shown). Contrary to the neocortex, the expression of PARV was reduced in the entorhinal cortex (ENT) and the hippocampus (Figure 5C–F) of conditional mutants. To quantify this result, we analyzed PARV levels by immunoblot (IB) (Figure 5G and H), and found indeed a significant reduction in forebrain lysates of P60–90 mutant compared to control mice. Moreover, in other brain regions, like the cerebellum, the expression of PARV was found to be normal by immunohistochemical (IHC) (Supplementary Figure 2A and B) and IB analysis (data not shown). To assay dendritic differentiation, we examined the expression of MAP2 and observed a dramatic alteration of the apical and basal cortical dendrites of trkBNesCre. While in the most superficial layers dendrites appeared fasciculated and poorly stained (Figure 5I and J), in the deepest layers (V–VI) staining of fibers and cell bodies was further reduced in trkBNesCre compared to controls (Figure 5K and L). To quantify this result, we examined cortical MAP2 levels by IB (Figure 5M and N) and found a significant reduction in lysates of P60–90 mutant compared to control mice. Moreover, in other brain regions, like the cerebellum (Figure 5M and N) and the hippocampus (Supplementary Figure 2C and D), the expression of MAP2, assayed by either IB or IHC, respectively, was found to be unchanged in trkBNesCre mice. These results clearly indicate that delayed neuronal migration upon removal of TrkB signaling during cortical development correlates with defects in neuronal differentiation.
Figure 5.
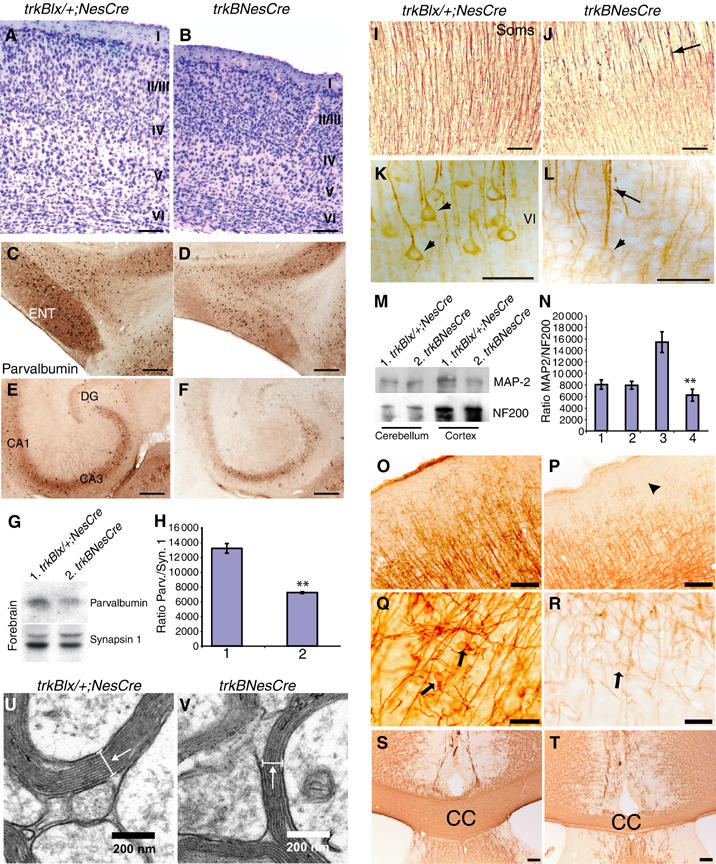
Neocortical development in trkBNesCre mice. (A, B) Coronal sections of 28-week-old brains of trkBNesCre and controls stained with Cresyl violet. (C–F) Sagital sections of control and mutant mice stained with anti-PARV Ab. (G, H) Analysis of PARV protein levels in forebrain lysates from control and trkBNesCre mice. To control for loading, the blot was reprobed with anti-synapsin Abs (G, lower panel). (H) Densitometric analyses of PARV levels in the forebrain of trkBNesCre compared to control mice (H, lane 2 compared with control lane 1), **P<0.002. (I–L) MAP2 IMS in trkBNesCre cortices (J, L) compared to controls (I, K); arrows in (J, L) indicate the apparent fasciculation of dendrites in trkBNesCre and arrowhead in (L) indicates reduced staining of MAP2 in cell bodies of trkBNesCre compared to controls (K). (M, N) Analysis of MAP-2 protein levels in either cerebellum or cortical lysates of control and trkBNesCre mice. To control for loading, the blot was reprobed with anti-NF200 Abs (M, lower panel). (N) Densitometric analyses of MAP2 levels in the cortex of trkBNesCre compared to control mice (N, lane 4 compared with control lane 3), **P<0.01, and in the cerebellum of trkBNesCre compared to control mice (N, lane 2 compared with control lane 1), P=0.9. (O–R) OL differentiation. Brain sections from control (O–Q) and trkBNesCre mice (P–R) immunostained with anti-CNPase Abs. (S, T) Immunoassays of brain sections using anti-MBP Abs reveal decreased staining and size of the CC in trkBNesCre (T) compared to control mice (S). (U, V) Ultrastructure of myelinated axons in the CC revealing reduced myelin sheaths and myelin thickness in trkBNescre (V) compared to controls (U) (indicated by an arrow). Scale bars: (A–F), 200 μm; (I, J), 100 μm; (K, L), 50 μm; (O, P), 100 μm; (Q, R), 20 μm; (S, T), 100 μm; (U, V), 200 nm.
CNS hypomyelination
In the CNS, axons are myelinated by oligodendrocytes (OLs), which allows for rapid axonal impulse propagation. As OLs primarily support axonal integrity and stabilize myelin structure (Lappe-Siefke et al, 2003), we investigated OL differentiation in trkBNesCre mice. IHC analysis of brain sections from P180 mice stained with CNPase, which differentiates OLs from neurons and astrocytes, revealed clearly diminished labeling of neuronal fibers (Figure 5O and P, and P arrowhead), and much lower levels of CNPase throughout the OL soma (Figure 5Q and R). CNPase IMS also revealed altered corpus callosum (CC) morphology in trkBNesCre mice compared to controls (see Supplementary Figure 2E and F). Moreover, IMS using an antibody (Ab) against OL membranes and CNS myelin (MAB328) showed weak staining and a reduction in the thickness of the CC in trkBNesCre compared to controls (data not shown). IMS also showed that myelin epitopes, such as MBP in the CC or in the cortex, were highly diminished in trkBNesCre compared to control mice (Figure 5S and T; data not shown). To quantify these results, we examined MBP and CNPase protein levels in cell lysates from the cortex and cerebellum of control and trkBNesCre mice at P60–90 (Supplementary Figure 2G). Densitometric analysis revealed a significant reduction in the expression of MBP (Supplementary Figure 2H, lane 1 versus 2, P=0.04) and CNPase (Supplementary Figure 2I, lane 1 versus 2, P=0.01) in the cortex of trkBNesCre compared to controls. Conversely, in the cerebellum, the levels of MBP (Supplementary Figure 2H, lane 3 versus 4, P=0.4) and CNPase (Supplementary Figure 2I, lane 3 versus 4, P=0.6) of control and trkBNesCre mice were similar. Since both full-length and truncated forms of TrkB have been detected in OLs (Condorelli et al, 1995), we wanted to determine the cellular basis for the observed hypomyelination. Quantification of myelinated axons in the stratum radiatum of the CA1 region of P120 trkBNesCre revealed a 17% decrease compared to control mice (Table II). A similar reduction has been previously observed in the trkBCaMKIICre mice (Minichiello et al, 1999). Since the myelination defects were most prevalent in the CC, we analysed the number of myelinated axons, and the number of myelin sheaths and myelin thickness per axon in the CC of trkBNesCre and control mice. We found a significant reduction in the number of myelinated axons (Table II), and a significant reduction in the number of myelin sheaths and the mean thickness of myelin per axon (Table III and Figure 5U and V) in the trkBNesCre compared to controls. Together these results indicate that TrkB signaling regulates myelination in the CNS and that this may occur, at least in part, through the regulation of OL differentiation.
Table 2.
Quantification of myelinated axons in the CA1 and Corpus callosum
|
CA1 |
Corpus callosum |
|||
|---|---|---|---|---|
| Genotype | Myelinated axons/mm2±s.e.m. | t-test | Myelinated axons/mm2±s.e.m. | t-test |
| trkBlx/+;NesCre | 3267±208 | 1 015 833±29 425 | ||
| trkBNesCre | 2700±149 | P=0.04 | 885 000±14 222 | P=0.006 |
| Two animals were analyzed per genotype. Counts were obtained from six semithin sections 20 μm apart per animal and area (see Materials and methods). | ||||
Table 3.
Quantification of myelin sheaths and myelin thickness per axon in the Corpus callosum
| Myelin sheaths | Myelin thickness | |||
|---|---|---|---|---|
| Mean±s.e.m. (number of axons analyzed) | t-test | Mean±s.e.m. (number of axons analyzed) | t-test | |
| trkBlx/+;NesCre | 12.22±0.33 | 126.9±3.75 | ||
| (n=46) | (n=35) | |||
| trkBNesCre | 10.94±0.34 | P=0.009 | 117.6±2.65 | P=0.04 |
| (n=48) | (n=43) | |||
| Two animals were analyzed per genotype. The analysis of myelin sheaths and myelin thickness has been carried on axons with a diameter of about 750 nm (see Materials and methods). | ||||
Functional consequences upon removal of TrkB signaling
Having discovered defects in the migration and differentiation of neurons and OLs upon removal of TrkB signaling during cortical development, we examined the functional consequences derived from these defects. Damage to the CNS causes the activation of both astrocytes and microglia (Streit et al, 1999). We therefore looked for reactive astrogliosis by IMS brain sections with an Ab against glial fibrillary acidic protein (GFAP). In the neocortex of control (Figure 6A–D) and trkBCaMKIICre mice (data not shown), the expression of GFAP was low as expected, whereas in trkBNesCre it was strongly increased already at P15, and remained elevated at all subsequent stages analyzed (P54 in Figure 6A–D). In addition, the GFAP-positive cells showed enlarged processes (Figure 6D and I), a typical morphology of reactive astrocytes. We then looked for the induction of microglia, which is also a normal response to CNS injury or neuronal death. Brain sections stained with an Ab directed against the macrophage surface antigen Mac1 showed no evidence of reactive microglia (data not shown). Interleukin 6 (IL6) protein levels, which are typically elevated in inflammatory responses (Van Wagoner and Benveniste, 1999), were measured in forebrain protein extracts obtained from control, trkBCaMKIICre, and trkBNesCre mice. As shown in Figure 6E, there was no statistically significant augment of IL6 protein levels in trkBNesCre compared to control or trkBCaMKIICre mice (45±5, 43±1, 39±4 pg/ml, respectively, P>0.6, n=4), suggesting a lack of an inflammatory response. To determine whether TrkB signaling was required for the survival of cortical neurons, we analysed brain sections of P42–70 trkBNesCre and control mice, and found no increase in apoptosis either by pyknotic nuclei detection using nissl staining, by TUNEL analysis, or by fluoro Jade staining (data not shown). The failure to detect increased neuronal apoptosis could, however, be due to the slow loss of cortical neurons over a longer period. Although we found no evidence of an increase in cell death, we did find an upregulation of immediate early genes (IEG), such as c-fos, that are known to regulate neuronal excitability and survival (Zhang et al, 2002) (Figure 6F and G). Double immunofluorescence (IMF) staining for c-Fos and GFAP confirmed that the upregulation of c-Fos occurred in neurons (Figure 6H and I). Moreover, IHC analysis of the inhibitory neurotransmitter GABA, or the enzyme that catalyzes its synthesis, GAD 67, and the excitatory neurotransmitter glutamate, revealed a reduction in their levels in the trkBNesCre compared to controls (Figure 6J–O). Together, these results suggest that the absence of TrkB signaling during neocortical development does not give rise to massive cell death or typical inflammatory responses, but does alter neuronal physiology, which likely results in the activation of astrocytes as an attempt by the CNS to restore homeostasis.
Figure 6.
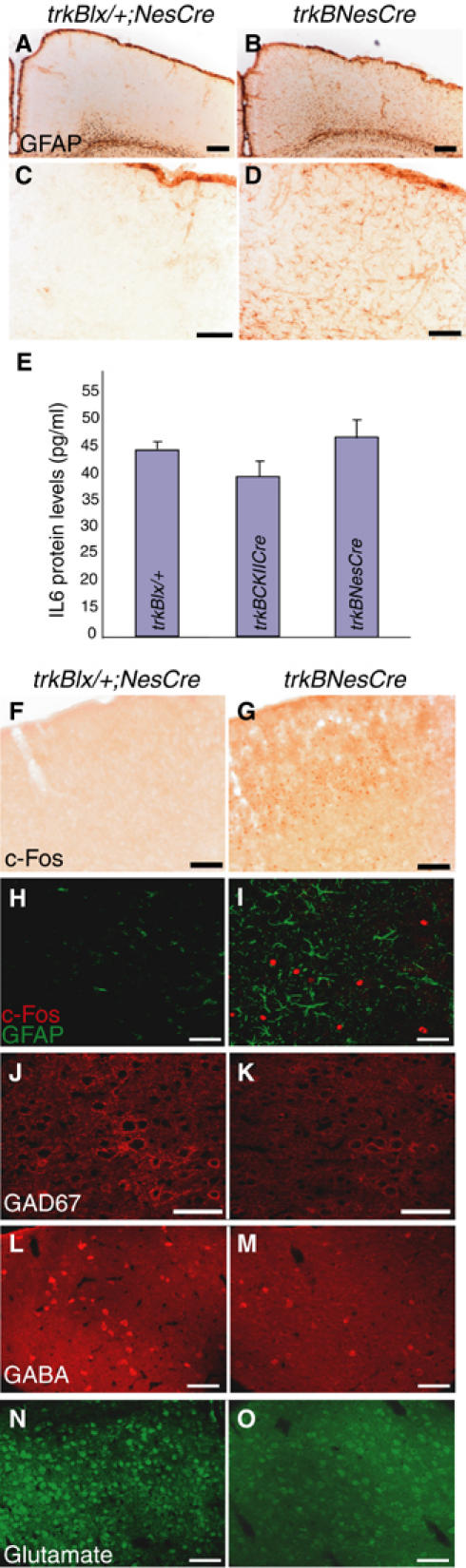
Functional consequences upon removal of TrkB signaling. (A–D) GFAP IMS of coronal brain sections in trkBNesCre mutant cortices (B, D) compared to control brains (A, C). (E) Levels of IL6 protein in the forebrains of control, trkBCKIICre, and trkBNesCre mice. (F, G) IMS for c-Fos expression and (H, I) double IMF using GFAP and c-Fos Abs in cortices of trkBNesCre and control mice. (J–O) IMF analysis of inhibitory and excitatory neurotransmitters in Soms cortices of trkBNesCre and control mice using Abs against GAD67 (J, K), GABA (L, M), and glutamate (N, O). Scale bars: (A, B), 200 μm; (C, D), 100 μm; (F, G), 200 μm: (H–O), 50 μm.
TrkB coordinates different functions through the Shc and PLCγ sites
To determine whether both the Shc and PLCγ sites are also required for TrkB-mediated differentiation of cortical neurons and OLs, we examined the morphology of the cortex of P15 trkBD/D mice, and observed that, similar to trkBNesCre described above, or of similar age (data not shown), neurons in layers II–III were tightly packed compared to the other layers and those of control mice (Figure 7A and B). We also examined the differentiation of various neuronal populations, as well as glial cells. Using MAP2, we observed an alteration in the distribution of apical and basal dendrites in the neocortex of trkBD/D compared to control mice. In the most superficial cortical layers of trkBD/D, dendrites appeared fasciculated and poorly stained, and in the deep layers the staining of fibers and cell bodies was further reduced (Figure 7C and D), as was observed in the adult trkBNesCre and trkBNesCre of a similar age (Supplementary Figure 3A and B). Western blot analysis confirmed that MAP2 levels were significantly reduced in the cortices of trkBD/D compared to controls, P=0.003 (Figure 7E and F). This phenotype has not been observed in either trkBPLC/PLC or trkBSHC/SHC mice (Minichiello et al, 1998, 2002). The patterns and levels of expression of calbindin, PARV and calretinin were similar to those seen in the trkBNesCre neocortex (data not shown). To examine OL differentiation, we immunostained brain sections obtained from P15 single-point mutants and trkBD/D for CNPase. The results revealed normal OL differentiation in the trkBPLC/PLC and trkBSHC/SHC (data not shown), but diminished labeling of neuronal fibers in the trkBD/D, similar to that observed in the adult trkBNesCre mice (Figure 7G and H) and trkBNesCre mice of a similar age (data not shown). To quantify this result, we examined CNPase levels in cortical cell lysates and found diminished levels in the trkBD/D compared to control mice (P=0.002, Figure 7I and J). Moreover, as in the adult trkBNesCre mice and trkBNesCre mice of a similar age, MBP IMS was reduced in the cortex of trkBD/D compared to control mice (Figure 7K and L; Supplementary Figure 3E and F). These results clearly indicate that the differentiation of cortical neurons and OLs depend to a major extent on signaling from both the Shc and PLCγ sites downstream of activated TrkB. In addition, the further reduced activation of the Ras/MAPK and PI3K/AKT pathways observed in the trkBD/D compared to that seen in the trkBSHC/SHC, which do not show developmental defects in the CNS, suggests that threshold levels of activation of these pathways are required for mediating TrkB-dependent CNS development. To study the functional consequences of the defects observed in the trkBD/D, we analysed brain sections of P15 single-point mutants and trkBD/D mice for GFAP expression. As with the adult trkBNesCre and trkBNesCre of a similar age (Supplementary Figure 3C and D), we found reactive astrocytes in the trkBD/D mice (Figure 7M and N), but normal GFAP expression in the single-point mutants (data not shown). Although we could not examine most of the other markers due to the early death of the trkBD/D mice, the above results further indicate that the Shc and the PLCγ sites together are responsible for mediating TrkB functions during cortical development.
Figure 7.
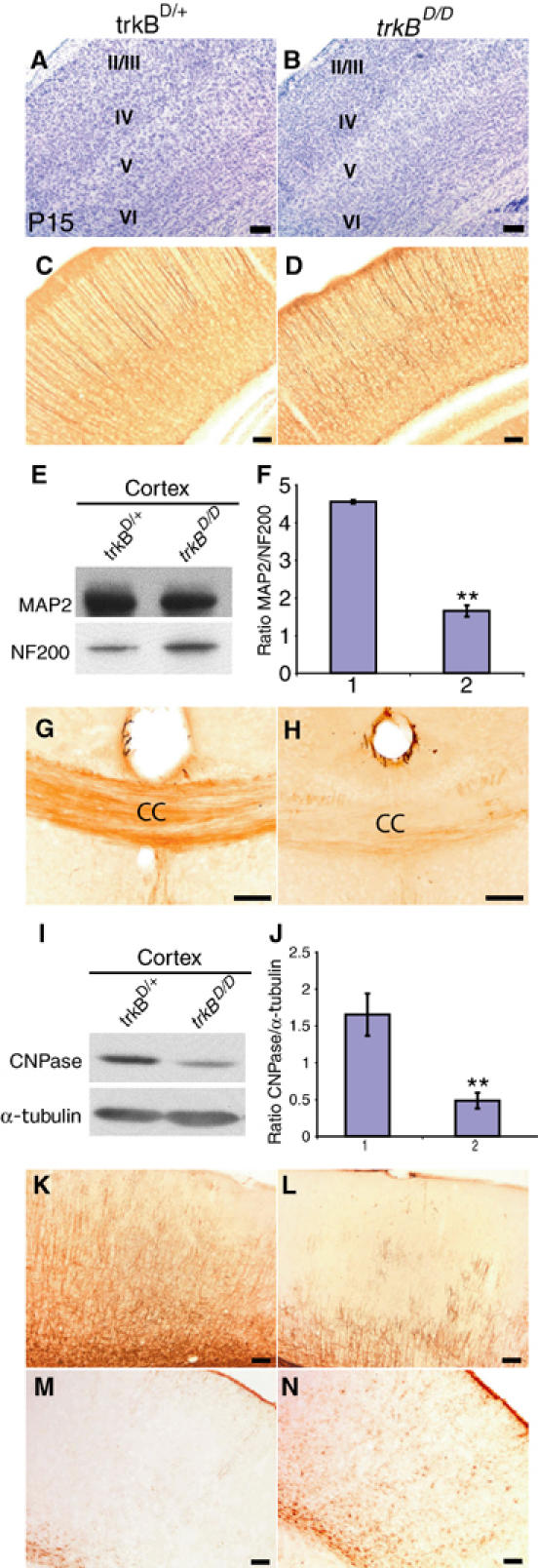
Differentiation of neurons and glia is also mediated through from the Shc and pLCγ sites of TrkB. (A, B) Cresyl violet staining of Soms cortices of trkBD/D and control mice. (C–L) Altered neuronal and OL differentiation in trkBD/D mutants. (C, D) MAP-2 IMS in Soms cortices of trkBD/D and controls at P15. (E) Representative western blot of MAP2 levels in P17 trkBD/D and littermate control cortices. To control for protein loading, blots were reprobed with anti-NF200 Abs. (F) Densitometric analyses of MAP2 levels in the cortex of trkBD/D mice compared to controls. **P=0.003. (G, H) CNPase IMS in the CC at P14 (G, H), and MBP in the cortex at P17 (K, L) of trkBD/D and controls. (I) Representative western blot of CNPase expression in P17 trkBD/D and control brains. To control for loading, blots were reprobed with anti-α-tubulin Abs. (J) Densitometric analyses of CNPase levels in the cortex of trkBD/D mice compared to controls. **P=0.002. (M, N) GFAP IMS in the Soms cortex of trkBD/D mice compared to controls at P15. Scale bars: (A–D, F, G, J, K, N–Q), 100 μm.
Discussion
Both the development and functioning of the CNS occur through precisely timed and interdependent events, some of which take place during embryogenesis, such as the proliferation and migration of neuronal and glia progenitors, while others continue after birth, such as cell differentiation, maturation, and functioning. Among the many genes involved in these different processes, we and others have shown that the TrkB neurotrophin receptor tyrosine kinase is an essential component of the protein kinase cascades that regulate synaptic plasticity and behavior involving forebrain structures such as the hippocampus and cerebral cortex (Minichiello et al, 1999, 2002; Xu et al, 2000). Moreover, TrkB regulates the survival and differentiation of different neuronal populations in both the CNS and PNS (Bibel and Barde, 2000). In this study, we asked whether and when TrkB signaling would be required during the formation of the cerebral cortex. Using different mouse models, such as trkB conditional mutants and trkB point mutant mice, we show that TrkB signaling has an essential role in the timing of migration of newly generated neurons, in the differentiation of diverse populations of neurons and glia in the neocortex, and in brain physiology. Our results further indicate that the Shc and PLCγ sites of TrkB are both required for proper neuronal migration and differentiation in the cerebral cortex, and that the Ras/MAPK and PI3K/AKT pathways are the likely mediators of these functions downstream of TrkB.
How does the absence of TrkB signaling interfere with neuronal migration?
Normal neuronal migration in the mouse cerebral cortex begins at a specific stage (E11) of development when the first cohort of postmitotic neurons leaves the ventricular zone, and eventually forms the preplate (PP). Subsequently, the cells that will ultimately create the deepest cortical layer, layer VI, migrate and reach the PP, which they penetrate and split into two layers: an upper marginal zone (MZ) and a lower subplate (SP). As successive layers of cortical neurons are born, they migrate through the SP and previous neuronal layers, stopping under the MZ. The end result of this process is the stratification of cortical neurons according to their developmental time of birth and start of migration. The oldest neurons are found in layers I and VI, whereas the remaining cortical layers are comprised of cells organized in an ‘inside out' manner with respect to their date of birth, with layer II/III containing the youngest cells (Gilmore and Herrup, 1997). The results of our BrdU experiments and studies of cortex morphology suggest that the absence of TrkB signaling during cortical development delays neuronal migration such that newly generated neurons that would normally form the deepest layers, for example, do not migrate through the SP at the correct time. This would result in enlarged superficial layers, since they are the last layers to be formed (see Figure 8A and B). Moreover, our results clearly indicate that it is the migration of postmitotic neuronal progenitors and not their proliferation that is altered in the absence of TrkB signaling during the formation of the cortical layers, since the total number of BrdU-positive cells observed at different embryonic stages and in different developing cortical layers are similar between control and trkB mutant cortices. In addition, BrdU incorporation into cells of the adult dentate gyrus, where neurogenesis persists and gives rise to mainly neuron-specific progenitors (Seaberg and van der Kooy, 2002), is similar between control and trkBNesCre mutant mice (data not shown), supporting our conclusion that neuronal migration is affected and not neurogenesis nor neuronal proliferation. Furthermore, our results are in agreement with those of Gates et al (2000), who observed defects in the migration of trkB−/− cortical neurons transplanted into a wild-type neocortex, and of Borghesani et al (2002), who showed that BDNF initiates the movement of cerebellar granule cells from the EGL to their final position in the IGL.
Figure 8.
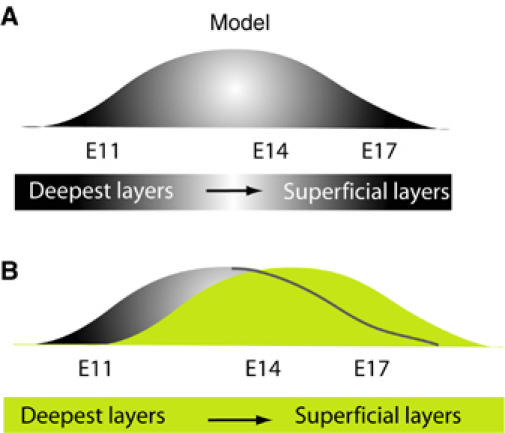
Model: removal of TrkB signaling during neocortical development affects neuronal stratification. (A) Neuronal stratification in different cortical layers of murine cortex (between E11 and E17) depends on the date of neuronal birth and proper migration. (B) Absence of TrkB signaling during cortical development causes a delay in neuronal migration, thereby altering neuronal stratification.
Does the delayed migration of cortical neurons affect their differentiation?
To study the early requirement of TrkB signaling during neocortical development, we have conditionally deleted trkB from the embryonic CNS to avoid conflicting interpretations due to the poor health of null mutants. We have carried out similar experiments on trkB null mutants, and found equivalent phenotypes to those reported here for the trkBNesCre mice. With this approach, we now show that TrkB signaling is required for proper migration of cortical neurons during the formation of the neocortex, and, in addition, is required for the differentiation of diverse populations of neurons in the neocortex and normal axonal myelination. Are these latter effects a consequence of altered migration? Evidence suggests that the differentiation of neurons is in part determined by the changing cellular environment through which they travel to reach their final location (Purves et al, 2001). Our observations in the conditional mutants lend support to this hypothesis, though are limited by the present genetic tools, which do not allow us to dissect the two functions. Thus, we cannot exclude the possibility that the differentiation defect is independent of the migration defect. We have previously reported defects in dendritic differentiation of cerebellar Purkinje cells and an abnormal rostro-caudal foliation pattern in P12 trkB null mutants (Minichiello et al, 1998). However, the premature death of these mice did not allow for further investigation of the phenotype. With the trkBNesCre conditional mutants, we can now conclude that absence of TrkB signaling causes a delay in cerebellar development that is overcome already at the third postnatal week when cerebella from both controls and mutants appeared similar (data not shown).
Is the myelination defect due to altered signaling in axons or myelinating glia?
It has recently been shown that NGF/TrkA signaling reduces myelination by OLs, and this effect is mediated by changes to the axonal signals that control myelination, rather than by direct action on myelinating glia (Chan et al, 2004). In the present study, where we have deleted TrkB during embryonic development from neurons and glial precursors of the CNS, we show that TrkB signaling promotes myelination, which may occur at least in part through the regulation of OL differentiation. Deletion of trkB at P20 does not affect OL differentiation nor CNS myelination as observed in the trkBCaMKIICre mutant mice (Minichiello et al, 1999), suggesting that TrkB signaling in axons regulates OL myelination before P20, and/or that TrkB signaling in OLs is sufficient to mediate their proper differentiation and myelination function. In order to discriminate these two effects, we are currently generating new genetic tools, which would allow removal of TrkB signaling specifically from OLs. This should help us to determine if TrkB signaling affects myelination either directly by acting on myelinating glia or indirectly by controlling axonal signals that influence OLs.
TrkB mediates important functions during neocortex development through the Shc and PLCγ sites
Having shown that TrkB signaling is required during neocortical development to mediate important events such as neuronal migration, differentiation, and myelination, and after development is complete, to regulate neuronal plasticity and behavior (Minichiello et al, 1999), we have asked which signaling pathways downstream of the TrkB receptor are responsible for neurotrophin-induced cell migration and differentiation. Thus far, these pathways have not been well defined. Recently, Polleux et al (2002), using an in vitro migration assay combined with pharmacological perturbations, showed that tangential migration is stimulated by BDNF and NT-4, and attenuated by the Trk-family inhibitor K252a, and the inhibition of PI3K. To better understand how TrkB regulates cortical development at the molecular level, we have used genetic tools that allow for the interference of single (Minichiello et al, 2002) or multiple phosphorylation sites in the intracellular region of TrkB. In this way, we have now been able to show that ablation of both the Shc and PLCγ sites cause abnormal cortical development. Moreover, biochemical analyses suggest that a threshold level of Ras/MAPK and PI3K/AKT activation is required for TrkB-mediated cell migration and differentiation. We cannot, however, exclude the possibility that additional, uncharacterized pathways activated through these two sites also contribute to TrkB-mediated functions during cortical development. Since mice carrying single-point mutations at either the Shc or PLCγ site show normal cortical development, it is attractive to speculate that both sites, besides activating independent pathways, stabilize the formation of a transductosome upon TrkB receptor activation.
Materials and methods
Transgenic animals
The methods used to generate the floxed trkB, CamKII-cre, and the transgenic NesCre mouse lines have been described previously (Minichiello et al, 1999; Tronche et al, 1999). Specifically, the trkBCaMKIICre and the trkBNesCre lines were generated by crossing the trkB floxed mouse to the CamKII-cre or the NesCre mouse line, respectively. The trkBD/D mouse line was created using a knockin gene-targeting strategy similar to that reported in Minichiello et al (2002). Additional details can be found in Supplementary data.
Biochemistry
Cortical neurons derived from the cerebral cortices of E15.5 mouse embryos expressing either +/+ or mutated forms of TrkB were treated for biochemical analysis as reported previously (Minichiello et al, 2002), whereas brain tissue biochemistry was carried out as described in Minichiello et al (1999). Specific Abs are described in Supplementary data.
Expression levels of protein
Densitometric analysis (NIH image) of imaged blots was used to compare the expression levels of proteins in mutant and control brains, and was performed on three different blots for each Ab. To control for loading, density values were corrected by dividing them by the values obtained for reference proteins. The ratios obtained for mutant and control samples were compared statistically using Student's t-tests.
Histology and EM
Histological and IHC analysis, Nissl, and Golgi stains, and electron microscopic analyses were carried out as described previously (Minichiello et al, 2002). Additional details and Abs are described in Supplementary data.
In vivo BrdU incorporation
Pregnant females were intraperitoneally injected at either E12.5 or E14.5 with BrdU (100 mg/kg body weight; Sigma) dissolved in PBS. The embryos were removed at E18.5, and their fixed brains were paraffin-embedded and sagittally sectioned (8 μm). Sections were dewaxed in xylene, rehydrated, and processed for antigen retrieval with a Retriever cooker (PickCell) following the manufacturer's instructions. Additional details can be found in Supplementary data.
Glial cultures
Astrocytes were isolated from P4 mice using the method described by McCarthy and de Vellis (1980). This method allows 98% purity of the culture.
IL6 ELISA
To detect IL6 protein levels in the brains of control and trkBNesCre mutant mice, we used the Cytoscreen Mouse IL6 kit (Biosource International) according to the manufacturer's instructions, using 1.5 mg of forebrain extract/well.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Data
Acknowledgments
We thank Drs AM Goffinet for providing the reelin Abs, W Birchmeier for providing the (GST)-Gab1plasmid to raise Gab1 Abs., Drs C Nerlov and C Serguera for critical reading of the manuscript, Dr R Klein for his support on the generation of the trkBD/D mice, Regeneron Pharmaceutical, Inc for providing BDNF, and E Perlas, B Brühl, and S Oberle for excellent technical assistance. DL Medina was supported by a Marie Curie fellowship; O von Bohlen und Halbach and K Unsicker are supported by the DFG (SFB636/A5).
References
- Akagi K, Sandig V, Vooijs M, Van der Valk M, Giovannini M, Strauss M, Berns A (1997) Cre-mediated somatic site-specific recombination in mice. Nucleic Acid Res 25: 1766–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibel M, Barde YA (2000) Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev 14: 2919–2937 [DOI] [PubMed] [Google Scholar]
- Borghesani P, Peyrin J, Klein R, Rubin J, Carter A, Schwartz P, Luster A, Corfas G, Segal R (2002) BDNF stimulates migration of cerebellar granule cells. Development 129: 1435–1442 [DOI] [PubMed] [Google Scholar]
- Chan JR, Watkins TA, Cosgaya JM, Zhang C, Chen L, Reichardt LF, Shooter EM, Barres BA (2004) NGF controls axonal receptivity to myelination by Schwann cells or oligodendrocytes. Neuron 43: 183–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Climent E, Sancho-Tello M, Minana R, Barettino D, Guerri C (2000) Astrocytes in culture express the full-length Trk-B receptor and respond to brain derived neurotrophic factor by changing intracellular calcium levels: effect of ethanol exposure in rats. Neurosci Lett 288: 53–56 [DOI] [PubMed] [Google Scholar]
- Condorelli D, Salin T, Dell' Albani P, Mudo G, Corsaro M, Timmusk T, Metsis M, Belluardo N (1995) Neurotrophins and their trk receptors in cultured cells of the glial lineage and in white matter of the central nervous system. J Mol Neurosci 6: 237–248 [DOI] [PubMed] [Google Scholar]
- Gates M, Tai C, Macklis J (2000) Neocortical neurons lacking the protein-tyrosine kinase B receptor display abnormal differentiation and process elongation in vitro and in vivo. Neuroscience 98: 437–447 [DOI] [PubMed] [Google Scholar]
- Gilmore E, Herrup K (1997) Cortical development: layers of complexity. Curr Biol 7: R231–R234 [DOI] [PubMed] [Google Scholar]
- Graus-Porta D, Blaess S, Senften M, Littlewood-Evans A, Damsky C, Huang Z, Orban P, Klein R, Schittny JC, Muller U (2001) Beta1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron 31: 367–379 [DOI] [PubMed] [Google Scholar]
- Klein R, Martin-Zanca D, Barbacid M, Parada L (1990) Expression of the tyrosine kinase receptor gene trkB is confined to the murine embryonic and adult nervous system. Development 109: 845–850 [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun P, Griffiths I, Nave K (2003) Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet 33: 366–374 [DOI] [PubMed] [Google Scholar]
- McCarthy K, de Vellis J (1980) Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol 85: 890–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L, Calella AM, Medina DL, Bonhoeffer T, Klein R, Korte M (2002) Mecchanisms of TrkB-mediated hippocampal long-term potentiation. Neuron 36: 121–137 [DOI] [PubMed] [Google Scholar]
- Minichiello L, Casagranda F, Tatche R, Stucky C, Postigo A, Lewin G, Davies A, Klein R (1998) Point mutation in trkB causes loss of NT4-dependent neurons without major effects on diverse BDNF responses. Neuron 21: 335–345 [DOI] [PubMed] [Google Scholar]
- Minichiello L, Korte M, Wolfer D, Kuhn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp H, Bonhoeffer T, Klein R (1999) Essential role for TrkB receptors in hippocampus-mediated learning. Neuron 24: 401–414 [DOI] [PubMed] [Google Scholar]
- Polleux F, Whitford K, Dijkhuizen P, Vitalis T, Ghosh A (2002) Control of cortical interneuron migration by neurotrophins and PI3-kinase signaling. Development 129: 3147–3160 [DOI] [PubMed] [Google Scholar]
- Postigo A, Calella AM, Fritzsch B, Knipper M, Katz D, Eilers A, Schimmang T, Lewin G, Klein R, Minichiello L (2002) Distinct requirements for TrkB and TrkC signaling in target innervation by sensory neurons. Genes Dev 16: 633–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purves D, Augustine GJ, Fitzpatrick D, Katz LC, LaMantia AS, McNamara JO, Williams SM (2001) Neuroscience, 2nd ed Sunderland (MA): Sinauer Associates Inc [Google Scholar]
- Seaberg R, van der Kooy D (2002) Adult rodent neurogenic regions: the ventricular subependyma contains neural stem cells, but the dentate gyrus contains restricted progenitors. J Neurosci 22: 1784–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit W, Walter S, Pennell N (1999) Reactive microgliosis. Prog Neurobiol 57: 563–581 [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban P, Bock R, Klein R, Schutz G (1999) Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 23: 99–103 [DOI] [PubMed] [Google Scholar]
- Van Wagoner N, Benveniste E (1999) Interleukin-6 expression and regulation in astrocytes. J Neuroimmunol 100: 124–139 [DOI] [PubMed] [Google Scholar]
- Xu B, Gottschalk W, Chow A, Wilson R, Schnell E, Zang K, Wang D, Nicoll R, Lu B, Reichardt L (2000) The role of brain-derived neurotrophic factor receptors in the mature hippocampus: modulation of long-term potentiation through a presynaptic mechanism involving TrkB. J Neurosci 20: 6888–6897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhang D, McQuade J, Behbehani M, Tsien J, Xu M (2002) c-fos regulates neuronal excitability and survival. Nat Genet 30: 416–420 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Data