

Abstract
Aging is a complex phenomenon and its impact is becoming more relevant due to the rising life expectancy and because aging itself is the basis for the development of age-related diseases such as cancer, neurodegenerative diseases and type 2 diabetes. Recent years of scientific research have brought up different theories that attempt to explain the aging process. So far, there is no single theory that fully explains all facets of aging. The damage accumulation theory is one of the most accepted theories due to the large body of evidence found over the years. Damage accumulation is thought to be driven, among others, by oxidative stress. This condition results in an excess attack of oxidants on biomolecules, which lead to damage accumulation over time and contribute to the functional involution of cells, tissues and organisms. If oxidative stress persists, cellular senescence is a likely outcome and an important hallmark of aging. Therefore, it becomes crucial to understand how senescent cells function and how they contribute to the aging process. This review will cover cellular senescence features related to the protein pool such as morphological and molecular hallmarks, how oxidative stress promotes protein modifications, how senescent cells cope with them by proteostasis mechanisms, including antioxidant enzymes and proteolytic systems. We will also highlight the nutritional status of senescent cells and aged organisms (including human clinical studies) by exploring trace elements and micronutrients and on their importance to develop strategies that might increase both, life and health span and postpone aging onset.
Abbreviations: AD, Alzheimer's Disease;; AGEs, advanced glycation endproducts; ALP, autophagy-lysosome pathway; ARF, alternate reading frame; ATGs, autophagy-related proteins; BAG3, Bcl2-associated athanogene 3; Bcl-2, B-cell lymphoma 2; BrdU, 5-bromodeoxyuridine;; CMA, chaperone-mediated Autophagy; Cu, copper; CVD, cardiovascular diseases;; Cys, cysteine; DDR, DNA damage response;; Fe, iron; GPx, glutathione peroxidase;; GSH, glutathione; GSSG, glutathione disulfide; H2O2, hydrogen peroxide; Hsc70, heat shock cognate 70; HSF1, heat shock factor protein 1; Hsps, heat shock proteins; IFN-γ, interferon gamma; IKK, IκB-Kinase; IL, Interleukins; LAMP1, lysosome-membrane associated protein 1; LAMP2a, lysosome-membrane associated protein type 2A; LC3, microtubule-associated protein light chain 3; MA, Macroautophagy; MAP2K3, mitogen-activated protein kinase kinase 3; MCI, mild cognitive impairment;; MiA, microautophagy; MiT/TFE, microphthalmia/transcription factor E; Mn, manganese; Msr, methionine sulfoxide reductase;; mTOR, mammalian target of rapamycin;; Nrf2, nuclear-erythroid factor 2; PARP1, poly (ADP-ribose) polymerase 1;; PD, Parkinson's Disease;; PINK1, PTEN-induced putative kinase 1 Prxs, peroxiredoxins; RA, retinoic acid;; Raf, rapidly accelerated fibrosarcoma; Ras, rat sarcoma;; RDA, recommended dietary allowance;; RNS, reactive nitrogen species; ROS, reactive oxygen species; SAHF, senescence-associated heterochromatic foci; SASP, senescence associated secretory phenotype; SA-β-Gal, senescence-associated β-galactosidase activity;; Se, selenium;; SIPS, stress induced premature senescence; Srx, sulfiredoxin;; O2∙−, superoxide; ∙OH, hydroxyl radical; TE, trace elements; TFEB, transcription factor EB; TGFβ, transforming-growth-factor-β; TNF-α, tumor necrosis factor alpha;; Ub, ubiquitin; ULK1, autophagy-initiating kinase ULK1; ULK1, unc-51 like kinase 1; Ump1, ubiquitin-mediated proteolysis 1;; UPS, ubiquitin proteasomal system;; Zn, zinc
Keywords: Aging, Senescence, Protein oxidation, Proteostasis, Trace elements, Antioxidants, Micronutrients
Graphical abstract
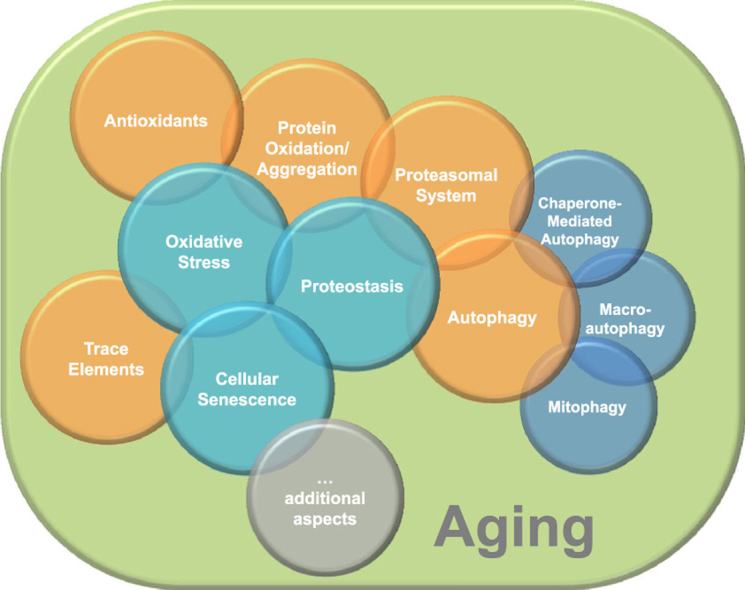
1. Introduction
In 1952 Peter Medawar published a paper entitled “An unsolved problem in biology”, originally presented as a lecture at University College of London, aging being the subject of his discussion. Nowadays, some mechanisms behind this “unsolved problem” have been unraveled, but it is still far from being solved. Aging is within the focus of research worldwide due to its increasing impact on the whole population. As we are confronted with an ever aging population in most of modern societies, the study of aging became a hot topic for scientific research and the number of publications regarding aging has steadily increased over the years (Fig. 1).
Fig. 1.

Publications per year related to “aging”. The search term “aging” was entered on PubMed on October 7th, 2016. Results were plotted as publications per year.
Recently, different hallmarks of aging have been identified and put together, cellular senescence and loss in proteostasis being two among several described [1]. But how can we explain aging from a cellular and molecular perspective?
In this review we aim to provide a state of the art literature on aged/senescent cells regarding how they arise, how they cope with oxidative stress and how this is related to the loss of proteostasis. This is of utmost importance to develop strategies that might extend not only life span but in particular health span. The review covers three main hallmarks of aging i) oxidative stress, ii) proteostasis, and iii) cellular senescence.
We provide a brief description of oxidative stress and antioxidant defense mechanisms and describe how the main proteostasis mechanisms related to protein turnover are affected in the course of aging. In addition we focus on cellular senescence and provide insights to nutritional issues such as trace elements and micronutrient status and how they can play a role in both, senescence-promotion and on senescence-avoidance features and how they influence proteostasis. We close the review with some insights on new ideas and therapies that might promote life and health span extension in the future.
It is not the intention of this review to extensively describe each protein, process or consequence, but rather to give a comprehensive overview of the different topics related to the field of oxidative stress, proteostasis and senescence/aging. This educational review and the accompanying illustrations are aimed to provide an introduction to the field of aging focusing on the above mentions hallmarks.
2. Aging, senescence and cellular damage
2.1. Aging and senescence
Aging is defined as the time-dependent persistent change of functionality and reproducibility, affecting all higher organisms. Biologically, it is considered a time-associated cellular (and also systemic) functional decline, related with an increased probability of morbidity and mortality. Since most cellular functions are performed by proteins, aging may be (in part) the consequence of a dysregulation/malfunction of the cellular proteome (proteostasis). This has also take into account that due to age-associated DNA damage not all proteins might be replaced accordingly. So, aging is characterized by the accumulation of cellular damage, in turn, leading to increased susceptibility to diseases including cancer, type 2 diabetes and cardiovascular disease and finally death. These diseases have their root on the aging process itself, since aging is known to be single highest risk factor for their development. Thus, it becomes crucial to understand aging mechanisms. The process of aging involves different interdependent hallmarks on cellular, molecular and organ level. One major contributor is the development of cellular senescence [1], [2]. The possible link between aging and senescence was first described by Leonard Hayflick and Paul Moorhead in 1961 after observing a limited proliferation capacity in cultured human primary fibroblasts [3]. This is known as the Hayflick limit and originates from the inability of telomeres to maintain their lengths due to the replication process and a decline in the protection systems against stressors during aging. Consequently, cells lose their proliferative capacity and enter a state of irreversible cell cycle arrest, termed replicative senescence [4], [5], [6]. In addition to shortened telomeres, various other triggers exist including non-telomeric and non-genotoxic stress generated by various physicochemical signals, such as mitochondrial deterioration and oxidative stress (further described in chapter Protein oxidation and aggregation in aging), DNA-replication stress or activated oncogenes such as rat sarcoma (Ras)- and rapidly accelerated fibrosarcoma (Raf)-proteins. All of them lead to senescence or, if triggered by an excess of stressors also summarized as premature cellular or damage-induced senescence [6], [7], [8], [9], [10]. These triggers stimulate various signaling pathways, including DNA damage response (DDR), transforming-growth-factor-β (TGFβ), alternate reading frames (ARF) or mitogen-activated protein kinase kinase 3 (MAP2K3), leading to an upregulation of cell cycle inhibitor p53, its downstream target p21 and the tumor suppressor protein p16INK4aINK4a, representing the cascade from pre- to acquired-senescence [11], [12], [13], [14], [15].
When senescence is established, cells undergo widespread changes and develop specific characteristics, serving as senescence markers. However, no marker identified so far is entirely specific for cellular senescence, but evaluating a series of these markers can help to define the senescent state (Fig. 2). Phenotypically, senescent cells increase in size and protein content [3], [16], [17], they develop enlarged nuclei and lysosomes which possess elevated senescence-associated β-galactosidase activity (SA-β-Gal). This marker is one of the most widely used for the identification of senescence in cells and tissues [16], [17], [18]. Moreover, based on the stable growth arrest, markers of proliferation like Ki-67 and 5-bromodeoxyuridine (BrdU) are other beneficial tools for the detection of cellular senescence [14], [19]. Additionally, the main regulators of the senescence program, p53/p21 and p16INK4aINK4a proteins are upregulated and quantifiable components for the characterization of the senescent state [12], [15], [20], [21] and are, consequently, more and more used as reliable senescence markers. Dependent on the expression of p16INK4aINK4a, senescence-associated heterochromatic foci (SAHF) are generated during the establishment of oncogene-induced senescence [22]. Another important feature of senescence is the formation of the so called senescence-associated secretory phenotype (SASP). SASP is characterized by an increased expression and secretion of pro-inflammatory cytokines and chemokines (e.g. the interleukins IL-6, IL-1 and IL-8), growth factors (IGF-binding proteins, their regulators and transforming growth-factors), components of extracellular matrix (matrix metalloproteinases, serine proteases), fibronectin and reactive oxygen/nitrogen species (ROS/RNS), leading to the activation of the immune response and elimination of senescent cells [23], [24], [25], [26], [27], [28], [29], [30], [31]. Due to intercellular communication of senescent cells, factors of the secretory phenotype may also induce the generation of senescence in functional normal neighboring cells in a paracrine manner. Altogether, the SASP can act both as a tumor suppressor or a promoter mechanism and alter cellular and tissue homeostasis [23], [32], [33], [34].
Fig. 2.

Features of senescent cells: Several markers were identified to characterize the senescent state in relation to morphology and proteostasis. During the development of senescence, cells show morphological changes by extension of their size and protein content or nuclei enlargement. Also, their lysosomes size and number increase resulting in an elevated activity of SA-β-Gal, the most widely used marker for senescence. The cells enter a proliferative arrest state, detected by cell cycle inhibitor levels such as p53/p21 and tumor suppressor p16INK4a, the latter is correlated with the formation of the SAHF. Other factors secreted during senescence are cytokines and chemokines, growth factors, proteases, fibronectin as well as ROS and RNS, altogether these are summarized as the SASP. Additionally, proteostasis changes during senescence shown by an increase in modified proteins, accumulation of protein aggregates and reduced functionality of the proteasomal and autophagy systems.
Besides this, other imbalanced factors, in particular deficiencies in proteostasis, such as the accumulation of oxidized proteins and declined protein turnover are involved in the induction and/or the development of senescence (Fig. 3, Fig. 4) and will be discussed in the following chapters.
Fig. 3.

Cellular and molecular features of young and aged cells. Comparing to young, aged cells often exhibit marked features. For example, it is known that their nucleus is often enlarged. Their proteolysis mechanisms suffer a major reduction in functionality, found by evaluating the activity of both proteasomal and lysosomal mechanisms. The consequence is the accumulation of oxidized proteins and the formation of insoluble material such as lipofuscin, a hallmark of aged cells as well and altogether contributing to a loss in cellular proteostasis. Furthermore, recent years have brought us evidence of a secretory phenotype acquired by senescent cells (SASP), which is characterized by the release of several inflammatory cytokines into the surrounding cells and tissues, resulting in low-grade chronic inflammation over time causing tissue and organism dysfunction.
Fig. 4.

Proteostasis changes in a senescent cell. The scheme shows the overall changes of the cellular systems maintaining protein homeostasis (proteostasis) and thus, cellular functionality during aging. The main proteolytic systems, responsible for recognition and degradation of un/misfolded or oxidatively damaged proteins are the proteasomal system, involving the ATP-dependent 26S proteasome as well as the ATP-independent 20S proteasome, and autophagy (including both autophagy (MA) as well as the chaperone mediated one (CMA)). Damaged proteins can be directly recognized as substrates by the 20S proteasome, resulting in proteolytic removal from the cell. Furthermore, they can be recognized by chaperones or heat shock proteins that keep their substrates in a soluble state, preventing the formation of aggregates. Another fate might be the formation of aggregates, driven by hydrophobic residues exposure. Such aggregates can be incorporated into an autophagosome and fusing with lysosomes, resulting in proteolytic degradation by lysosomal proteases, or they can be removed from the cell via excretion as exosome. Modified from [115] and according to [122].
3. Oxidative stress in aging and senescence
The consequences of oxidative stress have raised several theories proposed over the years to explain the aging phenomena, but there is not a prevailing one yet. The most widely accepted one is the The Free Radical Theory of Aging proposed by Denham Harman in 1956 [35], stating that “aging may be related to the deleterious side attacks of free radicals (which are generally produced during metabolic processes) on cell constituents”. Over the years, different updates, such as the mitochondrial free radical theory of aging proposed in the early 80s by Miquel and later updated in a paper entitled “An update on the oxygen stress-mitochondrial mutation theory of aging: genetic and evolutionary implications” [36], were proposed and the concept of free radicals as the cause of aging evolved. Helmut Sies, one of the founders of the oxidative stress definition, initially suggested oxidative stress as a likely cause of the age-related damage. “A disturbance in the pro-oxidant-antioxidant balance in favor of the former”, this was how oxidative stress was initially viewed, and known to promote damage in different cellular constituents [37]. However, it should be kept in mind, that recent insights on specific redox regulatory effects of oxidants support the idea that, when balanced, ROS play an important role in signal transduction cascades enhancing or suppressing cellular fates such as proliferation or differentiation [37], [38]. Recently, Sies reviewed and altered his own definition to “A disturbance in the prooxidant-antioxidant balance in favor of the former leading to a disruption in redox signaling” [37]. When an oxidative state is reached and if prolonged, senescence might arise, thus, ROS have been described as important mediators for cellular senescence progression [39]. In fact, when exogenous hydrogen peroxide (H2O2), a major intracellular ROS, was experimentally added, a strong senescent-phenotype established across different cell types, suggesting that H2O2 can act as a potent inducer of cellular senescence (Fig. 3, experimental set-up 3) [8]. Whereas senescence can be achieved by exogenously adding H2O2, endogenously formed ROS, such as superoxide (O2•−) and the highly reactive hydroxyl radical (•OH), can also contribute to the maintenance of the common senescence feature of irreversible growth-arrest. Under pathological levels, ROS have been implicated in an induction of senescence-like phenotypes as also found in oncogene-induced senescence and p16INK4aINK4A-induced senescence [39], [40], [41], [42].
ROS are among others mainly produced by mitochondria during the normal metabolism. Thus, mitochondria are considered the most prominent source of cellular ROS. Dysfunctional mitochondria leak electrons and generate O2•− as by-products, especially, on the complex I (NADH dehydrogenase) and complex III (Cytochrome bc1 complex) [43]. High ROS levels produced by dysfunctional mitochondria have been suggested as the main cause of aging [44], resulting from error accumulation impinged on biomolecules. Extensive description on the characteristics and different sources of free radicals is not within the scope of this review but can be found elsewhere [45].
The phenomenon of oxidative stress is associated with aging and senescence. In fact, this is supported by two lines of research. One is directed towards the analysis of increased levels of oxidative stress products in senescent cells, aged tissues or organisms. For example, Stadtman and Levine gathered data from several reports, showing that carbonylated proteins, a marker for severe and chronic oxidative stress, were found to be dramatically elevated in the last third of life analyzing different samples such as: human dermal fibroblasts in tissue culture, human lens, human brain obtained at autopsy, rat liver and whole flies [46]. The other line of research is based on challenging cells with oxidants in order to generate oxidative stress. In the model, named Stress-Induced Premature Senescence (SIPS) (Fig. 5), cells are challenged with sub-toxic concentrations of an oxidant, such as H2O2, or oxidant generators, such as paraquat [47], UV [48], iron or copper [49]. Thus, a chronic stress response is induced that culminates in a senescence-like phenotype, exhibiting common features of senescence such as p21 and p16INK4a overexpression, increased SA-β-Gal activity and increase in cellular volume and rounding. Moreover, submitting cells to hyperoxia, known to induce chronic oxidative stress [50], led to a similar gene expression pattern when compared to aged fibroblasts [51]. Another ways exist to reproduce single cellular aging hallmarks, e.g. as protein accumulation by exposing cells to artificial lipofuscin (described in chapter Protein modifications in aging) [52].
Fig. 5.

Cellular aging models. Primary cells can be isolated from young and old donors. (1.) Cells isolated from an old donor are called in vivo aged cells and can be investigated directly to study aging. In case of using cells from a young donor there a several options to study aging: (2.) After a multitude of subcultures, the young cells reach their replicative limit and stop dividing. These cells are called senescent or in vitro aged cells and are the most frequently used cellular aging model. (3.) Treatment of young cells with different oxidants or stressors for several days leads to the so-called stress-induced premature senescence (SIPS). After a short recovery phase these SIPS cells can be used for aging research. (4.) Finally young cells can be incubated with aggregates such as AGEs or lipofuscin. These cells mirror several important features of aged cells. Therefore, these aggregate-fed cells are an additional useful aging model.
So, several strategies were developed to investigate senescence cells (Fig. 5). Each of the models is useful according to the underlying scientific question.
Therefore, it seems to be established that oxidative stress is one of the key events in senescence progression and development. However, during evolution cells were equipped with antioxidant defense mechanisms that can prevent or recover cells from an oxidative to a reductive state.
3.1. Antioxidants
In the prevention of oxidative stress the organism and the cells are equipped with an antioxidative defense network. This network contains both endogenous and exogenous defense molecules (see Fig. 6). The endogenous defense consists of enzymatic antioxidants, such as thiol peroxidases, superoxide dismutases, catalase, and non-enzymatic antioxidants, such as glutathione. The exogenous antioxidant system comprises micronutrients, e.g. lipid- and water-soluble vitamins, as well as the trace elements zinc and selenium (involved in enzymatic functioning). Both systems act hand-in-hand and rely on each other to be effective.
Fig. 6.

Overview of endogenous and exogenous antioxidants. This figure gives a broad overview of antioxidants. Endogenous antioxidants comprise proteins, low-molecular weight molecules and enzymes, among others. Exogenous (dietary) sources of antioxidants include animal products, fruits, vegetables and grains (see chapter on micronutrients).
In the following, we will describe partially those particular classes of antioxidant enzymes named thiol peroxidases and methionine sulfoxide reductase (Msr), since these are involved in direct protection of the protein pool. The thiol peroxidases have recently gained special attention in the aging process. They essentially comprise two families of proteins: the glutathione peroxidase (GPx)-type enzymes and the peroxiredoxins (Prxs) [53]. Prxs are thiol-dependent peroxidases, present in all organisms. They all contain a conserved cysteine (Cys) residue that undergoes a cycle of peroxide-dependent oxidation and thiol-dependent reduction during catalysis. Mammalian cells express six isoforms of Prx (Prx I to VI), which are classified into three subgroups (2-Cys, atypical 2-Cys, and 1-Cys) based on the number and position of Cys residues that participate in the catalysis [54]. During the catalytic cycle, Prxs can be overoxidized to cysteine sulfinic acid, and be rescued by the specific enzymatic reduction by sulfiredoxin (Srx) in an ATP-dependent manner [55]. Increasing evidence suggest that Prx hyperoxidation is a potential mechanism to explain the age-related oxidative stress that disrupts normal physiological signaling. Mitochondrial-derived Prx III plays a major role in the control of the mitochondrial level of reactive oxygen species. In a mouse model, the knockout of Prx III causes an accelerated decline of physical strength at the age of 10 months [56] and overoxidized (inactive) Prx III was found to be accumulated in aged rat liver mitochondria [57]. Caloric restriction, an intervention that prolongs life span in different organisms, reduces age-related mortality in rhesus monkeys [58], stimulates Prx activity through augmented Srx1-repair and slows replicating aging [59]. Moreover, decreased levels of Prx I accelerate aging by promoting severe haemolytic anaemia and several malignant cancers in rodents [60]. In another study, comparing chondrocytes isolated from young adults to chondrocytes isolated from older adults, the old ones exhibited higher levels of Prx I-III hyperoxidation, basally and under conditions of oxidative stress.
Tsa1, the homolog of mammalian Prx I in yeast, is able to multimerize upon hyperoxidation, gaining molecular chaperone activity and inhibiting the aggregation of insulin in vitro [61]. Given its abundance and high reactivity towards peroxides, Prxs are preferential targets in H2O2-induced signaling [62]. In this context, it has been recently found that this enzyme is able to recruit the Hsp70 chaperones and the Hsp104 disaggregase to misfolded and aggregated proteins during aging, which reveals a new concept on peroxide signaling in proteostasis and life span control [63].
Glutathione peroxidases are selenium or sulfur-dependent peroxidases that reduce H2O2 to water, using glutathione (GSH, reduced form) as electron donors [53]. The fact that GPxs compete with Prxs for peroxides [53], their abundance and localization may at least indirectly impact in the aging process. Mammals have eight GPxs, including five selenoproteins. GPx1 is the first identified and one of the best studied selenoproteins. The naked mole rat, a rodent model of delayed aging because of its unusually long life span (>28 years), is characterized by the reduced utilization of selenium due to a specific defect in GPx1 expression. The reducing substrate for these enzymes, GSH, is the most abundant although slow reacting towards peroxides [62]) intracellular antioxidant and plays a role in signal transduction, gene expression and apoptosis [64], [65]. GSH in whole blood of healthy subjects (18–84 years) declined while GSSG (oxidized glutathione) increased with age [65], leading to a decreased antioxidative capacity [66]. In mice, increased hepatic GSH concentrations resulted in a protection against apoptosis [67].
The other antioxidant enzymes protecting the protein pool directly are the methionine sulfoxide reductases (Msr). Cysteine and methionine oxidation into cysteinyl derivatives and methionine sulfoxide, respectively, are known to be involved in several cellular processes [68], [69], [70], [71]. This view is supported by the high number of enzymes that can reverse the oxidation state back to its reduced form, to enable continuous pathway activation/deactivation according to required processes. Cysteinyl derivatives can be reverted back to cysteine by assistance of enzymes such as glutaredoxin or thioredoxin [72] and methionine sulfoxide to methionine by the methionine sulfoxide reductases [73]. In fact, Picot and colleagues have shown that the gene expression of the two isoforms MsrA and MsrB2, is decreased during replicative senescence of WI-38 fibroblasts, and this decline was related to modifications in its catalytic activity and accumulation in oxidized proteins [74].
However, if the system is not sufficient, oxidative stress and the accumulation of oxidized proteins can occur. The following chapter will be devoted to protein oxidation and how can this lead to the accumulation of insoluble non-degradable protein aggregates commonly seen in aging.
3.2. Protein modifications in aging
The harmful effects of imbalanced ROS levels on proteins during oxidative stress are a main research topic for the scientific community. But why, amongst different biomolecules prone to damage such as lipids and DNA, should proteins be considered so relevant? Practically, every cellular process requires the involvement of a protein. Therefore, studies on how oxidative stress can modulate proteins, either reversibly or irreversibly became an attractive and important field of study. How does oxidative stress modulate protein function? Remarkably, depending on several factors such as the type of ROS, their concentration and half-life, and also the affected amino acid, different outcomes can occur [75].
As discussed before, ROS play a crucial role in redox signaling [37], and it might be dependent on reversible redox switches by amino acids oxidation/reduction cycles.
In contrast to reversible methionine and cysteine oxidation as discussed above, there is no enzymatic or non-enzymatic way to revert types of protein oxidation such as protein carbonylation. In fact, due to its irreversibility, it has been used as a marker for severe chronic oxidative damage [76], [77], [78] as those found in many disorders that impair cellular function and survival [46], [79] and in experimental conditions upon cells challenged with oxidants such as H2O2 [80], [81]. Thus it is not surprising to find a positive correlation of carbonylated proteins with oxidative stress, age, and severity of disease [46], [82]. Protein carbonyls can be formed by the direct oxidant attack mediated by a metal-catalyzed oxidation (Fenton reaction) on proline, threonine, lysine and arginine. The most abundant products from this reaction on proteins are glutamic semialdehyde for proline and aminoadipic semialdehyde for lysine [83]. Increased carbonylation leads to protein unfolding followed by exposure of hydrophobic residues and decreasing solubility, resulting in an increased risk for aggregation [76], [83], [84]. Secondary reactions generating carbonyls on proteins can also occur. For example, protein carbonylation can result from modified aldehydes (from lipid peroxidation) such as 4-hydroxynonenal (4-HNE) [85]. This is thought to play a major role in metabolic diseases displaying increased levels of oxidative stress [86], [87], [88]. The evaluation of protein carbonyl content in several cells, tissues and organisms has shown that in the last third of life, these oxidative modified proteins increase, dramatically [82]. In fact, muscle adult stem cells accumulate carbonylated proteins in several, crucial cellular pathways such as carbohydrate metabolism, protein maintenance, cellular motility and protein homeostasis [89]. Moreover, Baraibar and Friguet have shown that carbonylation of enzymes affects intermediate metabolism in several tissues [90]. There is a large body of evidence supporting the accumulation of oxidized proteins in senescent cells and aged tissues. The risk for their accumulation lies in the formation of insoluble protein aggregates. Therefore, carbonylated proteins and other irreversibly modified proteins must be degraded in order to prevent them from forming aggregates.
Protein aggregates result from the accumulation of abnormal or oxidized proteins. Their accumulation can result from exceeding formation compared to degradation rates, e.g. by inhibition of one of the major proteolytic turnover mechanism, the proteasomal system. Over the years, it has been distinctly shown that the proteasome deals efficiently with oxidized/abnormal/misfolded protein turnover [91], [92], [93]. However, it may be the case that the formation of damaged proteins overwhelms their degradation and contributes to their accumulation. In fact, we have shown that upon oxidative stress, part of carbonylated actin is degraded by the proteasome, but depending on the intensity or duration of stimulus, carbonylated actin can form protein aggregates that inhibit the proteasome [80], [94]. Oxidative modifications in proteins such as protein carbonyls, increase the surface hydrophobicity of proteins, dramatically [95]. Carbonylated proteins are more prone to unfold and expose their hydrophobic core, usually concealed inside a folded native protein. At this point, oxidized proteins can interact amongst each other, contributing to an increase in insoluble protein aggregates [76], [95], [96], [97]. The formation of a Schiff-base, resulting from the reaction of a carbonyl group of a carbonylated protein with an amino group of another protein, can further contribute to aggregate formation. This demonstrates an interesting feature of aggregate enlargement without the involvement of any further oxidation [94]. Taken together, these events contribute to the aggregation-proneness of carbonylated proteins.
Protein aggregates and their relation to aging have been known for more than a century and were first described by Hannover in 1842, when visualizing the cytosol of old neurons. These aging-related protein aggregates are named “lipofuscin” from the Greek “lipo” meaning fat (with other words hydrophobic) and “fuscus” dark [98], [99]. Other names used include “age fluorophore” or “age pigment” [100], [101]. This insoluble material is composed by highly oxidized crossed-linked material, such as proteins, lipids and sugars. Moreover, transition metal ions can bind to lipofuscin and further mediate ROS production by Fenton reaction. This can extend the damage caused by lipofuscin as it becomes an intracellular ROS source itself. However, cells normally engulf this material by macroautophagy to prevent, at least partially, aggregate-toxicity.
Moreover, the idea that protein aggregates are functionally inert is long gone. Recent studies have shown that protein aggregates have the ability to change gene expression dynamics, resulting in a regulation at transcriptional level [102], [103]. Interestingly, protein aggregates have been described to be highly involved in aging progression across different species, emphasizing their role as a hallmark of aging [104]. This has been shown in bacteria [105], [106], yeast [107], C. elegans [108] and mammalian cells [109].
The presence of lipofuscin has been detected in several tissues including heart, liver, kidney and skin. In fact, different tissues comprising heart, liver, cerebellum, skeletal muscle and testis from old rats were found to display more lipofuscin content comparing to young rats [110], [111], lipofuscin can also be seen in aged and SIPS fibroblasts models comparing to young cells (Fig. 7). A fact that is more pronounced in post-mitotic tissue such as in neurons or muscle cells, since they no longer divide and are able to dilute the accumulated damage. Remarkably, hearing loss induced by cochlear degeneration in the aging mouse model SAMP-8 (senescence accelerated mouse prone 8) was found to be related to the accumulation of lipofuscin in spiral ganglion neurons [112].
Fig. 7.

Lipofuscin accumulation in young, aged and SIPS fibroblasts. One hallmark of aged cells is the formation of lipofuscin, which consists of highly oxidized proteins and lipids. A special characteristic of lipofuscin is its stable autofluorescence which can be used for the detection as well as quantification. Aged human dermal fibroblasts, obtained from an 81-year old donor (panel B) are marked by a strong accumulation of lipofuscin compared to fibroblasts from a 1-year old donor (panel A). To investigate the aging process in cell culture systems the so-called model of stress-induced premature senescence (SIPS) can be used. During SIPS, cells are treated chronically with a low dose of an oxidant to generate cells with a senescent phenotype. Panel C shows fibroblasts which were incubated with 40 µM paraquat as stressor for 10 days leading to the accumulation of lipofuscin. Lipofuscin autofluorescence was measured at 408 nm excitation and 420 nm emission wavelengths using a laser scanning microscope. Fibroblasts are cultivated as described in König et al. [314].
Furthermore, age-related neurological disorders, such as Alzheimer's (AD) and Parkinson's Disease (PD), are mostly characterized by the accumulation of insoluble protein aggregates [113]. The accumulation of the proteins amyloid-β and tau in AD and α-synuclein in PD is toxic to the hippocampus and Substantia nigra neurons, respectively, inducing cell death and corresponding cognitive ability loss. Taken together, these studies clearly demonstrate a link between organism aging and the formation/accumulation of protein aggregates/lipofuscin, suggesting a loss in proteostasis mechanisms.
4. Proteostasis in aging
Proteostasis refers to a balanced and functional cellular proteome, meaning that the requirement of proteins of a cell is optimized to each situation, either by re-localization or by fine-tuned cycles of protein synthesis/turnover. During aging, there is an increased risk of protein damage, either by oxidation or misfolding, that requires either their refolding or degradation. Proteostasis is maintained by an array of cellular mechanisms such as proteasomal degradation, autophagy clearance and molecular chaperones. However, aging correlates with a marked loss in proteostasis, evidenced by several age-related diseases exhibiting the accumulation of dysfunctional proteins into large insoluble aggregates. This suggests that the mechanisms decrease their efficiency allowing the formation of toxic protein aggregates. The loss of proteostasis is therefore a likely and strong contributor to senescence and aging [114], [115], [116] (Fig. 4).
The following chapters will explore how aging affects proteostasis in terms of proteasomal system and autophagy-lysosome-pathway (ALP).
4.1. Proteasome and aging
The most important cellular machinery catalyzing the degradation of proteins that are no longer needed, (oxidatively) damaged or even un-/misfolded proteins with reduced function is the proteasomal system [117], [118].
The proteasomal system is composed of different parts with various functions. The central part is the 20S core proteasome, a large cylindrical protease, containing an overall of six different proteolytic centers with different specificities. Several regulator proteins can bind to the 20S core, changing its activity or substrate specificity. Since the 20S is not able to degrade natively folded proteins, its main substrates are already unfolded dysfunctional proteins. These proteins are recognized as substrates via damage-exposed hydrophobic structures that are normally buried inside globular soluble proteins. The 20S-mediated proteolysis of an (partially) unfolded substrate does not consume any ATP. In order to enable also the degradation of natively folded substrates, the proteasome can bind a 19S regulator – forming the so-called 26S proteasome – that enables substrate degradation in an ATP-dependent and -consuming manner. In this case ATP is not necessary for proteolysis; it rather delivers the energy necessary for unfolding of the substrate. In order to label functional proteins for 26S proteasomal degradation, the substrate must be marked by a short chain of ubiquitin (Ub)-molecules. This is realized in a very specific manner by the so-called ubiquitin-system a highly complex machinery, that recognized substrates and labels them for terminal proteolysis [119].
Another important form of the 20S proteasome is the immunoproteasome (i20S) that can be induced by tumor necrosis factor alpha (TNF-α), interferon gamma (IFN-γ), lipopolysaccharides or other forms of stress. On the one hand, it plays an important role in the production of short oligopeptides that can be presented by the major histocompatibility complex-I (MHC-I) on the cell's surface during the immune-response. On the other hand, i20S shows higher proteolytic activity towards oxidized proteins compared to 20S [120]. In inflammatory processes, always accompanied by oxidative stress and therefore increased protein oxidation, the immunoproteasome and its co-expressed 11S regulator contribute significantly to preserving functional proteostasis [121].
In order to prevent an accumulation of oxidatively damaged proteins in phases of (mild) oxidative stress, the so-called “heat shock proteins” (Hsps) are induced, that are able to prevent accumulation of damaged proteins. One of these proteins is Hsp70, able to keep damaged proteins soluble and to interact with the 20S proteasome at the same time. Substrate recognition is assumed to be similar to the proteasome: exposed hydrophobic protein-sequences. Reeg et al. [122] demonstrated an interaction of Hsp70 with both, oxidatively damaged substrate proteins and the 20S proteasome, indicating a role of Hsp70 in the proteasomal degradation after mild oxidative stress. Besides this, Hsp70 is involved in proper protein folding during de novo synthesis. After binding to Hsp70, an (partially) unfolded substrate may either refold into native conformation and is released or remains bound to 20S, kept in a soluble state, unable to form aggregates with other unfolded proteins. Interestingly, in phases of stress detachment of the 19S regulator cap from the 20S “core” proteasome is mediated by Hsp70 binding to the 19S, resulting in a decline of 26S-proteolytic capacity, while the pool of free 20S is increased resulting in enhanced degradation of unfolded proteins [123].
Another heat shock protein involved in response to oxidative stress is Hsp90. One of its functions is the protection of 20S from inactivation by oxidative damage [124], other functions are the assistance in the late stages of proper protein folding, especially on signaling proteins involved in development and cell division: substrates include steroid hormone receptors, kinases, as well as key oncogenic proteins such as p53 [125], a group termed as “Hsp90 client proteins”. Furthermore, Hsp90 also assists the proteasomal degradation of substrates [126].
Therefore, molecular chaperones play a decisive role in proteostasis and life extension, but are known to decline in aging [116], [127]. Interestingly, the amount of i20S increases during aging. In mice a 3- to 6-fold increased amount was found, as well as significant shifts in the 20S to i20S ratio in rat livers [128]; similar changes were found in astrocytes, neurons and endothelial cells in the aged human hippocampus (average 70 years) compared to a younger control (average 42 years) group. Also i20S accumulates with aging in tissues that contain normally only the constitutive form of 20S – especially postmitotic aging tissues such as muscles and nerves are affected. Whether this results in a changed cleaving preference of the core proteasome or a changed activity is still under discussion. Also, an overall age-dependent decrease of proteasomal activity was found in different tissues: spinal cord, liver (up to 50% decrease [129]), adipose tissue, heart, brain (cortex and hippocampus), while in other regions such as brain stem or cerebellum, proteasomal activity did not decrease [130]). During the aging process, both the 20S and the 26S proteasome were affected, and in extraordinary long-lived species such as the naked mole-rat a significantly higher proteasomal activity (both 20S and 26S), higher amount of the 19S regulator and more immunoproteasomes were found compared to mice, which are of comparable size but have a much shorter life span [131].
Though, even if proteasomal activity declines, the reasons are still discussed. According to some authors, the amount of proteasomes found in the aged cell declines, as well as specific proteasomal activities – the reasons given are changes in its composition and/or structure, partly mediated by oxidative damage; furthermore an increase of both oxidatively damaged and polyubiquitinated substrate proteins was found [132]. This increase may be due to declined proteasomal activity, another cause may be the intracellular accumulation of oxidized protein aggregates over the whole lifetime of the organism, resulting in structures, that are recognized as proteasomal substrates, but that became resistant to proteolytic degradation by covalent ROS-mediated cross-linking, thus, distracting proteasomal activity. In turn, the resulting decrease of proteasomal activity as well as an age-related increase in cellular ROS contributes to enhanced formation of such proteasomal pseudo-substrates.
An age related decrease of proteasomal activity was shown in several different tissues like rat liver (−50% in the peptidylglutamyl hydrolase) and brain: decrease of chymotrypsin like activity in cortex, hippocampus and spinal cord of 12 month old animals, while no change was detected in brain stem or cerebellum, while at the same time an increase in protein oxidation was accompanied by reduced proteasomal activity in this areas [130]. In rat muscle an upregulation of the immunoproteasome was found, as well as increased oxidation of proteasomal subunit oxidation. The induction of proteasomal activity by 19S and proteolysis of polyubiquitinated substrates were also reduced. In the hind limb muscles from 30 month old Sprague-Dawley rats the levels of both beta1 and beta5 subunits were found to be increased up to three-fold like the 19S regulator subunits Rpt5 and Rpt6, though, the proteasomal activity was not measured in this study, suggesting a response to the increased amounts of oxidatively damaged proteins [19]. In contrast, other studies using F344 rats also revealed significant decrease in proteasomal activity (−30% for chymotrypsin like activity) for both 20S and 26S, but no reduction of the proteasomal amount compared to young rats [133]. In contrast, in the hearts of senescent rats both a loss of proteasomal amount as well as loss of proteolytic capacity were found [132]. Similar decrease of all three proteasomal activities was also found in human BJ fibroblasts [134], [135]. Detailed investigation of proteasomal subunits in human fibroblasts (WI 38) also revealed that only the expression of catalytic beta subunits was reduced, as well as proper proteasomal assembly, since a considerable amount of alpha subunits was detected in a free state [136]. Interestingly early works demonstrated that the 20S proteasome is inhibited by cross-linked substrate proteins [137], [138]. Furthermore, the reduction of proteasomal activity found in aged retina cells was shown to be reproducible by exposing young cells to N-ethyl-maleimide (NEM), that modifies the sulfhydryl-group of cysteine-residues: after NEM-exposure of young retina cells the chymotrypsin-like activity decreased to about 65%, the caspase-like activity to about 80% compared to untreated ones [139].
Thus in recent studies, despite of decreased activity, in many cases also reduced expression and/or amount of UPS-subunits was detected during the aging process [130], [140].
Also in aging, an increased formation of Ubiquitin-B+1 (Ubb+1), a misframed mutant of ubiquitin protein is found, leading to accumulation of polyubiquitinated proteins, causing proteasome malfunction [141], apoptosis and likely playing a role in AD progression [142] and other age-related diseases [143]. Furthermore, a variety of different enzymes, involved in substrate-polyubiquitination are affected by aging [144], thus influencing several essential cellular functions in favor of the progression of neurodegenerative diseases, accelerated senescence [145], reduced life span, carcinogenesis, genomic instability, increased susceptibility to (oxidative) stress, formation of protein aggregates [146] such as lipofuscin. As described before, lipofuscin is a highly oxidized material containing covalently cross-linked proteins, lipids and sugars that is very resistant to mammalian proteases and accumulates especially in post-mitotic cells over time, showing a strong negative correlation with the remaining life span [119], [147], [148], [149]. Furthermore, lipofuscin was shown to inhibit the proteasome and contribute to intracellular ROS-formation as described before [52], [94]. Besides lipofuscin, the formation of the so-called advanced glycation end-products (AGEs, glycated proteins and lipids) is a life span limiting factor, since AGEs showed similar effects on the UPS as lipofuscin [150]. Further discussed causes for decreased UPS-function are oxidative damage to UPS-subunits, defective expression of regulatory subunits, reduced assembly of 26S that may originate from a decreased availability of ATP, as well as a shifted cellular redox state, that may induce posttranslational modifications of UPS-subunits (e.g. S-glutathionylation or phosphorylation), inducing changes in activity [151] and activity of the poly (ADP-ribose) polymerase 1 (PARP1) [152], [153].
Taken these results together, induction/restoring of the UPS and its compounds were suggested as possible strategies to counteract the aging process [145], [154], [155]. Overexpression of single catalytic proteasomal subunits also increased the amount of other active subunits (via a hitherto unknown regulatory loop) [136]. Cells, stable overexpressing active proteasomal subunits, showed reduced amounts of protein aggregation after oxidative stress [136], improved resistance to oxidative stress (H2O2, menadione, 4-HNE), enhanced viability, and even an increase of replicative life span by 15–20% (human fibroblasts [156]), while the restoration of proteasomal activity in senescent cells (via a lentivirus gene-delivery system) decreased the amount of aging-markers SA-β-Gal and p21 [157]. Similar results – an overall increase of proteasomal activity – were achieved by overexpression of the proteasome maturation enzyme ubiquitin-mediated proteolysis 1 (Ump1) in both yeast and mammalian cells [158], [159]. Also, activation of the redox-sensible transcription factor “nuclear-erythroid factor 2” (Nrf2), which induces the expression of several antioxidative enzymes, amongst other subunits of the UPS. Nrf2 turned out to be downregulated by about 45–65% in (replicative) senescent cells [160]; together with induction of Nrf2 by 18α-Glycyrrhetinic acid (18α-GA), proteasomal activity was also enhanced, however this shift in proteasomal activity (and also enhanced resistance to oxidative stress and life span extension) was massively suppressed by Nrf2-knockdown via the according siRNA [161].
Finally, induction of the UPS in (even terminal) senescent cells may not increase life span dramatically, but it may restore the ability of cells to respond/adapt to changed (environmental) conditions in an appropriate manner, resulting in decreased apoptosis and prolonged “healthy aging” (also in animals). This will indirectly affect life span, and focusing on the role of the UPS in maintaining cellular proteostasis. Moreover, “healthy aging” may – at least partially – result from enhanced regulation of the proteome and thus, cellular functionality.
4.2. Age-related changes in autophagy
Together with the UPS, the ALP belongs to the main intracellular degradation systems, responsible for the removal of dysfunctional cellular constituents and their recycling. One part of the ALP is autophagy which initiates the degradation of long-lived proteins and damaged organelles (mitophagy, peroxiphagy) by the delivery of the cargo into lysosomes. To maintain the cellular equilibrium by autophagy three different forms exist: chaperone-mediated autophagy (CMA), microautophagy (MiA) and macroautophagy (MA) (reviewed in [162]). CMA can be activated by different stressors, such as starvation, oxidative stress or exposure to toxic compounds. Subsequently, substrate proteins interact with the constitutive heat shock cognate 70 (Hsc70) [163], [164], [165] followed by the binding of the chaperone-substrate complex to lysosome-membrane associated protein type 2A (Lamp2A), a receptor protein on the lysosomal membrane. After unfolding, the substrate can enter through the lysosomal membrane. CMA is limited to soluble proteins, so no organelles can be degraded. Regarding MiA less information is currently available [166]. During MiA cytosolic proteins are directly sequestered by the lysosomes, followed by their degradation in the lumen. The lack of specific markers to measure MiA makes it difficult to follow this process and its changes during aging. MA, often simply referred to as autophagy, is also considered to be a stress-inducible form, sequestering cytoplasmic constituents by a double membrane-bound vacuole, the so called autophagosome. The autophagosome further fuses with the lysosome, generating a single-membrane-bound vacuole, the autolysosome, where the degradation by acid hydrolases takes place. Initiation of the autophagosomal membrane, elongation, maturation to the autophagosome and its fusion with the lysosome are mediated by different autophagy-related proteins (ATGs) [163], [167]. Further causes of age-related changes in autophagy will be more specified in the following paragraph by summarizing the current facts about autophagy and aging.
4.2.1. Decline of CMA, macroautophagy and mitophagy during aging
During aging, in contrast to other lysosomal membrane proteins, such as Lysosomal-associated membrane protein 1 (LAMP1), levels of lysosomal protein receptor Lamp2A decrease in such extent that CMA activity can no longer be preserved [168]. Initially, the decrease of Lamp2A can be compensated by the increasing number of lysosomes during senescence. Immunofluorescence detection and immunoblot analysis of Lamp1 have confirmed the rising number of lysosomes in aged cells [168], [169], [170]. Regarding the lysosomal activity several studies postulated that lysosomal activity increases during cellular aging [17], [147], [169], while others reported the opposite direction [171], [172]. The limitation in CMA is that only Hcs70 containing lysosomes, responsible for substrate translocation, are active for CMA [165]. In the middle-aged state, CMA is maintained by increasing chaperone-levels while in advanced age, levels of Lamp2A are so low that they can no longer be compensated by chaperones [164], [173]. However, other studies also described a decline in Hsc70 protein expression, resulting in an insufficient content of the chaperone and unfeasible lysosomes for CMA. If instability of the receptor or impaired recovery from the lumen is responsible for the decrease of Lamp2A needs to be further investigated. In the case of MA the decreased formation and clearance of autophagosomes may be one reason for the decline of autophagy during aging [174]. Several studies have shown that i) formation of autophagosomes is decreased due to an overall decline in autophagy-related proteins (ATGs) but also that ii) clearance of autophagosomes is impaired by, either inability of autophagosome to fuse with lysosomes or a decrease in lysosomal activity [135], [175]. Additionally, mammalian target of rapamycin (mTOR), particularly mTORC1 has been described to inhibit the initiation of autophagy due to ATG1/ULK1-2 phosphorylation [176] (Fig. 8). Furthermore, it was reported that mTORC1 activity is enhanced in aged tissue and linked to mitochondrial dysfunction independent of growth factor signaling [177], [178]. ALP-related proteins, demonstrating reduced MA in aging are summarized in Table 1.
Fig. 8.

Age-related changes in the autophagy-lysosome pathway. The autophagy-lysosome pathway (ALP) is one of the main intracellular degradation systems, responsible for the removal of dysfunctional cell constituents and their recycling. One part of the ALP, macroautophagy can be subdivided into different phases, each of them can be negatively affected in aging. Phase I includes different autophagy-related genes (ATGs), which are mainly responsible for the initiation and development of the autophagophore. During the aging process, ATGs, such as Beclin-1, ULK1, ATG5 and ATG12 decrease; resulting in a decline in the initial steps of autophagy. In addition up-regulation of Bcl-2 and mammalian target of rapamycin (mTOR), enhanced by decreased levels of Sestrin1, involved in AMPK activation, aggravate the impaired initiation of autophagy. The potential decrease of p62 and Parkin, both involved in the delivery of either dysfunctional, ubiquitinated proteins or Pink1-tagged mitochondria, can support the accumulation of dysfunctional proteins and organelles in aging. Finally, reduced conversion of unbound LC3-I into the membrane-bound LC3-II demonstrates the impairment of the initiation phase. In the second phase, the number of autophagosomes and lysosomes increase in aging, reported by several studies, analyzing lysosomal-associated membrane protein 1 (LAMP1) and the autophagosomal marker monodansylcadaverine (MDC), related to total cellular protein. But an increased autophagosome and lysosome number is not able to explain decreased protein degradation and increased protein aggregation as well as accumulation in aging, assuming that fusion of both is likely to be impaired, a process which is not known yet and needs further investigation. In addition to impaired mito- and macroautophagy also chaperone-mediated autophagy is reduced in aging, demonstrated by reduced levels of Lamp2a and HSC70.
Table 1.
Age-related changes in protein levels of the Autophagy Lysosomal Proteolysis.
| Protein | Expression in aging | Function | Transcription-factors | Ref. |
|---|---|---|---|---|
| Sirtuin1 | ↓ | NAD-dependent protein deacetylase, Autophagy induction | – | [[315], [316], [317]] |
| ATG5 | ↓ | Autophagosome formation | MiT/TFE, AFT4, CHOP | [[318], [319], [320], [321]] |
| Beclin 1 | ↓ | Initiation of Autophagy | NF-κB, E2F, JUN | [318],[[322], [323], [324]] |
| Bcl-2 | ↑ | Anti-apoptotic, inhibition of autophagy via interacting/suppressing of Beclin 1 | NF-κB, | [325] |
| LAMP2a | ↓ | Substrate uptake into lysosomes during CMA | TFEB | [168,326] |
| Hsc70 | ↓ | Chaperone, responsible for substrate translocation in CMA | HSF1 | [165] |
| LC3 | ↓ | Formation autophagosomal membrane | FOXO3, GATA1, AFT4, CHOP, JUN | [321,322,324,327,328] |
| ULK1 | ↓ | Initiation of Autophagy | FOXO3, AFT4 | [322,329] |
| P62 | ↓↑ | Binds LC3, promotes selective autophagy, associates with protein aggregates in several neurodegenerative diseases | Nrf2 | [[330], [331], [332], [333], [334]] |
| Sestrin1 | ↓ | Stress-inducible, inhibitor of mTORC1 | FOXO, p53 | [335,336] |
| BAG3 | ↑ | Interaction with p62, co-chaperone of HSP70 | HSF1 | [[337], [338], [339], [340]] |
| mTORC1 | ↑ | Suppression of autophagy | ATF5 | [341] |
| PINK1 | ↓ | Targeting mitochondria for mitophagy | NRF2 | [342] |
In addition aging is accompanied by the slow accumulation of dysfunctional mitochondria [179], [180], [181], [182], [183]. Since dysfunctional mitochondria contribute to an elevated intracellular ROS formation, their adequate degradation is essential for cellular homeostasis. The process of mitochondrial degradation is performed by the lysosomal system, through a special form of MA termed mitophagy. However, before the lysosomal system can catabolize mitochondria, several steps are necessary for the recognition of damaged organelles and their further targeting for the autophagy-machinery. In this context two proteins play a key role: PTEN-induced putative kinase 1 (PINK1) and Parkin. PINK1 is responsible for the identification of damaged mitochondria. In healthy mitochondria PINK1 is continuously cleaved and degraded by different proteases keeping low endogenous PINK1 levels under normal conditions. In contrast, damaged mitochondria are marked by an accumulation of PINK1 at the outer mitochondrial membrane. This stabilization of PINK1 leads to the recruitment of the ubiquitin ligase Parkin which ubiquitinates several outer membrane proteins and initiates, therefore, the engulfment of mitochondria by the autophagosome [184]. MA in general but also mitophagy in particular have been shown to decline during aging [185], [186], [187], [188]. However, the reasons for this reduction in mitophagy seem to be diverse. Interestingly, it has been shown that PINK1 expression decreases with age in murine lung tissue [186], [189] and PINK1 deficiency is marked by dysfunctional and swollen mitochondria [189], [190], a mitochondrial phenotype which can also be found in aged cells [191]. Furthermore, it was shown that PINK1 knockdown in HeLa cells affected the translocation of Parkin to mitochondria resulting in interrupted mitochondrial clearance [192]. Nevertheless, it was also shown that this effect can be rescued by the overexpression of Parkin [193]. Furthermore, Parkin overexpression experiments indicate a special relevance of this protein during aging. Rana et al. investigated the effects of Parkin overexpression on different aging parameters in Drosophila. They found a reduced protein aggregate formation in the brain and in flight muscles as well as an increased mitochondrial function in aged Parkin-overexpressing flies. Moreover, Parkin overexpression was able to extend the life span of these flies [194]. These data are consistent with the finding that Parkin-null mice have a reduced life span and accumulate more tau protein aggregates in the brain compared to wild-type animals [195].
5. The role of trace elements and micronutrients in aging
5.1. Trace elements in aging
Trace elements (TE) are one of the key regulators of both metabolic and physiological pathways and are also known to be altered during the mammalian aging process. In order to preserve the cellular homeostasis and thus functionality, an optimal intake of TEs is required. Consequently, TE deficiencies but also overloads are found to be associated with functional changes, increasing the risk of incidence and severity of several (also age associated) diseases.
TE act on age-affected processes such as functionality of the immune system (selenium (Se), zinc (Zn), copper (Cu)) [196] and oxidative stress (iron (Fe), manganese (Mn), Zn, Se, Cu) [197]. The ability of TEs to both ameliorate oxidative damage and to promote repair processes and recovery is mediated by their function as essential co-factors for various antioxidative enzymes such as Cu, Zn-superoxide dismutase, Mn-superoxide dismutase, catalase (Cu, Fe), and different types of glutathione peroxidases (Se). These antioxidative enzymes are necessary for limiting lipid peroxidation, assisting the repair of oxidative base-modifications as well as restoring the functionality of (oxidatively) modified proteins that accompany both, several diseases (especially the ones associated with inflammation) and the aging-process. Since TEs can also act pro-oxidative in high concentrations, a delicate balance is crucial for cellular function.
During the aging process low concentrations of Se and Zn, and high concentrations of Cu have been reported in several studies [198]. Furthermore, serum Cu/Zn ratio was found to be significantly increased in senescent individuals compared to middle-aged adults [199]. Interestingly, the ratio of Cu/Zn was significantly increased in elderly patients suffering from age-related diseases compared to the healthy ones. The ratio found in healthy elderly was due to high copper values, whereas in the patients, as well high amounts of copper and low amounts of zinc were found in the serum. The Cu/Zn ratio was significantly and positively correlated to products of lipid peroxidation, suggesting a relationship between the Cu/Zn ratio and (increased) systemic oxidative stress. Furthermore, the intracellular accumulation of iron is widely accepted as an important feature of the aging process particularly in post-mitotic tissues [200], whereas both heme-Fe and heme biosynthesis are known to decline significantly with age [201]. Additionally, low serum Se levels seem to be a useful predictor of mortality as shown in two independent studies in elderly people [202], [203].
Though, until now only a few analyses have investigated the connection between trace elements and aging/senescence at a cellular level. An extension of replicative life span in bovine adrenocortical cells after selenium supplementation (20 nM) has been reported by Hornsby et al. [204]. Also, in hepatocytes high doses of Se (0.5 and 2.5 μM) were found to be able to extend telomere length, prolonging the cellular life span, too [205]. This may be mediated by incorporation of Se into selenoproteins with antioxidative function, since the balance between oxidative stress and a performing antioxidative defense preserves proteostasis and cellular function and also affects the rates of aging and telomere shortening amongst others.
However, already in the nanomolar range, Se concentrations should be sufficient to optimize the expression of selenoproteins. Furthermore, it seems that the levels of selenium affect both the regulation of entry into replicative senescence and are able to modulate characteristic markers of senescence: while selenium-supplementation (45 nM) increases the number of population doublings, its deficiency (3 nM) impairs significantly the proliferative capacity as found in WI-38 fibroblasts [206]. Also, the long-term deficiency of Se causes a decrease in the cellular antioxidative capacity by dysregulation of selenoprotein-expression.
After an incubation period for two passages in Se-depleted medium, both a decrease of almost all selenoproteins as well as an increase in senescent characteristics was found in cells. On the other hand, in MRC-5 fibroblasts treated with methylselenic acid (up to 10 μM), it was revealed that Se compounds can also induce senescence mediated by a DNA damage response and increased amounts of SA-β-Gal were found [207]. Furthermore, sodium selenite treatment (20 µM) attenuates the interaction of the heat shock protein Hsp90 with the IκB-Kinase (IKK) in tumor cells – thus promoting inactivation of the NF-κB pathway and inhibition of autophagy via downregulation of Beclin 1 expression, inducing a cell signaling switch from autophagy to apoptosis [208].
Growing evidence points to zinc as a positive regulator of autophagy. In vitro studies have consistently shown that Zn is critical for both basal as well as induced autophagy [209], [210], [211]. High doses of Zn in the culture medium 20–200 μM) have shown to induce autophagy in MCF-7 breast cancer cells [209], in astrocytes [211] and also in human hepatoma cells [210]. Zn depletion either caused by treatment with the cell permeable Zn chelators TPEN or Chelex-100 was able to suppress basal and induced autophagy [209], [211]. Also, a possible role of Zn as modulator of the SASP was suggested [212]. The fact that a Zn deficiency develops during aging raises the suspicion that there may be a possible connection between Zn and senescence.
In colon cancer cells cultured in a low Zn environment for six weeks, morphological changes and also typical markers of senescence were detectable [213], while in contrast addition of Zn induced increased formation of ROS, causing senescence in vascular smooth muscle cells. Zn also decreased the cells’ antioxidative capacity by downregulation of catalase expression [214]. Furthermore, an imbalanced intracellular Zn homeostasis mediates both oxidative damage and neuronal cell death in neurodegenerative diseases [215], [216]. Thus, oxidative damage and cellular senescence may be promoted by unbuffered regulatory mechanisms of intracellular/cytosolic Zn.
Copper is another TE that is known to induce (premature) senescence. It plays an important role as a cofactor of different enzymes, but free ionic copper is cytotoxic, because it mediates the formation of the highly reactive hydroxyl radical (•OH) via Fenton reaction, that is virtually capable of damaging any cellular biomolecule. Recent studies in cellular models showed, that Cu can induce the appearance of senescent characteristics [49]. Copper can also inhibit the proteasomal β5-subunit (resulting in a decrease of proteasomal activity) and induce apoptosis in human cancer cells [217].
The available literature regarding Mn and aging mainly addresses the notion of Mn as a risk factor for Parkinsons’ disease (PD), yet the mechanism of manganese neurotoxicity is still unclear. Dysfunction of the ubiquitin proteasomal system (UPS) was shown to play a fundamental role in PD pathogenesis, as well as in increased oxidative stress (chronic low-grade inflammation), that may be induced by Mn. In PC12 cells treated with different concentrations of MnCl2, the proteasomal activity decreased with increasing concentrations of MnCl2 implying that the proteasomal dysfunction may be associated with Mn-induced cytotoxicity [218], [219].
An accumulation of Fe is known as a function of age in several tissues in vivo and is associated with the pathology of several age-related diseases. The according changes may be caused by a dysregulation of iron homeostasis at a cellular level, but the mechanism is still poorly understood. In IMR-90 fibroblasts the total iron content was shown to increase exponentially during cellular senescence, resulting in 10-fold higher levels of Fe compared to young cells. Furthermore, low-dose exposure to hydrogen peroxide (H2O2) also induced early senescence in IMR-90 cells and accelerated the senescence-associated accumulation of Fe [220]. This accumulation may also contribute to the increased amounts of oxidative stress and the decrease of cellular function that characterizes senescent cells.
However, the association between TE status, age-related diseases and cellular senescence is only poorly described. Additionally, there is limited knowledge about the interactions of different TEs especially during aging. Cellular responses to changes in both single TE and TE patterns and how these (synergetic) changes affect senescent cells, organs or the whole organism are less investigated. Finally, it is important to focus on the mechanisms of signaling driving these changes.
5.2. The human situation: protein carbonylation in age-related diseases and its possible prevention by antioxidant micronutrients
Human intervention studies assessing the effect of nutritional antioxidants on biomarkers of protein oxidation are scarce, with few studies showing a positive effect (reduction of protein carbonyls) [221], [222], [223], and some showing no effect [224], [225], [226]. Similarly to TE, micronutrients are also crucial for cellular homeostasis. Micronutrients are nutritional compounds distinct from the macronutrients (protein, carbohydrates and fat). They comprise numerous substances which are only required in small quantities and include vitamins, trace elements, minerals and other diet-derived compounds such as flavonoids and carotenoids. Although micronutrients play key roles as antioxidants and in physiological maintenance their impact on preventing oxidative stress in human trials has been disappointing.
Elevated levels of protein carbonyls in serum, plasma, and tissues have been observed in various age-related diseases and in aging in general. Whether they are the cause or consequence is under discussion and may vary depending on the type of disease.
In Fig. 9, we give an overview of (age-related) diseases associated with protein oxidation. Most evidence exists for neurodegenerative diseases such as Alzheimer's disease [227], [228], [229], [230], [231], [232], [233], [234], [235], [236] and Parkinson's disease [237], where carbonylation of proteins has been demonstrated in brains of AD patients [238], [239], dementia patients with Lewy bodies [240], and whole brain of PD patients [241]. Furthermore, protein carbonyls were found in various diseases such as acute/adult respiratory distress syndrome [242], [243], chronic lung disease [244], [245], [246], [247], amyotrophic lateral sclerosis [248], [249], rheumatoid arthritis and juvenile chronic arthritis [250], [251], severe sepsis [252], [253], cystic fibrosis [254], [255], cataractogenesis [256], age-related macular degeneration [257], chronic renal failure, uremia [258], [259], [260], [261], Diabetes mellitus Type I and Type II [262], [263], [264], [265], [266], inflammatory bowel disease [267], ischemia-reperfusion [268], systemic amyloidosis [269], and essential arterial hypertension [270].
Fig. 9.

Protein oxidation in age-related diseases. This figure shows the numerous diseases in which protein oxidation has been demonstrated so far. Protein oxidation may be the cause or consequence of these diseases which affect nearly all organ systems.
The prevention of age-related diseases by dietary antioxidants seems plausible since one way to increase health span is by having a healthy life style which includes physical activity and a healthy diet. It seems that a healthy diet rich in micronutrients is an easy, safe, cheap and effective way to reduce the risk for different age-related diseases. The cellular micronutrient status depends only on the diet or nutritional supplements; in cell culture, the status is dependent on the medium. Studies on the impact of different micronutrient concentrations and their role in young compared to senescent cells are rare. For this reason, in this part of the review we will focus on the role of micronutrients in aging, cognition and cardiovascular diseases (CVD) in animal studies and human trials.
The assessment and interpretation of the micronutrient status of older individuals is quite difficult since inter-individual differences increase with age and the group of aged people is highly heterogeneous in terms of physical and mental health status, nutritional habits, weight, anthropometrics, life style etc. Various human studies show that there is a decline and/or an inadequacy of most micronutrients during aging concerning both, intake as well as the status [271], [272], [273]. These changes are likely due to altered dietary habits with an inadequate intake of micronutrient-rich foods such as fruits, vegetables and whole grains but also due to a reduced bioavailability and an increased turnover of micronutrients. Lipid-soluble vitamins (A, D, E, and K) are affected by these changes as are the water-soluble vitamins (C, B1, B2, B6, B12, and folate). The recommended dietary allowance (RDA) and sources of these vitamins for elderly people ≥65 years in relation to adults 18–65 years are shown in Table 2, collected from [274], [275], [276], [277].
Table 2.
| Vitamin | Dietary sources |
RDA (≥65 years) |
Recommendation for persons aged ≥65 years (in percent to adults) | |
|---|---|---|---|---|
| Men | Women | |||
| Thiamin (B1) | meat, especially pork, grains | 1.2 mg | 1.1 mg | 100 |
| Riboflavin (B2) | dairy products, eggs, whole grain, leafy vegetables | 1.3 mg | 1.1 mg | 100 |
| Pyridoxine (B6) | milk, meat, grains | 1.7 mg | 1.5 mg | 131 |
| Folate | green leafy vegetables, grains and liver | 400 µg | 400 µg | 100 |
| Cobalamin (B12) | animal products, fortified breakfast cereals (USA) | 2.4 µg | 2.4 µg | 100 |
| Ascorbic Acid (C) | citrus fruits, green vegetables | 90 mg | 75 mg | 100 |
| Vitamin A (Retinol) | animal products | 900 µg | 700 µg | 100 |
| Vitamin D | fatty fish, fish liver oil and egg yolk, fortified milk (USA) | 20 µg | 20 µg | 133 |
| Vitamin E (α-Toc.) | vegetable oils, whole grain, nuts, fruits, vegetables, meat | 12 mg | 12 mg | 80 |
| Vitamin Ka | green vegetables, broccoli, Brussels sprouts | 120 µg | 90 µg | 100 |
Adequate intake.
Vitamin E (α-tocopherol) exhibits antioxidant and anti-inflammatory properties and modulates signaling pathways involved in neurodegeneration thus acting neuroprotective and being crucial for normal neurological function [278]. α-Tocotrienol has been shown to attenuate glutamate-induced activation of phospholipase A2 arachidonic acid release in murine hippocampal neural cells [279]. In humans, low concentrations of tocopherols and tocotrienols are associated with AD and mild cognitive impairment (MCI) [278]. AD-, MCI- and control-patients (>70 years) had significantly different plasma vitamin E concentrations with higher tocopherols, tocotrienols and total vitamin E being associated with a reduced prevalence of AD [280], and all 8 vitamin E isoforms were lower in subjects with MCI or AD compared to healthy subjects [281]. Cognitive decline during 18 months was less pronounced in older persons consuming higher amounts of vitamin E [282].
Vitamin A (retinol) and its biological active form retinoic acid (RA) are essential for brain development and cognitive functions, such as the memory process, by modulating synaptic plasticity and improving neurogenesis [283]. Treatment with RA reduced the number of senescent cells, the expression of p21 and the inflammatory signaling as well as led to an extension of the life span in a senescent mouse model [284]. Young rats displayed better memory than old rats and retinol supplementation of old rats improved memory; in addition there was a positive correlation between serum retinol and spatial memory in young and old rats [283]. In an aging rat model, plasma retinol was significantly reduced in old compared to young and middle-aged animals [285]. In humans, some studies demonstrated a significant decline in plasma vitamins A and E with age, but in contrast, significantly higher vitamins A and E were observed in healthy centenarians compared to younger subjects, implicating a possible role of these vitamins in longevity [286]. A meta-analysis showed significantly lower vitamin A and E in AD patients compared to healthy controls [287]. In a case-control study, plasma vitamin E and β-carotene in patients with AD and multi-infarct dementia as well as plasma vitamin A concentrations of AD patients were significantly lower than in healthy patients [288]. Elderly MCI and AD patients had significantly lower plasma vitamin A and E as well as some carotenoids (lutein, α-carotene, zeaxanthin and β-cryptoxanthin (only for AD)) compared to healthy subjects [289]. The consumption of a diet rich in luteinandzeaxanthin was associated with a slower age-related cognitive decline [290]. Carotenoids are plant-derived antioxidants and anti-inflammatory compounds suggested to prevent age-related diseases including CVD, cognitive impairments and different forms of cancers [291]. Lutein and zeaxanthin are the most prominent carotenoids in the central nervous system and in the human brain tissue with proposed neuroprotective function which is due to decreased oxidative stress and activation of anti-inflammatory pathways [290].
Vitamin D, which can be synthesized endogenously by UV-B radiation, exerts its function by its biological active form 1,25(OH)2 vitamin D3. It functions as steroid hormone, in the regulation of the immune system, seems to be involved in the cell cycle regulation, in the protection against free radicals in the central nervous system and thus may play a role in senescence and aging [292]. Vitamin D receptor knockout mice develop signs of premature aging, i.e. infertility, muscle atrophy, reduced immune function, and osteoporosis and have a shorter life span [293]. Vitamin D deficiency, which is highly prevalent especially in house-bound or hospitalized elderly, is linked to age-related disease such as neurodegenerative diseases and CVD [273], [292].
Water-soluble vitamins are also related to cognitive function. Vitamin B1 (thiamin) plays an important role as a cofactor in several biochemical pathways and especially high concentrations are found in heart, liver, kidney, brain and skeletal muscle. Inadequate thiamin has been observed in older subjects [294] and abnormal thiamin-dependent processes appear in AD [295]. Vitamin B2 (riboflavin) is a cofactor in metabolic redox reactions, amino acid and lipid metabolism. Furthermore it functions as an antioxidant, due to its involvement in the glutathione redox cycle (in glutathione reductase), the prevention of lipid peroxidation and by affecting antioxidant enzymes [296]. In animal studies, riboflavin deficiency increased lipid peroxidation and the administration of riboflavin decreased MDA and protein carbonyls [297]. In humans riboflavin intake and plasma MDA were negatively correlated [298]. Pyridoxine (vitamin B6) helps to reduce homocysteine concentrations thus protecting against CVD. Homocysteine is a non-proteinogenic amino acid which occurs under normal physiologic conditions. It is an essential intermediate in protein synthesis as it accepts a methyl-group from methyl-tetrahydrofolate (via methionine synthase, which contains cobalamin) to form methionine, the starter amino acid. A high homocysteine blood concentration is an important risk factor for atherosclerosis and cardiovascular diseases, since some human studies have shown a clear correlation between serum homocysteine and the incidence of carotid, coronary, and peripheral vascular disease [299], yet the mechanism behind this remains to be fully elucidated.
Furthermore, pyridoxine has been shown to act as an antioxidant in isolated rat hepatocytes by inhibiting iron-induced O2•− production, thus preventing lipid peroxidation, protein oxidation, and DNA damage [300]. Rats receiving a diet deficient in pyridoxine for one year developed abnormal walking-track patterns, however, when these rats received pyridoxine they partially recovered [301].
Tetrahydrofolate (THF), the biological active form of folic acid is involved in purine and pyrimidine synthesis, namely in 1-carbon transfer reactions and helps to reduce high homocysteine concentrations [302] which, together with reduced folate and vitamin B2 were associated with an increased risk for coronary artery disease [303]. In aged rats, memory and also oxidative stress status, assessed as the activity of SOD and catalase, the glutathione concentration as well as MDA, significantly improved in folic acid supplemented animals [304]. In another study, folate deficiency in mice was associated with hearing loss and significantly increased homocysteine concentrations [305]. Vitamin B12 (cobalamin) functions as a cofactor for both the methylmalonyl CoA mutase and the methionine synthase which is involved in homocysteine re-methylation and folate cycle [306]. Subsequently vitamin B12 deficiency leads to elevated homocysteine, and impaired folate metabolism [306]. Decreased vitamin B12 concentrations have been demonstrated in postmortem human frontal cortex from patients with autism and schizophrenia, as well as to decline with age (range from 19 weeks pregnancy to 80 years) [307].
Vitamin C (ascorbic acid) and dehydroascorbic acid form a redox system functioning as antioxidant which recycles vitamin E and tetrahydrobiopterin. It is required for carnitine biosynthesis and the synthesis of neurotransmitters. In the central nervous system, intracellular ascorbate acts as an antioxidant, and it is necessary for peptide amidation, myelin formation, and in protecting against glutamate toxicity [308]. More amyloid-β plaque deposits have been observed in mice with moderate intracellular vitamin C deficiency [309]. Thus it has been suggested that populations at increased risk for epilepsy and seizures, such as in AD should avoid vitamin C deficiency [310]. It is known that aged individuals consume poor amounts of vitamin C [294], furthermore it declines with age and individuals with lowest intake seem to have the highest stroke mortality rate [311].
Since micronutrients play key roles in cellular homeostasis, in general physiologic maintenance and in the protection against oxidative stress, they can help to prevent or ameliorate detrimental age-related changes. Some, but not all, micronutrients possess antioxidant properties which are responsible for their beneficial actions. Antioxidants seem to influence cerebral functioning by reducing ROS-induced damage and stabilizing membranes [287], are positively associated with an improved cognitive function in healthy elderly adults and were decreased in patients with MCI and AD [290]. Epidemiological studies imply that individuals with a high intake of fruits and vegetables, and thus high antioxidant consumption have a reduced risk of CVD [312], AD and cognitive impairments [287], [313]. Therefore, it is highly recommended to consume a lifelong diversified diet including an abundance of foods rich in antioxidant nutrients.
6. Conclusions and future directions
This educational review focuses on the main proteostasis mechanisms current knowledge and their changes during aging. Due to the frequency of disturbances in proteostasis during aging, resulted in modified protein accumulation, one can only imagine the effects of attempts to improve the age-related failure of protein maintenance mechanisms. Especially in post-mitotic and slowly dividing tissues, this should keep a working cellular metabolism for substantially longer time, therefore, postponing senescence hallmarks and premature aging. Furthermore, from the organismal side, this would lead to the preservation of organ functions, including brain, skeletal and cardiac muscle, and, therefore, prolong healthy aging.
The attempts to achieve this are quite limited, although some studies on micronutrients and trace elements are promising to follow this line further. Future strategies to improve the selective and efficient removal of damaged or aggregated proteins need to be developed, which might also include the use of hormones or endocrine factors. Besides animal and human studies, in order to reveal the mechanism of action of these interventions in aged cells, more suitable models have to be employed. Regardless, of the importance to do so, only a few groups have been used such as postmitotic cells or developed aging models to study proteostasis maintenance.
References
- 1.C. López-Otín, M. Blasco, L. Partridge, M. Serrano, G. Kroemer, The Hallmarks of Aging. Cell153(6):1194–1217 [DOI] [PMC free article] [PubMed]
- 2.Flatt T. A new definition of aging? Front. Genet. 2012;3:148. doi: 10.3389/fgene.2012.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hayflick L., Moorhead P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 4.Harley C.B., Futcher A.B., Greider C.W. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345(6274):458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 5.As I.J., Greider C.W. Short telomeres induce a DNA damage response in Saccharomyces cerevisiae. Mol. Biol. cell. 2003;14(3):987–1001. doi: 10.1091/mbc.02-04-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuilman T., Michaloglou C., Mooi W.J., Peeper D.S. The essence of senescence. Genes Dev. 2010;24(22):2463–2479. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maruyama J., Naguro I., Takeda K., Ichijo H. Stress-activated MAP kinase cascades in cellular senescence. Curr. Med. Chem. 2009;16(10):1229–1235. doi: 10.2174/092986709787846613. [DOI] [PubMed] [Google Scholar]
- 8.Ben-Porath I., Weinberg R.A. The signals and pathways activating cellular senescence. Int. J. Biochem. Cell Biol. 2005;37(5):961–976. doi: 10.1016/j.biocel.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 9.Zhu J., Woods D., McMahon M., Bishop J.M. Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev. 1998;12(19):2997–3007. doi: 10.1101/gad.12.19.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serrano M., Lin A.W., McCurrach M.E., Beach D., Lowe S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88(5):593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi A. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat. Cell Biol. 2006;8(11):1291–1297. doi: 10.1038/ncb1491. [DOI] [PubMed] [Google Scholar]
- 12.Beausejour C.M. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003;22(16):4212–4222. doi: 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Munoz-Espin D., Serrano M. Cellular senescence: from physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014;15(7):482–496. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- 14.Munoz-Espin D. Programmed cell senescence during mammalian embryonic development. Cell. 2013;155(5):1104–1118. doi: 10.1016/j.cell.2013.10.019. [DOI] [PubMed] [Google Scholar]
- 15.Helman A. p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat. Med. 2016;22(4):412–420. doi: 10.1038/nm.4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Young A.R. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23(7):798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurz D.J., Decary S., Hong Y., Erusalimsky J.D. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J. Cell Sci. 2000;113:3613–3622. doi: 10.1242/jcs.113.20.3613. [DOI] [PubMed] [Google Scholar]
- 18.Lee B.Y. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell. 2006;5(2):187–195. doi: 10.1111/j.1474-9726.2006.00199.x. [DOI] [PubMed] [Google Scholar]
- 19.Freund A., Laberge R.M., Demaria M., Campisi J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell. 2012;23(11):2066–2075. doi: 10.1091/mbc.E11-10-0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nielsen G.P. Immunohistochemical survey of p16INK4A expression in normal human adult and infant tissues. Lab. Investig. J. Tech. Methods Pathol. 1999;79(9):1137–1143. [PubMed] [Google Scholar]
- 21.LeBrasseur N.K., Tchkonia T., Kirkland J.L. Cellular senescence and the biology of aging, Nestle Nutr. Inst. Workshop Ser. 2015;83:11–18. doi: 10.1159/000382054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Narita M. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113(6):703–716. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- 23.Kuilman T. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133(6):1019–1031. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 24.Coppe J.P. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garfinkel S., Brown S., Wessendorf J.H., Maciag T. Post-transcriptional regulation of interleukin 1 alpha in various strains of young and senescent human umbilical vein endothelial cells. Proc. Natl. Acad. Sci. USA. 1994;91(4):1559–1563. doi: 10.1073/pnas.91.4.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coppe J.P., Desprez P.Y., Krtolica A., Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang S., Moerman E.J., Jones R.A., Thweatt R., Goldstein S. Characterization of IGFBP-3, PAI-1 and SPARC mRNA expression in senescent fibroblasts. Mech. Ageing Dev. 1996;92(2–3):121–132. doi: 10.1016/s0047-6374(96)01814-3. [DOI] [PubMed] [Google Scholar]
- 28.West M.D., Pereira-Smith O.M., Smith J.R. Replicative senescence of human skin fibroblasts correlates with a loss of regulation and overexpression of collagenase activity. Exp. Cell Res. 1989;184(1):138–147. doi: 10.1016/0014-4827(89)90372-8. [DOI] [PubMed] [Google Scholar]
- 29.Blasi F., Carmeliet P. uPAR: a versatile signalling orchestrator. Nat. Rev. Mol. Cell Biol. 2002;3(12):932–943. doi: 10.1038/nrm977. [DOI] [PubMed] [Google Scholar]
- 30.Sato I. Reduction of nitric oxide producing activity associated with in vitro aging in cultured human umbilical vein endothelial cell. Biochem. Biophys. Res. Commun. 1993;195(2):1070–1076. doi: 10.1006/bbrc.1993.2153. [DOI] [PubMed] [Google Scholar]
- 31.Lee A.C. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J. Biol. Chem. 1999;274(12):7936–7940. doi: 10.1074/jbc.274.12.7936. [DOI] [PubMed] [Google Scholar]
- 32.Kuilman T., Peeper D.S. Senescence-messaging secretome: SMS-ing-ing cellular stress. Nature Rev. Cancer. 2009;9(2):81–94. doi: 10.1038/nrc2560. [DOI] [PubMed] [Google Scholar]
- 33.Coppe J.P., Kauser K., Campisi J., Beausejour C.M. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J. Biol. Chem. 2006;281(40):29568–29574. doi: 10.1074/jbc.M603307200. [DOI] [PubMed] [Google Scholar]
- 34.Hoenicke L., Zender L. Immune surveillance of senescent cells--biological significance in cancer- and non-cancer pathologies. Carcinogenesis. 2012;33(6):1123–1126. doi: 10.1093/carcin/bgs124. [DOI] [PubMed] [Google Scholar]
- 35.Harman D. Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 1956;11(3):298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 36.Miquel J. An update on the oxygen stress-mitochondrial mutation theory of aging: genetic and evolutionary implications. Exp. Gerontol. 1998;33(1–2):113–126. doi: 10.1016/s0531-5565(97)00060-0. [DOI] [PubMed] [Google Scholar]
- 37.Sies H. Oxidative stress: a concept in redox biology and medicine. Redox Biol. 2015;4:180–183. doi: 10.1016/j.redox.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Castro J.P., Grune T., Speckmann B. The two faces of ROS in adipocyte function and dysfunction. Biol. Chem. 2016 doi: 10.1515/hsz-2015-0305. [DOI] [PubMed] [Google Scholar]
- 39.Colavitti R., Finkel T. Reactive oxygen species as mediators of cellular senescence. IUBMB life. 2005;57(4–5):277–281. doi: 10.1080/15216540500091890. [DOI] [PubMed] [Google Scholar]
- 40.Passos J.F. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010;6:347. doi: 10.1038/msb.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Passos J.F. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007;5(5):e110. doi: 10.1371/journal.pbio.0050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moiseeva O., Bourdeau V., Roux A., Deschenes-Simard X., Ferbeyre G. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol. Cell. Biol. 2009;29(16):4495–4507. doi: 10.1128/MCB.01868-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fisher-Wellman K.H., Neufer P.D. Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol. Metab.: TEM. 2012;23(3):142–153. doi: 10.1016/j.tem.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barja G. The mitochondrial free radical theory of aging. Prog. Mol. Biol. Transl. Sci. 2014;127:1–27. doi: 10.1016/B978-0-12-394625-6.00001-5. [DOI] [PubMed] [Google Scholar]
- 45.Valko M. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007;39(1):44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 46.Stadtman E.R., Levine R.L. Protein oxidation. Ann. N.Y. Acad. Sci. 2000;899:191–208. doi: 10.1111/j.1749-6632.2000.tb06187.x. [DOI] [PubMed] [Google Scholar]
- 47.Höhn A., Sittig A., Jung T., Grimm S., Grune T. Lipofuscin is formed independently of macroautophagy and lysosomal activity in stress-induced prematurely senescent human fibroblasts. Free Radic. Biol. Med. 2012;53(9):1760–1769. doi: 10.1016/j.freeradbiomed.2012.08.591. [DOI] [PubMed] [Google Scholar]
- 48.Tavana O. Absence of p53-dependent apoptosis leads to UV radiation hypersensitivity, enhanced immunosuppression and cellular senescence. Cell Cycle. 2010;9(16):3328–3336. doi: 10.4161/cc.9.16.12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matos L., Gouveia A., Almeida H. Copper ability to induce premature senescence in human fibroblasts. Age (Dordr.,Neth.) 2012;34(4):783–794. doi: 10.1007/s11357-011-9276-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jamieson D., Chance B., Cadenas E., Boveris A. The relation of free radical production to hyperoxia. Annu. Rev. Physiol. 1986;48:703–719. doi: 10.1146/annurev.ph.48.030186.003415. [DOI] [PubMed] [Google Scholar]
- 51.Saretzki G., Feng J., von Zglinicki T., Villeponteau B. Similar gene expression pattern in senescent and hyperoxic-treated fibroblasts. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 1998;53(6):B438–B442. doi: 10.1093/gerona/53a.6.b438. [DOI] [PubMed] [Google Scholar]
- 52.Hohn A., Jung T., Grimm S., Grune T. Lipofuscin-bound iron is a major intracellular source of oxidants: role in senescent cells. Free Radic. Biol. Med. 2010;48(8):1100–1108. doi: 10.1016/j.freeradbiomed.2010.01.030. [DOI] [PubMed] [Google Scholar]
- 53.Flohe L. The impact of thiol peroxidases on redox regulation. Free Radic. Res. 2016;50(2):126–142. doi: 10.3109/10715762.2015.1046858. [DOI] [PubMed] [Google Scholar]
- 54.Rhee S.G., Chae H.Z., Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic. Biol. Med. 2005;38(12):1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 55.Biteau B., Labarre J., Toledano M.B. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425(6961):980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 56.Zhang Y.G. Featured article: accelerated decline of physical strength in peroxiredoxin-3 knockout mice. Exp. Biol. Med. (Maywood) 2016;241(13):1395–1400. doi: 10.1177/1535370216642039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Musicco C. Accumulation of overoxidized Peroxiredoxin III in aged rat liver mitochondria. Biochim. Biophys. Acta. 2009;1787(7):890–896. doi: 10.1016/j.bbabio.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 58.Colman R.J. Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat. Commun. 2014;5:3557. doi: 10.1038/ncomms4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nystrom T., Yang J., Molin M. Peroxiredoxins, gerontogenes linking aging to genome instability and cancer. Genes Dev. 2012;26(18):2001–2008. doi: 10.1101/gad.200006.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Neumann C.A. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature. 2003;424(6948):561–565. doi: 10.1038/nature01819. [DOI] [PubMed] [Google Scholar]
- 61.Jang H.H. Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell. 2004;117(5):625–635. doi: 10.1016/j.cell.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 62.Randall L.M., Ferrer-Sueta G., Denicola A. Peroxiredoxins as preferential targets in H2O2-induced signaling. Methods Enzym. 2013;527:41–63. doi: 10.1016/B978-0-12-405882-8.00003-9. [DOI] [PubMed] [Google Scholar]
- 63.Hanzen S. Lifespan control by. Cell. 2016;166(1):140–151. doi: 10.1016/j.cell.2016.05.006. [DOI] [PubMed] [Google Scholar]
- 64.Sies H. Glutathione and its role in cellular functions. Free Radic. Biol. Med. 1999;27(9–10):916–921. doi: 10.1016/s0891-5849(99)00177-x. [DOI] [PubMed] [Google Scholar]
- 65.Gil L. Age-associated analysis of oxidative stress parameters in human plasma and erythrocytes. Free Radic. Res. 2006;40(5):495–505. doi: 10.1080/10715760600592962. [DOI] [PubMed] [Google Scholar]
- 66.Wu G., Fang Y.Z., Yang S., Lupton J.R., Turner N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004;134(3):489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- 67.Cazanave S. High hepatic glutathione stores alleviate Fas-induced apoptosis in mice. J. Hepatol. 2007;46(5):858–868. doi: 10.1016/j.jhep.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 68.Hensley K., Robinson K.A., Gabbita S.P., Salsman S., Floyd R.A. Reactive oxygen species, cell signaling, and cell injury. Free Radic. Biol. Med. 2000;28(10):1456–1462. doi: 10.1016/s0891-5849(00)00252-5. [DOI] [PubMed] [Google Scholar]
- 69.Cho S.H. Redox regulation of PTEN and protein tyrosine phosphatases in H(2)O(2) mediated cell signaling. FEBS Lett. 2004;560(1–3):7–13. doi: 10.1016/s0014-5793(04)00112-7. [DOI] [PubMed] [Google Scholar]
- 70.Scherz-Shouval R. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26(7):1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang D.D., Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003;23(22):8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rodriguez-Manzaneque M.T., Tamarit J., Belli G., Ros J., Herrero E. Grx5 is a mitochondrial glutaredoxin required for the activity of iron/sulfur enzymes. Mol. Biol. Cell. 2002;13(4):1109–1121. doi: 10.1091/mbc.01-10-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cabreiro F., Picot C.R., Friguet B., Petropoulos I. Methionine sulfoxide reductases: relevance to aging and protection against oxidative stress. Ann. N.Y. Acad. Sci. 2006;1067:37–44. doi: 10.1196/annals.1354.006. [DOI] [PubMed] [Google Scholar]
- 74.Picot C.R., Perichon M., Cintrat J.-C., Friguet B., Petropoulos I. The peptide methionine sulfoxide reductases, MsrA and MsrB (hCBS-1), are downregulated during replicative senescence of human WI-38 fibroblasts. FEBS Lett. 2004;558(1–3):74–78. doi: 10.1016/S0014-5793(03)01530-8. [DOI] [PubMed] [Google Scholar]
- 75.Hohn A., Konig J., Grune T. Protein oxidation in aging and the removal of oxidized proteins. J. Proteom. 2013;92:132–159. doi: 10.1016/j.jprot.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 76.Nyström T. Role of oxidative carbonylation in protein quality control and senescence. EMBO J. 2005;24(7):1311–1317. doi: 10.1038/sj.emboj.7600599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dalle-Donne I., Giustarini D., Colombo R., Rossi R., Milzani A. Protein carbonylation in human diseases. Trends Mol. Med. 2003;9(4):169–176. doi: 10.1016/s1471-4914(03)00031-5. [DOI] [PubMed] [Google Scholar]
- 78.Dalle-Donne I., Rossi R., Giustarini D., Milzani A., Colombo R. Protein carbonyl groups as biomarkers of oxidative stress. Clin. Chim. Acta Int. J. Clin. Chem. 2003;329(1–2):23–38. doi: 10.1016/s0009-8981(03)00003-2. [DOI] [PubMed] [Google Scholar]
- 79.Castro J.P., Jung T., Grune T., Almeida H. Actin carbonylation: from cell dysfunction to organism disorder. J. Proteom. 2013;92:171–180. doi: 10.1016/j.jprot.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 80.Castro J.P., Ott C., Jung T., Grune T., Almeida H. Carbonylation of the cytoskeletal protein actin leads to aggregate formation. Free Radic. Biol. Med. 2012;53(4):916–925. doi: 10.1016/j.freeradbiomed.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 81.Voss P. Irradiation of GAPDH: a model for environmentally induced protein damage. Biol. Chem. 2007;388(6):583–592. doi: 10.1515/BC.2007.068. [DOI] [PubMed] [Google Scholar]
- 82.Levine R.L., Stadtman E.R. Oxidative modification of proteins during aging. Exp. Gerontol. 2001;36(9):1495–1502. doi: 10.1016/s0531-5565(01)00135-8. [DOI] [PubMed] [Google Scholar]
- 83.Møller I.M., Rogowska-Wrzesinska A., Rao R.S.P. Protein carbonylation and metal-catalyzed protein oxidation in a cellular perspective. J. Proteom. 2011;74(11):2228–2242. doi: 10.1016/j.jprot.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 84.Höhn A., Jung T., Grune T. Pathophysiological importance of aggregated damaged proteins. Free Radic. Biol. Med. 2014;71(0):70–89. doi: 10.1016/j.freeradbiomed.2014.02.028. [DOI] [PubMed] [Google Scholar]
- 85.Castro J.P., Jung T., Grune T., Siems W. 4-Hydroxynonenal (HNE) modified proteins in metabolic diseases. Free Radic. Biol. Med. 2016 doi: 10.1016/j.freeradbiomed.2016.10.497. [DOI] [PubMed] [Google Scholar]
- 86.Curtis J.M. Downregulation of adipose glutathione S-transferase A4 leads to increased protein. Diabetes. 2010;59(5):1132–1142. doi: 10.2337/db09-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Grimsrud P.A., Picklo M.J., Sr., Griffin T.J., Bernlohr D.A. Carbonylation of adipose proteins in obesity and insulin resistance: identification of adipocyte fatty acid-binding protein as a cellular target of 4-hydroxynonenal. Mol. Cell. Proteom.: MCP. 2007;6(4):624–637. doi: 10.1074/mcp.M600120-MCP200. [DOI] [PubMed] [Google Scholar]
- 88.Frohnert B.I., Bernlohr D.A. Protein carbonylation, mitochondrial. Adv. Nutr.: Int. Rev. J. 2013;4(2):157–163. doi: 10.3945/an.112.003319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Baraibar M.A. Oxidative stress-induced proteome alterations target different cellular pathways in human myoblasts. Free Radic. Biol. Med. 2011;51(8):1522–1532. doi: 10.1016/j.freeradbiomed.2011.06.032. [DOI] [PubMed] [Google Scholar]
- 90.Baraibar M.A., Friguet B. Oxidative proteome modifications target specific cellular pathways during oxidative stress, cellular senescence and aging. Exp. Gerontol. 2013;48(7):620–625. doi: 10.1016/j.exger.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 91.Grune T., Merker K., Sandig G., Davies K.J. Selective degradation of oxidatively modified protein substrates by the proteasome. Biochem. Biophys. Res. Commun. 2003;305(3):709–718. doi: 10.1016/s0006-291x(03)00809-x. [DOI] [PubMed] [Google Scholar]
- 92.Grune T., Reinheckel T., Davies K.J. Degradation of oxidized proteins in K562 human hematopoietic cells by proteasome. J. Biol. Chem. 1996;271(26):15504–15509. doi: 10.1074/jbc.271.26.15504. [DOI] [PubMed] [Google Scholar]
- 93.Shringarpure R., Grune T., Davies K.J. Protein oxidation and 20S proteasome-dependent proteolysis in mammalian cells. Cell. Mol. Life Sci.: CMLS. 2001;58(10):1442–1450. doi: 10.1007/PL00000787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hohn A. Lipofuscin inhibits the proteasome by binding to surface motifs. Free Radic. Biol. Med. 2011;50(5):585–591. doi: 10.1016/j.freeradbiomed.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 95.Petrov D., Zagrovic B. Microscopic analysis of protein oxidative damage: effect of carbonylation on structure, J. Am. Chem. Soc. 2011;133(18):7016–7024. doi: 10.1021/ja110577e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Grune T., Jung T., Merker K., Davies K.J.A. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and ‘aggresomes' during oxidative stress, aging, and disease. Int. J. Biochem. Cell Biol. 2004;36(12):2519–2530. doi: 10.1016/j.biocel.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 97.Jung T., Bader N., Grune T. Lipofuscin: formation, distribution, and metabolic consequences. Ann. N.Y. Acad. Sci. 2007;1119:97–111. doi: 10.1196/annals.1404.008. [DOI] [PubMed] [Google Scholar]
- 98.Terman A., Brunk U.T. Oxidative stress, accumulation of biological 'garbage', and aging. Antioxid. Redox Signal. 2006;8(1–2):197–204. doi: 10.1089/ars.2006.8.197. [DOI] [PubMed] [Google Scholar]
- 99.Terman A., Brunk U.T. Lipofuscin. Int J. Biochem. Cell Biol. 2004;36(8):1400–1404. doi: 10.1016/j.biocel.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 100.Koistinaho J., Hartikainen K., Hatanpaa K., Hervonen A. Age pigments in different populations of peripheral neurons in vivo and in vitro. Adv. Exp. Med. Biol. 1989;266:49–59. doi: 10.1007/978-1-4899-5339-1_4. [DOI] [PubMed] [Google Scholar]
- 101.Gutteridge J.M. Age pigments: role of iron and copper salts in the formation of fluorescent lipid complexes. Mech. Ageing Dev. 1984;25(1–2):205–214. doi: 10.1016/0047-6374(84)90141-6. [DOI] [PubMed] [Google Scholar]
- 102.Catalgol B. The proteasome is an integral part of solar ultraviolet a radiation-induced gene expression. J. Biol. Chem. 2009;284(44):30076–30086. doi: 10.1074/jbc.M109.044503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kastle M., Woschee E., Grune T. Histone deacetylase 6 (HDAC6) plays a crucial role in p38MAPK-dependent induction of heme oxygenase-1 (HO-1) in response to proteasome inhibition. Free Radic. Biol. Med. 2012;53(11):2092–2101. doi: 10.1016/j.freeradbiomed.2012.09.023. [DOI] [PubMed] [Google Scholar]
- 104.Grune T., Jung T., Merker K., Davies K.J. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and 'aggresomes' during oxidative stress, aging, and disease. Int J. Biochem. Cell Biol. 2004;36(12):2519–2530. doi: 10.1016/j.biocel.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 105.Dukan S., Nyström T. Bacterial senescence: stasis results in increased and differential oxidation of cytoplasmic proteins leading to developmental induction of the heat shock regulon. Genes Dev. 1998;12(21):3431–3441. doi: 10.1101/gad.12.21.3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lindner A.B., Madden R., Demarez A., Stewart E.J., Taddei F. Asymmetric segregation of protein aggregates is associated with cellular aging and rejuvenation. Proc. Natl. Acad. Sci. USA. 2008;105(8):3076–3081. doi: 10.1073/pnas.0708931105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mirzaei H., Regnier F. Identification of yeast oxidized proteins: chromatographic top-down approach for identification of carbonylated, fragmented and cross-linked proteins in yeast. J. Chromatogr. A. 2007;1141(1):22–31. doi: 10.1016/j.chroma.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 108.David D.C. Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol. 2010;8(8):e1000450. doi: 10.1371/journal.pbio.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sitte N. Proteasome inhibition by lipofuscin/ceroid during postmitotic aging of fibroblasts. FASEB J.: Off. Publ. Fed. Am. Soc. Exp. Biol. 2000;14(11):1490–1498. doi: 10.1096/fj.14.11.1490. [DOI] [PubMed] [Google Scholar]
- 110.Ikeda H., Tauchi H., Sato T. Fine structural analysis of lipofuscin in various tissues of rats of different ages. Mech. Ageing Dev. 1985;33(1):77–93. doi: 10.1016/0047-6374(85)90110-1. [DOI] [PubMed] [Google Scholar]
- 111.Ikeda H., Tauchi H., Shimasaki H., Ueta N., Sato T. Age and organ difference in amount and distribution of autofluorescent granules in rats. Mech. Ageing Dev. 1985;31(2):139–146. doi: 10.1016/s0047-6374(85)80024-5. [DOI] [PubMed] [Google Scholar]
- 112.Menardo J. Oxidative stress, inflammation, and autophagic stress as the key mechanisms of premature age-related hearing loss in SAMP8 mouse cochlea. Antioxid. Redox Signal. 2012;16(3):263–274. doi: 10.1089/ars.2011.4037. [DOI] [PubMed] [Google Scholar]
- 113.Soto C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003;4(1):49–60. doi: 10.1038/nrn1007. [DOI] [PubMed] [Google Scholar]
- 114.Labbadia J., Morimoto R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015;84:435–464. doi: 10.1146/annurev-biochem-060614-033955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kaushik S., Cuervo A.M. Proteostasis and aging. Nat. Med. 2015;21(12):1406–1415. doi: 10.1038/nm.4001. [DOI] [PubMed] [Google Scholar]
- 116.M.S. Hipp, S.-H. Park, F.U. Hartl, Proteostasis impairment in protein-misfolding and -aggregation diseases, Trends Cell Biol., 24(9), pp. 506–514. [DOI] [PubMed]
- 117.Jung T., Hohn A., Grune T. The proteasome and the degradation of oxidized proteins: Part II – protein oxidation and proteasomal degradation. Redox Biol. 2013;2C:99–104. doi: 10.1016/j.redox.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jung T., Grune T. The proteasome and the degradation of oxidized proteins: part I-structure of proteasomes. Redox Biol. 2013;1(1):178–182. doi: 10.1016/j.redox.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jung T., Catalgol B., Grune T. The proteasomal system. Mol. Asp. Med. 2009;30(4):191–296. doi: 10.1016/j.mam.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 120.Pickering A.M. The immunoproteasome, the 20S proteasome and the PA28alphabeta proteasome regulator are oxidative-stress-adaptive proteolytic complexes. Biochem. J. 2010;432(3):585–594. doi: 10.1042/BJ20100878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Johnston-Carey H.K., Pomatto L.C., Davies K.J. The immunoproteasome in oxidative stress, aging, and disease. Crit. Rev. Biochem. Mol. Biol. 2015;51(4):268–281. doi: 10.3109/10409238.2016.1172554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Reeg S. The molecular chaperone Hsp70 promotes the proteolytic removal of oxidatively damaged proteins by the proteasome. Free Radic. Biol. Med. 2016 doi: 10.1016/j.freeradbiomed.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Szabo E., Kovacs I., Grune T., Haczku A., Virag L. PARP-1: a new player in the asthma field? Allergy. 2011;66(7):811–814. doi: 10.1111/j.1398-9995.2011.02551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Conconi M. Protection from oxidative inactivation of the 20S proteasome by heat-shock protein 90. Biochem. J. 1998;333:407–415. doi: 10.1042/bj3330407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Saibil H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 2013;14(10):630–642. doi: 10.1038/nrm3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Castro J.P., Reeg S., Botelho V., Almeida H., Grune T. HSP90 cleavage associates with oxidized proteins accumulation after oxidative stress. Free Radic. Biol. Med. 2014;75(Suppl. 1):S24–S25. doi: 10.1016/j.freeradbiomed.2014.10.743. [DOI] [PubMed] [Google Scholar]
- 127.Calderwood S.K., Murshid A., Prince T. The shock of aging: molecular chaperones and the heat shock response in longevity and aging – a Mini-review. Gerontology. 2009;55(5):550–558. doi: 10.1159/000225957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gohlke S. Molecular alterations in proteasomes of rat liver during aging result in altered proteolytic activities. Age (Dordr.) 2014;36(1):57–72. doi: 10.1007/s11357-013-9543-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Conconi M., Szweda L.I., Levine R.L., Stadtman E.R., Friguet B. Age-related decline of rat liver multicatalytic proteinase activity and protection from oxidative inactivation by heat-shock protein 90. Arch. Biochem. Biophys. 1996;331(2):232–240. doi: 10.1006/abbi.1996.0303. [DOI] [PubMed] [Google Scholar]
- 130.Baraibar M.A., Friguet B. Changes of the proteasomal system during the aging process. Prog. Mol. Biol. Transl. Sci. 2012;109:249–275. doi: 10.1016/B978-0-12-397863-9.00007-9. [DOI] [PubMed] [Google Scholar]
- 131.Rodriguez K.A., Edrey Y.H., Osmulski P., Gaczynska M., Buffenstein R. Altered composition of liver proteasome assemblies contributes to enhanced proteasome activity in the exceptionally long-lived naked mole-rat. PLoS One. 2012;7(5):e35890. doi: 10.1371/journal.pone.0035890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bulteau A.L., Szweda L.I., Friguet B. Age-dependent declines in proteasome activity in the heart. Arch. Biochem. Biophys. 2002;397(2):298–304. doi: 10.1006/abbi.2001.2663. [DOI] [PubMed] [Google Scholar]
- 133.Hayashi T., Goto S. Age-related changes in the 20S and 26S proteasome activities in the liver of male F344 rats. Mech. Ageing Dev. 1998;102(1):55–66. doi: 10.1016/s0047-6374(98)00011-6. [DOI] [PubMed] [Google Scholar]
- 134.Sitte N., Merker K., Von Zglinicki T., Davies K.J., Grune T. Protein oxidation and degradation during cellular senescence of human BJ fibroblasts: part II – aging of nondividing cells. FASEB J.: Off. Publ. Fed. Am. Soc. Exp. Biol. 2000;14(15):2503–2510. doi: 10.1096/fj.00-0210com. [DOI] [PubMed] [Google Scholar]
- 135.Sitte N., Merker K., Von Zglinicki T., Grune T., Davies K.J. Protein oxidation and degradation during cellular senescence of human BJ fibroblasts: part I – effects of proliferative senescence. FASEB J.: Off. Publ. Fed. Am. Soc. Exp. Biol. 2000;14(15):2495–2502. doi: 10.1096/fj.00-0209com. [DOI] [PubMed] [Google Scholar]
- 136.Chondrogianni N. Central role of the proteasome in senescence and survival of human fibroblasts: induction of a senescence-like phenotype upon its inhibition and resistance to stress upon its activation. J. Biol. Chem. 2003;278(30):28026–28037. doi: 10.1074/jbc.M301048200. [DOI] [PubMed] [Google Scholar]
- 137.Farout L., Mary J., Vinh J., Szweda L.I., Friguet B. Inactivation of the proteasome by 4-hydroxy-2-nonenal is site specific and dependant on 20S proteasome subtypes. Arch. Biochem. Biophys. 2006;453(1):135–142. doi: 10.1016/j.abb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 138.Friguet B., Szweda L.I. Inhibition of the multicatalytic proteinase (proteasome) by 4-hydroxy-2-nonenal cross-linked protein. FEBS Lett. 1997;405(1):21–25. doi: 10.1016/s0014-5793(97)00148-8. [DOI] [PubMed] [Google Scholar]
- 139.Kapphahn R.J., Bigelow E.J., Ferrington D.A. Age-dependent inhibition of proteasome chymotrypsin-like activity in the retina. Exp. Eye Res. 2007;84(4):646–654. doi: 10.1016/j.exer.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kastle M., Grune T. Protein oxidative modification in the aging organism and the role of the ubiquitin proteasomal system. Curr. Pharm. Des. 2011;17(36):4007–4022. doi: 10.2174/138161211798764898. [DOI] [PubMed] [Google Scholar]
- 141.Fischer D.F. Long-term proteasome dysfunction in the mouse brain by expression of aberrant ubiquitin. Neurobiol. Aging. 2009;30(6):847–863. doi: 10.1016/j.neurobiolaging.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 142.Chen X., Petranovic D. Role of frameshift ubiquitin B protein in Alzheimer's disease. Wiley Inter. Rev. Syst. Biol. Med. 2016;8(4):300–313. doi: 10.1002/wsbm.1340. [DOI] [PubMed] [Google Scholar]
- 143.Tank E.M., True H.L. Disease-associated mutant ubiquitin causes proteasomal impairment and enhances the toxicity of protein aggregates. PLoS Genet. 2009;5(2):e1000382. doi: 10.1371/journal.pgen.1000382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tsakiri E.N., Trougakos I.P. The amazing ubiquitin-proteasome system: structural components and implication in aging. Int Rev. Cell Mol. Biol. 2015;314:171–237. doi: 10.1016/bs.ircmb.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 145.Reeg S., Grune T. Protein oxidation in aging: does It play a role in aging progression? Antioxid. Redox Signal. 2015;23(3):239–255. doi: 10.1089/ars.2014.6062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Nowotny K., Jung T., Grune T., Hohn A. Reprint of Accumulation of modified proteins and aggregate formation in aging. Exp. Gerontol. 2014;59:3–12. doi: 10.1016/j.exger.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 147.Hohn A., Grune T. Lipofuscin: formation, effects and role of macroautophagy. Redox Biol. 2013;1:140–144. doi: 10.1016/j.redox.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hohn A., Sittig A., Jung T., Grimm S., Grune T. Lipofuscin is formed independently of macroautophagy and lysosomal activity in stress-induced prematurely senescent human fibroblasts. Free Radic. Biol. Med. 2012;53(9):1760–1769. doi: 10.1016/j.freeradbiomed.2012.08.591. [DOI] [PubMed] [Google Scholar]
- 149.Jung T., Hohn A., Grune T. Lipofuscin: detection and quantification by microscopic techniques. Methods Mol. Biol. 2010;594:173–193. doi: 10.1007/978-1-60761-411-1_13. [DOI] [PubMed] [Google Scholar]
- 150.Tsakiri E.N. Diet-derived advanced glycation end products or lipofuscin disrupts proteostasis and reduces life span in Drosophila melanogaster. Free Radic. Biol. Med. 2013;65:1155–1163. doi: 10.1016/j.freeradbiomed.2013.08.186. [DOI] [PubMed] [Google Scholar]
- 151.Hohn T.J., Grune T. The proteasome and the degradation of oxidized proteins: part III-redox regulation of the proteasomal system. Redox Biol. 2014;2:388–394. doi: 10.1016/j.redox.2013.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bakondi E. Age-related loss of stress-induced nuclear proteasome activation is due to low PARP-1 activity. Free Radic. Biol. Med. 2011;50(1):86–92. doi: 10.1016/j.freeradbiomed.2010.10.700. [DOI] [PubMed] [Google Scholar]
- 153.Catalgol B. Chromatin repair after oxidative stress: role of PARP-mediated proteasome activation. Free Radic. Biol. Med. 2010;48(5):673–680. doi: 10.1016/j.freeradbiomed.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 154.Saez I., Vilchez D. The mechanistic links between proteasome activity, aging and age-related diseases. Curr. Genom. 2014;15(1):38–51. doi: 10.2174/138920291501140306113344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Pickering A.M., Lehr M., Miller R.A. Lifespan of mice and primates correlates with immunoproteasome expression. J. Clin. Investig. 2015;125(5):2059–2068. doi: 10.1172/JCI80514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chondrogianni N. Overexpression of proteasome beta5 assembled subunit increases the amount of proteasome and confers ameliorated response to oxidative stress and higher survival rates. J. Biol. Chem. 2005;280(12):11840–11850. doi: 10.1074/jbc.M413007200. [DOI] [PubMed] [Google Scholar]
- 157.Hwang J.S., Hwang J.S., Chang I., Kim S. Age-associated decrease in proteasome content and activities in human dermal fibroblasts: restoration of normal level of proteasome subunits reduces aging markers in fibroblasts from elderly persons. J. Gerontol. A Biol. Sci. Med. Sci. 2007;62(5):490–499. doi: 10.1093/gerona/62.5.490. [DOI] [PubMed] [Google Scholar]
- 158.Chen Q., Thorpe J., Dohmen J.R., Li F., Keller J.N. Ump1 extends yeast lifespan and enhances viability during oxidative stress: central role for the proteasome? Free Radic. Biol. Med. 2006;40(1):120–126. doi: 10.1016/j.freeradbiomed.2005.08.048. [DOI] [PubMed] [Google Scholar]
- 159.Chondrogianni N., Gonos E.S. Overexpression of hUMP1/POMP proteasome accessory protein enhances proteasome-mediated antioxidant defence. Exp. Gerontol. 2007;42(9):899–903. doi: 10.1016/j.exger.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 160.Kapeta S., Chondrogianni N., Gonos E.S. Nuclear erythroid factor 2-mediated proteasome activation delays senescence in human fibroblasts. J. Biol. Chem. 2010;285(11):8171–8184. doi: 10.1074/jbc.M109.031575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Tsakiri E.N. Proteasome dysfunction in Drosophila signals to an Nrf2-dependent regulatory circuit aiming to restore proteostasis and prevent premature aging. Aging Cell. 2013;12(5):802–813. doi: 10.1111/acel.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Klionsky D.J. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cuervo A.M. Autophagy and aging: the importance of maintaining "clean" cells. Autophagy. 2005;1(3):131–140. doi: 10.4161/auto.1.3.2017. [DOI] [PubMed] [Google Scholar]
- 164.Massey A., Kiffin R., Cuervo A.M. Pathophysiology of chaperone-mediated autophagy. Int. J. Biochem. Cell Biol. 2004;36(12):2420–2434. doi: 10.1016/j.biocel.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 165.Agarraberes F.A., Terlecky S.R., Dice J.F. An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J. Cell Biol. 1997;137(4):825–834. doi: 10.1083/jcb.137.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ahlberg J., Glaumann H. Uptake–microautophagy–and degradation of exogenous proteins by isolated rat liver lysosomes. Effects of pH, ATP, and inhibitors of proteolysis. Exp. Mol. Pathol. 1985;42(1):78–88. doi: 10.1016/0014-4800(85)90020-6. [DOI] [PubMed] [Google Scholar]
- 167.Wesselborg S., Stork B. Autophagy signal transduction by ATG proteins: from hierarchies to networks. Cell Mol. Life Sci. 2015;72(24):4721–4757. doi: 10.1007/s00018-015-2034-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Cuervo A.M., Dice J.F. Age-related decline in chaperone-mediated autophagy. J. Biol. Chem. 2000;275(40):31505–31513. doi: 10.1074/jbc.M002102200. [DOI] [PubMed] [Google Scholar]
- 169.Ott C., Konig J., Hohn A., Jung T., Grune T. Reduced autophagy leads to an impaired ferritin turnover in senescent fibroblasts. Free Radic. Biol. Med. 2016 doi: 10.1016/j.freeradbiomed.2016.10.492. [DOI] [PubMed] [Google Scholar]
- 170.Hashimoto T. Age-dependent increase in lysosome-associated membrane protein 1 and early-onset behavioral deficits in APPSL transgenic mouse model of Alzheimer's disease. Neurosci. Lett. 2010;469(2):273–277. doi: 10.1016/j.neulet.2009.12.015. [DOI] [PubMed] [Google Scholar]
- 171.Zhao H., Brunk U.T., Garner B. Age-related lysosomal dysfunction: an unrecognized roadblock for cobalamin trafficking? Cell Mol. Life Sci. 2011;68(24):3963–3969. doi: 10.1007/s00018-011-0861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hsu C.Y., Chuang Y.L., Chan Y.P. Changes in cellular degradation activity in young and old worker honeybees (Apis mellifera) Exp. Gerontol. 2014;50:128–136. doi: 10.1016/j.exger.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 173.Majeski A.E., Dice J.F. Mechanisms of chaperone-mediated autophagy. Int. J. Biochem. Cell Biol. 2004;36(12):2435–2444. doi: 10.1016/j.biocel.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 174.Bergamini C.M., Signorini M. Exploring the catalytic mechanism of skeletal muscle udp-glucose pyrophosphorylase: identification of a hyperreactive cysteine at the enzyme active site. Int.J. Biochem. 1991;23(1):123–127. doi: 10.1016/0020-711x(91)90018-i. [DOI] [PubMed] [Google Scholar]
- 175.Kenessey A., Banay-Schwartz M., DeGuzman T., Lajtha A. Increase in cathepsin D activity in rat brain in aging. J. Neurosci. Res. 1989;23(4):454–456. doi: 10.1002/jnr.490230412. [DOI] [PubMed] [Google Scholar]
- 176.Kim J., Kundu M., Viollet B., Guan K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011;13(2):132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Nacarelli T., Azar A., Sell C. Aberrant mTOR activation in senescence and aging: a mitochondrial stress response? Exp. Gerontol. 2015;68:66–70. doi: 10.1016/j.exger.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ott C., Konig J., Hohn A., Jung T., Grune T. Macroautophagy is impaired in old murine brain tissue as well as in senescent human fibroblasts. Redox Biol. 2016;10:266–273. doi: 10.1016/j.redox.2016.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hepple R.T. Impact of aging on mitochondrial function in cardiac and skeletal muscle. Free Radic. Biol. Med. 2016;98:177–186. doi: 10.1016/j.freeradbiomed.2016.03.017. [DOI] [PubMed] [Google Scholar]
- 180.Kudryavtseva A.V. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget. 2016 doi: 10.18632/oncotarget.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sugiyama S., Takasawa M., Hayakawa M., Ozawa T. Changes in skeletal muscle, heart and liver mitochondrial electron transport activities in rats and dogs of various ages. Biochem. Mol. Biol. Int. 1993;30(5):937–944. [PubMed] [Google Scholar]
- 182.Terman A., Brunk U.T. The aging myocardium: roles of mitochondrial damage and lysosomal degradation. Heart Lung Circ. 2005;14(2):107–114. doi: 10.1016/j.hlc.2004.12.023. [DOI] [PubMed] [Google Scholar]
- 183.Leduc-Gaudet J.P. Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget. 2015;6(20):17923–17937. doi: 10.18632/oncotarget.4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Jin S.M., Youle R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012;125(Pt 4):795–799. doi: 10.1242/jcs.093849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sun N. Measuring in vivo mitophagy. Mol. Cell. 2015;60(4):685–696. doi: 10.1016/j.molcel.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Sosulski M.L. Deregulation of selective autophagy during aging and pulmonary fibrosis: the role of TGFbeta1. Aging Cell. 2015;14(5):774–783. doi: 10.1111/acel.12357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Diot A. A novel quantitative assay of mitophagy: combining high content fluorescence microscopy and mitochondrial DNA load to quantify mitophagy and identify novel pharmacological tools against pathogenic heteroplasmic mtDNA. Pharm. Res. 2015;100:24–35. doi: 10.1016/j.phrs.2015.07.014. [DOI] [PubMed] [Google Scholar]
- 188.Hoshino A. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun. 2013;4:2308. doi: 10.1038/ncomms3308. [DOI] [PubMed] [Google Scholar]
- 189.Bueno M. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015;125(2):521–538. doi: 10.1172/JCI74942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Gautier C.A., Kitada T., Shen J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. USA. 2008;105(32):11364–11369. doi: 10.1073/pnas.0802076105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Terman A., Kurz T., Navratil M., Arriaga E.A., Brunk U.T. Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid. Redox Signal. 2010;12(4):503–535. doi: 10.1089/ars.2009.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Geisler S. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010;12(2):119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 193.Exner N. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J. Neurosci. 2007;27(45):12413–12418. doi: 10.1523/JNEUROSCI.0719-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Rana A., Rera M., Walker D.W. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc. Natl. Acad. Sci. USA. 2013;110(21):8638–8643. doi: 10.1073/pnas.1216197110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Rodriguez-Navarro J.A. Mortality, oxidative stress and tau accumulation during ageing in parkin null mice. J. Neurochem. 2007;103(1):98–114. doi: 10.1111/j.1471-4159.2007.04762.x. [DOI] [PubMed] [Google Scholar]
- 196.Maggini S., Wintergerst E.S., Beveridge S., Hornig D.H. Selected vitamins and trace elements support immune function by strengthening epithelial barriers and cellular and humoral immune responses. Br. J. Nutr. 2007;98(Suppl 1):S29–S35. doi: 10.1017/S0007114507832971. [DOI] [PubMed] [Google Scholar]
- 197.Sies H. Oxidative stress: oxidants and antioxidants. Exp. Physiol. 1997;82(2):291–295. doi: 10.1113/expphysiol.1997.sp004024. [DOI] [PubMed] [Google Scholar]
- 198.Madaric A., Ginter E., Kadrabova J. Serum copper, zinc and copper/zinc ratio in males: influence of aging. Physiol. Res./Acad. Sci. Bohemoslov. 1994;43(2):107–111. [PubMed] [Google Scholar]
- 199.Mezzetti A. Copper/zinc ratio and systemic oxidant load: effect of aging and aging-related degenerative diseases. Free Radic. Biol. Med. 1998;25(6):676–681. doi: 10.1016/s0891-5849(98)00109-9. [DOI] [PubMed] [Google Scholar]
- 200.Bulvik B.E. Aging is an organ-specific process: changes in homeostasis of iron and redox proteins in the rat. Age (Dordr., Neth.) 2012;34(3):693–704. doi: 10.1007/s11357-011-9268-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Cook C.I., Yu B.P. Iron accumulation in aging: modulation by dietary restriction. Mech. Ageing Dev. 1998;102(1):1–13. doi: 10.1016/s0047-6374(98)00005-0. [DOI] [PubMed] [Google Scholar]
- 202.Ray A.L. Low serum selenium and total carotenoids predict mortality among older women living in the community: the women's health and aging studies. J. Nutr. 2006;136(1):172–176. doi: 10.1093/jn/136.1.172. [DOI] [PubMed] [Google Scholar]
- 203.Akbaraly N.T. Selenium and mortality in the elderly: results from the EVA study. Clin. Chem. 2005;51(11):2117–2123. doi: 10.1373/clinchem.2005.055301. [DOI] [PubMed] [Google Scholar]
- 204.Hornsby P.J., Harris S.E. Oxidative damage to DNA and replicative lifespan in cultured adrenocortical cells. Exp. Cell Res. 1987;168(1):203–217. doi: 10.1016/0014-4827(87)90429-0. [DOI] [PubMed] [Google Scholar]
- 205.Liu Q. Effects of trace elements on the telomere lengths of hepatocytes L-02 and hepatoma cells SMMC-7721. Biol. Trace Elem. Res. 2004;100(3):215–227. doi: 10.1385/BTER:100:3:215. [DOI] [PubMed] [Google Scholar]
- 206.Legrain Y., Touat-Hamici Z., Chavatte L. Interplay between selenium levels, selenoprotein expression, and replicative senescence in WI-38 human fibroblasts. J. Biol. Chem. 2014;289(9):6299–6310. doi: 10.1074/jbc.M113.526863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Wu M., Wu R.T., Wang T.T., Cheng W.H. Role for p53 in selenium-induced senescence. J. Agric. Food Chem. 2011;59(21):11882–11887. doi: 10.1021/jf203012a. [DOI] [PubMed] [Google Scholar]
- 208.Jiang Q. Heat shock protein 90-mediated inactivation of nuclear factor-kappaB switches autophagy to apoptosis through becn1 transcriptional inhibition in selenite-induced NB4 cells. Mol. Biol. Cell. 2011;22(8):1167–1180. doi: 10.1091/mbc.E10-10-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Hwang J.J. Zinc(II) ion mediates tamoxifen-induced autophagy and cell death in MCF-7 breast cancer cell line. Biometals: Int. J. Met. Ions Biol. Biochem. Med. 2010;23(6):997–1013. doi: 10.1007/s10534-010-9346-9. [DOI] [PubMed] [Google Scholar]
- 210.Liuzzi J.P., Yoo C. Role of zinc in the regulation of autophagy during ethanol exposure in human hepatoma cells. Biol. Trace Elem. Res. 2013;156(1–3):350–356. doi: 10.1007/s12011-013-9816-3. [DOI] [PubMed] [Google Scholar]
- 211.Lee S.J., Koh J.Y. Roles of zinc and metallothionein-3 in oxidative stress-induced lysosomal dysfunction, cell death, and autophagy in neurons and astrocytes. Mol. Brain. 2010;3(1):30. doi: 10.1186/1756-6606-3-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Mocchegiani E. Metallothioneins, ageing and cellular senescence: a future therapeutic target. Curr. Pharm. Des. 2013;19(9):1753–1764. [PubMed] [Google Scholar]
- 213.Rudolf E., Rudolf K. Low zinc environment induces stress signaling, senescence and mixed cell death modalities in colon cancer cells. Apoptosis: Int. J. Program. Cell death. 2015;20(12):1651–1665. doi: 10.1007/s10495-015-1182-5. [DOI] [PubMed] [Google Scholar]
- 214.Patrushev N., Seidel-Rogol B., Salazar G. Angiotensin II requires zinc and downregulation of the zinc transporters ZnT3 and ZnT10 to induce senescence of vascular smooth muscle cells. PLoS One. 2012;7(3):e33211. doi: 10.1371/journal.pone.0033211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.McCord M.C., Aizenman E. The role of intracellular zinc release in aging, oxidative stress, and Alzheimer's disease. Front. Aging Neurosci. 2014;6:77. doi: 10.3389/fnagi.2014.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Granzotto A., Sensi S.L. Intracellular zinc is a critical intermediate in the excitotoxic cascade. Neurobiol. Dis. 2015;81:25–37. doi: 10.1016/j.nbd.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 217.Xiao Y. Molecular study on copper-mediated tumor proteasome inhibition and cell death. Int. J. Oncol. 2010;37(1):81–87. doi: 10.3892/ijo_00000655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Li H., Wu S., Shi N., Lian S., Lin W. Nrf2/HO-1 pathway activation by manganese is associated with reactive oxygen species and ubiquitin-proteasome pathway, not MAPKs signaling. J. Appl. Toxicol.: JAT. 2011;31(7):690–697. doi: 10.1002/jat.1654. [DOI] [PubMed] [Google Scholar]
- 219.Cai T. Proteasome inhibition is associated with manganese-induced oxidative injury in PC12 cells. Brain Res. 2007;1185:359–365. doi: 10.1016/j.brainres.2007.09.075. [DOI] [PubMed] [Google Scholar]
- 220.Killilea D.W., Wong S.L., Cahaya H.S., Atamna H., Ames B.N. Iron accumulation during cellular senescence. Ann. N.Y. Acad. Sci. 2004;1019:365–367. doi: 10.1196/annals.1297.063. [DOI] [PubMed] [Google Scholar]
- 221.Anraku M. Antioxidant effects of a dietary supplement: reduction of indices of oxidative stress in normal subjects by water-soluble chitosan. Food Chem. Toxicol. 2009;47(1):104–109. doi: 10.1016/j.fct.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 222.Freese R., Dragsted L.O., Loft S., Mutanen M. No effect on oxidative stress biomarkers by modified intakes of polyunsaturated fatty acids or vegetables and fruit. Eur. J. Clin. Nutr. 2008;62(9):1151–1153. doi: 10.1038/sj.ejcn.1602865. [DOI] [PubMed] [Google Scholar]
- 223.O'Byrne D.J., Devaraj S., Grundy S.M., Jialal I. Comparison of the antioxidant effects of Concord grape juice flavonoids alpha-tocopherol on markers of oxidative stress in healthy adults. Am. J. Clin. Nutr. 2002;76(6):1367–1374. doi: 10.1093/ajcn/76.6.1367. [DOI] [PubMed] [Google Scholar]
- 224.Jacob K., Periago M.J., Bohm V., Berruezo G.R. Influence of lycopene and vitamin C from tomato juice on biomarkers of oxidative stress and inflammation. Br. J. Nutr. 2008;99(1):137–146. doi: 10.1017/S0007114507791894. [DOI] [PubMed] [Google Scholar]
- 225.Jacob R.A. Moderate antioxidant supplementation has no effect on biomarkers of oxidant damage in healthy men with low fruit and vegetable intakes. J. Nutr. 2003;133(3):740–743. doi: 10.1093/jn/133.3.740. [DOI] [PubMed] [Google Scholar]
- 226.Carty J.L. The effects of vitamin C supplementation on protein oxidation in healthy volunteers. Biochem. Biophys. Res. Commun. 2000;273(2):729–735. doi: 10.1006/bbrc.2000.3014. [DOI] [PubMed] [Google Scholar]
- 227.Korolainen M.A., Goldsteins G., Alafuzoff I., Koistinaho J., Pirttila T. Proteomic analysis of protein oxidation in Alzheimer's disease brain. Electrophoresis. 2002;23(19):3428–3433. doi: 10.1002/1522-2683(200210)23:19<3428::AID-ELPS3428>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 228.Smith C.D. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc. Natl. Acad. Sci. USA. 1991;88(23):10540–10543. doi: 10.1073/pnas.88.23.10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Castegna A. Proteomic identification of oxidatively modified proteins in Alzheimer's disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic. Biol. Med. 2002;33(4):562–571. doi: 10.1016/s0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- 230.Butterfield D.A., Lauderback C.M. Lipid peroxidation and protein oxidation in Alzheimer's disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic. Biol. Med. 2002;32(11):1050–1060. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- 231.Hensley K. Brain regional correspondence between Alzheimer's disease histopathology and biomarkers of protein oxidation. J. Neurochem. 1995;65(5):2146–2156. doi: 10.1046/j.1471-4159.1995.65052146.x. [DOI] [PubMed] [Google Scholar]
- 232.Aksenov M., Aksenova M., Butterfield D.A., Markesbery W.R. Oxidative modification of creatine kinase BB in Alzheimer's disease brain. J. Neurochem. 2000;74(6):2520–2527. doi: 10.1046/j.1471-4159.2000.0742520.x. [DOI] [PubMed] [Google Scholar]
- 233.Aksenov M.Y., Aksenova M.V., Butterfield D.A., Geddes J.W., Markesbery W.R. Protein oxidation in the brain in Alzheimer's disease. Neuroscience. 2001;103(2):373–383. doi: 10.1016/s0306-4522(00)00580-7. [DOI] [PubMed] [Google Scholar]
- 234.Castegna A. Proteomic identification of oxidatively modified proteins in Alzheimer's disease brain. Part II: dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. J. Neurochem. 2002;82(6):1524–1532. doi: 10.1046/j.1471-4159.2002.01103.x. [DOI] [PubMed] [Google Scholar]
- 235.Conrad C.C. Oxidized proteins in Alzheimer's plasma. Biochem. Biophys. Res. Commun. 2000;275(2):678–681. doi: 10.1006/bbrc.2000.3356. [DOI] [PubMed] [Google Scholar]
- 236.Choi J., Malakowsky C.A., Talent J.M., Conrad C.C., Gracy R.W. Identification of oxidized plasma proteins in Alzheimer's disease. Biochem. Biophys. Res. Commun. 2002;293(5):1566–1570. doi: 10.1016/S0006-291X(02)00420-5. [DOI] [PubMed] [Google Scholar]
- 237.Floor E., Wetzel M.G. Increased protein oxidation in human substantia nigra pars compacta in comparison with basal ganglia and prefrontal cortex measured with an improved dinitrophenylhydrazine assay. J. Neurochem. 1998;70(1):268–275. doi: 10.1046/j.1471-4159.1998.70010268.x. [DOI] [PubMed] [Google Scholar]
- 238.Pamplona R. Proteins in human brain cortex are modified by oxidation, glycoxidation, and lipoxidation. Effects of Alzheimer disease and identification of lipoxidation targets. J. Biol. Chem. 2005;280(22):21522–21530. doi: 10.1074/jbc.M502255200. [DOI] [PubMed] [Google Scholar]
- 239.Markesbery W.R. Oxidative stress hypothesis in Alzheimer's disease. Free Radic. Biol. Med. 1997;23(1):134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 240.Lyras L. Oxidative damage to proteins, lipids, and DNA in cortical brain regions from patients with dementia with Lewy bodies. J. Neurochem. 1998;71(1):302–312. doi: 10.1046/j.1471-4159.1998.71010302.x. [DOI] [PubMed] [Google Scholar]
- 241.Alam Z.I. A generalised increase in protein carbonyls in the brain in Parkinson's but not incidental Lewy body disease. J. Neurochem. 1997;69(3):1326–1329. doi: 10.1046/j.1471-4159.1997.69031326.x. [DOI] [PubMed] [Google Scholar]
- 242.Lenz A.G. Oxidatively modified proteins in bronchoalveolar lavage fluid of patients with ARDS and patients at-risk for ARDS. Eur. Respir. J. 1999;13(1):169–174. doi: 10.1034/j.1399-3003.1999.13a31.x. [DOI] [PubMed] [Google Scholar]
- 243.Quinlan G.J., Evans T.W., Gutteridge J.M. Oxidative damage to plasma proteins in adult respiratory distress syndrome. Free Radic. Res. 1994;20(5):289–298. doi: 10.3109/10715769409145628. [DOI] [PubMed] [Google Scholar]
- 244.Gladstone I.M., Jr., Levine R.L. Oxidation of proteins in neonatal lungs. Pediatrics. 1994;93(5):764–768. [PubMed] [Google Scholar]
- 245.Schock B.C., Sweet D.G., Halliday H.L., Young I.S., Ennis M. Oxidative stress in lavage fluid of preterm infants at risk of chronic lung disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2001;281(6):L1386–L1391. doi: 10.1152/ajplung.2001.281.6.L1386. [DOI] [PubMed] [Google Scholar]
- 246.Buss I.H., Darlow B.A., Winterbourn C.C. Elevated protein carbonyls and lipid peroxidation products correlating with myeloperoxidase in tracheal aspirates from premature infants. Pediatr. Res. 2000;47(5):640–645. doi: 10.1203/00006450-200005000-00014. [DOI] [PubMed] [Google Scholar]
- 247.Ramsay P.L. Clara cell secretory protein oxidation and expression in premature infants who develop bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2001;164(1):155–161. doi: 10.1164/ajrccm.164.1.2008022. [DOI] [PubMed] [Google Scholar]
- 248.Ferrante R.J. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 1997;69(5):2064–2074. doi: 10.1046/j.1471-4159.1997.69052064.x. [DOI] [PubMed] [Google Scholar]
- 249.Siciliano G. Antioxidant capacity and protein oxidation in cerebrospinal fluid of amyotrophic lateral sclerosis. J. Neurol. 2007;254(5):575–580. doi: 10.1007/s00415-006-0301-1. [DOI] [PubMed] [Google Scholar]
- 250.Mantle D., Falkous G., Walker D. Quantification of protease activities in synovial fluid from rheumatoid and osteoarthritis cases: comparison with antioxidant and free radical damage markers. Clin. Chim. Acta Int. J. Clin. Chem. 1999;284(1):45–58. doi: 10.1016/s0009-8981(99)00055-8. [DOI] [PubMed] [Google Scholar]
- 251.Renke J., Popadiuk S., Korzon M., Bugajczyk B., Wozniak M. Protein carbonyl groups' content as a useful clinical marker of antioxidant barrier impairment in plasma of children with juvenile chronic arthritis. Free Radic. Biol. Med. 2000;29(2):101–104. doi: 10.1016/s0891-5849(00)00288-4. [DOI] [PubMed] [Google Scholar]
- 252.Winterbourn C.C. Protein carbonyl measurements show evidence of early oxidative stress in critically ill patients. Crit. Care Med. 2000;28(1):143–149. doi: 10.1097/00003246-200001000-00024. [DOI] [PubMed] [Google Scholar]
- 253.Abu-Zidan F.M., Plank L.D., Windsor J.A. Proteolysis in severe sepsis is related to oxidation of plasma protein. Eur. J. Surg. 2002;168(2):119–123. doi: 10.1080/11024150252884359. [DOI] [PubMed] [Google Scholar]
- 254.McGrath L.T. Oxidative stress during acute respiratory exacerbations in cystic fibrosis. Thorax. 1999;54(6):518–523. doi: 10.1136/thx.54.6.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Range S.P., Dunster C., Knox A.J., Kelly F.J. Treatment of pulmonary exacerbations of cystic fibrosis leads to improved antioxidant status. Eur. Respir. J. 1999;13(3):560–564. doi: 10.1183/09031936.99.13356099. [DOI] [PubMed] [Google Scholar]
- 256.Boscia F., Grattagliano I., Vendemiale G., Micelli-Ferrari T., Altomare E. Protein oxidation and lens opacity in humans. Investig. Ophthalmol. Vis. Sci. 2000;41(9):2461–2465. [PubMed] [Google Scholar]
- 257.Totan Y. Oxidative macromolecular damage in age-related macular degeneration. Curr. Eye Res. 2009;34(12):1089–1093. doi: 10.3109/02713680903353772. [DOI] [PubMed] [Google Scholar]
- 258.Himmelfarb J., McMonagle E. Albumin is the major plasma protein target of oxidant stress in uremia. Kidney Int. 2001;60(1):358–363. doi: 10.1046/j.1523-1755.2001.00807.x. [DOI] [PubMed] [Google Scholar]
- 259.Himmelfarb J., McMonagle E., McMenamin E. Plasma protein thiol oxidation and carbonyl formation in chronic renal failure. Kidney Int. 2000;58(6):2571–2578. doi: 10.1046/j.1523-1755.2000.00443.x. [DOI] [PubMed] [Google Scholar]
- 260.Lim P.S., Cheng Y.M., Wei Y.H. Increase in oxidative damage to lipids and proteins in skeletal muscle of uremic patients. Free Radic. Res. 2002;36(3):295–301. doi: 10.1080/10715760290019318. [DOI] [PubMed] [Google Scholar]
- 261.Miyata T., van Ypersele de Strihou C., Kurokawa K., Baynes J.W. Alterations in nonenzymatic biochemistry in uremia: origin and significance of "carbonyl stress" in long-term uremic complications. Kidney Int. 1999;55(2):389–399. doi: 10.1046/j.1523-1755.1999.00302.x. [DOI] [PubMed] [Google Scholar]
- 262.Dominguez C., Ruiz E., Gussinye M., Carrascosa A. Oxidative stress at onset and in early stages of type 1 diabetes in children and adolescents. Diabetes Care. 1998;21(10):1736–1742. doi: 10.2337/diacare.21.10.1736. [DOI] [PubMed] [Google Scholar]
- 263.Cakatay U., Telci A., Salman S., Satman L., Sivas A. Oxidative protein damage in type I diabetic patients with and without complications. Endocr. Res. 2000;26(3):365–379. doi: 10.3109/07435800009066174. [DOI] [PubMed] [Google Scholar]
- 264.Telci A. Oxidative protein damage in plasma of type 2 diabetic patients. Horm. Metab. Res. 2000;32(1):40–43. doi: 10.1055/s-2007-978584. [DOI] [PubMed] [Google Scholar]
- 265.Telci A., Cakatay U., Salman S., Satman I., Sivas A. Oxidative protein damage in early stage Type 1 diabetic patients. Diabetes Res. Clin. Pract. 2000;50(3):213–223. doi: 10.1016/s0168-8227(00)00197-2. [DOI] [PubMed] [Google Scholar]
- 266.Grattagliano I. Oxidative retinal products and ocular damages in diabetic patients. Free Radic. Biol. Med. 1998;25(3):369–372. doi: 10.1016/s0891-5849(98)00059-8. [DOI] [PubMed] [Google Scholar]
- 267.Lih-Brody L. Increased oxidative stress and decreased antioxidant defenses in mucosa of inflammatory bowel disease. Dig. Dis. Sci. 1996;41(10):2078–2086. doi: 10.1007/BF02093613. [DOI] [PubMed] [Google Scholar]
- 268.Pantke U. Oxidized proteins as a marker of oxidative stress during coronary heart surgery. Free Radic. Biol. Med. 1999;27(9–10):1080–1086. doi: 10.1016/s0891-5849(99)00144-6. [DOI] [PubMed] [Google Scholar]
- 269.Miyata T., Ueda Y., Saito A., Kurokawa K. 'Carbonyl stress' and dialysis-related amyloidosis. Nephrol. Dial. Transpl. 2000;15(Suppl 1):25–28. doi: 10.1093/oxfordjournals.ndt.a027959. [DOI] [PubMed] [Google Scholar]
- 270.Kedziora-Kornatowska K. The markers of oxidative stress and activity of the antioxidant system in the blood of elderly patients with essential arterial hypertension. Cell Mol. Biol. Lett. 2004;9(4A):635–641. [PubMed] [Google Scholar]
- 271.Troesch B., Eggersdorfer M., Weber P. 100 years of vitamins: adequate intake in the elderly is still a matter of concern. J. Nutr. 2012;142(6):979–980. doi: 10.3945/jn.112.157826. [DOI] [PubMed] [Google Scholar]
- 272.Fabian E., Bogner M., Kickinger A., Wagner K.H., Elmadfa I. Vitamin status in elderly people in relation to the use of nutritional supplements. J. Nutr. Health Aging. 2012;16(3):206–212. doi: 10.1007/s12603-011-0159-5. [DOI] [PubMed] [Google Scholar]
- 273.Granado-Lorencio F. Determinants of fat-soluble vitamin status in patients aged 65 years and over. Eur. J. Clin. Nutr. 2013;67(12):1325–1327. doi: 10.1038/ejcn.2013.198. [DOI] [PubMed] [Google Scholar]
- 274.Anonymous . Institute of Medicine, NIH. ed Medicine Io; Washington, D.C.: 1998. Dietary Reference Intakes for Calcium and Vitamin D. [Google Scholar]
- 275.Anonymous . Institute of Medicine, NIH; Washington, D.C.: 1998. Dietary Reference Intakes for Folate and other B Vitamins. [Google Scholar]
- 276.Anonymous . Institute of Medicine, NIH. ed Medicine Io; Washington, D.C.: 1998. Dietary Reference Intakes for Vitamins A, K and Trace Elements. [Google Scholar]
- 277.Anonymous . Institute of Medicine, NIH. ed Medicine Io; Washington, D.C.: 1998. Dietary Reference Intakes for Vitamins C, E, Selenium and Carotenoids. [Google Scholar]
- 278.Mangialasche F. Classification and prediction of clinical diagnosis of Alzheimer's disease based on MRI and plasma measures of alpha-/gamma-tocotrienols and gamma-tocopherol. J. Intern. Med. 2013;273(6):602–621. doi: 10.1111/joim.12037. [DOI] [PubMed] [Google Scholar]
- 279.Khanna S. Nanomolar vitamin E alpha-tocotrienol inhibits glutamate-induced activation of phospholipase A2 and causes neuroprotection. J. Neurochem. 2010;112(5):1249–1260. doi: 10.1111/j.1471-4159.2009.06550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Mangialasche F. High plasma levels of vitamin E forms and reduced Alzheimer's disease risk in advanced age. J. Alzheimers Dis. 2010;20(4):1029–1037. doi: 10.3233/JAD-2010-091450. [DOI] [PubMed] [Google Scholar]
- 281.Mangialasche F. Tocopherols and tocotrienols plasma levels are associated with cognitive impairment. Neurobiol. Aging. 2012;33(10):2282–2290. doi: 10.1016/j.neurobiolaging.2011.11.019. [DOI] [PubMed] [Google Scholar]
- 282.Morris M.C., Evans D.A., Bienias J.L., Tangney C.C., Wilson R.S. Vitamin E and cognitive decline in older persons. Arch. Neurol. 2002;59(7):1125–1132. doi: 10.1001/archneur.59.7.1125. [DOI] [PubMed] [Google Scholar]
- 283.Touyarot K. A mid-life vitamin A supplementation prevents age-related spatial memory deficits and hippocampal neurogenesis alterations through CRABP-I. PLoS One. 2013;8(8):e72101. doi: 10.1371/journal.pone.0072101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Gutierrez-Fernandez A. Loss of MT1-MMP causes cell senescence and nuclear defects which can be reversed by retinoic acid. EMBO J. 2015;34(14):1875–1888. doi: 10.15252/embj.201490594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.van der Loo B. Age-related changes of vitamin A status. J. Cardiovasc. Pharm. 2004;43(1):26–30. doi: 10.1097/00005344-200401000-00005. [DOI] [PubMed] [Google Scholar]
- 286.Mecocci P. Plasma antioxidants and longevity: a study on healthy centenarians. Free Radic. Biol. Med. 2000;28(8):1243–1248. doi: 10.1016/s0891-5849(00)00246-x. [DOI] [PubMed] [Google Scholar]
- 287.Lopes da Silva S. Plasma nutrient status of patients with Alzheimer's disease: systematic review and meta-analysis. Alzheimers Dement. 2014;10(4):485–502. doi: 10.1016/j.jalz.2013.05.1771. [DOI] [PubMed] [Google Scholar]
- 288.Zaman Z. Plasma concentrations of vitamins A and E and carotenoids in Alzheimer's disease. Age Ageing. 1992;21(2):91–94. doi: 10.1093/ageing/21.2.91. [DOI] [PubMed] [Google Scholar]
- 289.Rinaldi P. Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer's disease. Neurobiol. Aging. 2003;24(7):915–919. doi: 10.1016/s0197-4580(03)00031-9. [DOI] [PubMed] [Google Scholar]
- 290.Johnson E.J. A possible role for lutein and zeaxanthin in cognitive function in the elderly. Am. J. Clin. Nutr. 2012;96(5):1161S–1165S. doi: 10.3945/ajcn.112.034611. [DOI] [PubMed] [Google Scholar]
- 291.Woodside J.V., McGrath A.J., Lyner N., McKinley M.C. Carotenoids and health in older people. Maturitas. 2015;80(1):63–68. doi: 10.1016/j.maturitas.2014.10.012. [DOI] [PubMed] [Google Scholar]
- 292.Pusceddu I. The role of telomeres and vitamin D in cellular aging and age-related diseases. Clin. Chem. Lab Med. 2015;53(11):1661–1678. doi: 10.1515/cclm-2014-1184. [DOI] [PubMed] [Google Scholar]
- 293.Keisala T. Premature aging in vitamin D receptor mutant mice. J. Steroid Biochem. Mol. Biol. 2009;115(3–5):91–97. doi: 10.1016/j.jsbmb.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 294.Chapman K.M., Ham J.O., Pearlman R.A. Longitudinal assessment of the nutritional status of elderly veterans. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 1996;51(4):B261–B269. doi: 10.1093/gerona/51a.4.b261. [DOI] [PubMed] [Google Scholar]
- 295.Gibson G.E. Abnormal thiamine-dependent processes in Alzheimer's Disease. Lessons from diabetes. Mol. Cell Neurosci. 2013;55:17–25. doi: 10.1016/j.mcn.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Ashoori M., Saedisomeolia A. Riboflavin (vitamin B(2)) and oxidative stress: a review. Br. J. Nutr. 2014;111(11):1985–1991. doi: 10.1017/S0007114514000178. [DOI] [PubMed] [Google Scholar]
- 297.Wang G., Li W., Lu X., Zhao X. Riboflavin alleviates cardiac failure in Type I diabetic cardiomyopathy. Heart Int. 2011;6(2):e21. doi: 10.4081/hi.2011.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Das B.S. Increased plasma lipid peroxidation in riboflavin-deficient, malaria-infected children. Am. J. Clin. Nutr. 1990;51(5):859–863. doi: 10.1093/ajcn/51.5.859. [DOI] [PubMed] [Google Scholar]
- 299.Okura T. Hyperhomocysteinemia is one of the risk factors associated with cerebrovascular stiffness in hypertensive patients, especially elderly males. Sci. Rep. 2014;4:5663. doi: 10.1038/srep05663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Mehta R., Dedina L., O'Brien P.J. Rescuing hepatocytes from iron-catalyzed oxidative stress using vitamins B1 and B6. Toxicol. Vitr. 2011;25(5):1114–1122. doi: 10.1016/j.tiv.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 301.Dellon A.L., Dellon E.S., Tassler P.L., Ellefson R.D., Hendrickson M. Experimental model of pyridoxine (B6) deficiency-induced neuropathy. Ann. Plast. Surg. 2001;47(2):153–160. doi: 10.1097/00000637-200108000-00008. [DOI] [PubMed] [Google Scholar]
- 302.Bazzano L.A. Dietary intake of folate and risk of stroke in US men and women: NHANES I epidemiologic follow-up study. National Health and Nutrition Examination Survey. Stroke. 2002;33(5):1183–1188. doi: 10.1161/01.str.0000014607.90464.88. [DOI] [PubMed] [Google Scholar]
- 303.Robinson K. Low circulating folate and vitamin B6 concentrations: risk factors for stroke, peripheral vascular disease, and coronary artery disease. European COMAC Group. Circulation. 1998;97(5):437–443. doi: 10.1161/01.cir.97.5.437. [DOI] [PubMed] [Google Scholar]
- 304.Singh R., Kanwar S.S., Sood P.K., Nehru B. Beneficial effects of folic acid on enhancement of memory and antioxidant status in aged rat brain. Cell Mol. Neurobiol. 2011;31(1):83–91. doi: 10.1007/s10571-010-9557-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Martinez-Vega R. Folic acid deficiency induces premature hearing loss through mechanisms involving cochlear oxidative stress and impairment of homocysteine metabolism. FASEB J.: Off. Publ. Fed. Am. Soc. Exp. Biol. 2015;29(2):418–432. doi: 10.1096/fj.14-259283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Hughes C.F., Ward M., Hoey L., McNulty H. Vitamin B12 and ageing: current issues and interaction with folate. Ann. Clin. Biochem. 2013;50(Pt 4):315–329. doi: 10.1177/0004563212473279. [DOI] [PubMed] [Google Scholar]
- 307.Zhang Y. Decreased brain levels of vitamin B12 in. PLoS One. 2016;11(1):e0146797. doi: 10.1371/journal.pone.0146797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Chawla J., Kvarnberg D. Hydrosoluble vitamins. Handb. Clin. Neurol. 2014;120:891–914. doi: 10.1016/B978-0-7020-4087-0.00059-0. [DOI] [PubMed] [Google Scholar]
- 309.Dixit S. Vitamin C deficiency in the brain impairs cognition, increases amyloid accumulation and deposition, and oxidative stress in APP/PSEN1 and normally aging mice. ACS Chem. Neurosci. 2015;6(4):570–581. doi: 10.1021/cn500308h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Warner T.A., Kang J.Q., Kennard J.A., Harrison F.E. Low brain ascorbic acid increases susceptibility to seizures in mouse models of decreased brain ascorbic acid transport and Alzheimer's disease. Epilepsy Res. 2015;110:20–25. doi: 10.1016/j.eplepsyres.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Gale C.R., Martyn C.N., Winter P.D., Cooper C. Vitamin C and risk of death from stroke and coronary heart disease in cohort of elderly people. BMJ. 1995;310(6994):1563–1566. doi: 10.1136/bmj.310.6994.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Gaziano J.M. Vitamin E and cardiovascular disease: observational studies. Ann. N.Y. Acad. Sci. 2004;1031:280–291. doi: 10.1196/annals.1331.028. [DOI] [PubMed] [Google Scholar]
- 313.Kang J.H., Ascherio A., Grodstein F. Fruit and vegetable consumption and cognitive decline in aging women. Ann. Neurol. 2005;57(5):713–720. doi: 10.1002/ana.20476. [DOI] [PubMed] [Google Scholar]
- 314.Konig J. Quantification of age-related changes of alpha-tocopherol in lysosomal membranes in murine tissues and human fibroblasts. BioFactors (Oxf. Engl.) 2016;42(3):307–315. doi: 10.1002/biof.1274. [DOI] [PubMed] [Google Scholar]
- 315.de Kreutzenberg S.V. Downregulation of the longevity-associated protein sirtuin 1 in insulin resistance and metabolic syndrome: potential biochemical mechanisms. Diabetes. 2010;59(4):1006–1015. doi: 10.2337/db09-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Zheng T., Lu Y. Changes in SIRT1 expression and its downstream pathways in age-related cataract in humans. Curr. Eye Res. 2011;36(5):449–455. doi: 10.3109/02713683.2011.559301. [DOI] [PubMed] [Google Scholar]
- 317.Sirotkin A.V., Dekanova P., Harrath A.H., Alwasel S.H., Vasicek D. Interrelationships between sirtuin 1 and transcription factors p53 and NF-kappaB (p50/p65) in the control of ovarian cell apoptosis and proliferation. Cell Tissue Res. 2014;358(2):627–632. doi: 10.1007/s00441-014-1940-7. [DOI] [PubMed] [Google Scholar]
- 318.Lipinski M.M. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer's disease. Proc. Natl. Acad. Sci. USA. 2010;107(32):14164–14169. doi: 10.1073/pnas.1009485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Codogno P., Meijer A.J. Atg5: more than an autophagy factor. Nat. Cell Biol. 2006;8(10):1045–1047. doi: 10.1038/ncb1006-1045. [DOI] [PubMed] [Google Scholar]
- 320.Nezich C.L., Wang C., Fogel A.I., Youle R.J. MiT/TFE transcription factors are activated during mitophagy downstream of Parkin and Atg5. J. Cell Biol. 2015;210(3):435–450. doi: 10.1083/jcb.201501002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Rouschop K.M. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2010;120(1):127–141. doi: 10.1172/JCI40027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Carames B., Taniguchi N., Otsuki S., Blanco F.J., Lotz M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010;62(3):791–801. doi: 10.1002/art.27305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Kang R., Zeh H.J., Lotze M.T., Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18(4):571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Sun T. c-Jun NH2-terminal kinase activation is essential for up-regulation of LC3 during ceramide-induced autophagy in human nasopharyngeal carcinoma cells. J. Transl. Med. 2011;9:161. doi: 10.1186/1479-5876-9-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Salminen A., Kaarniranta K., Kauppinen A. Beclin 1 interactome controls the crosstalk between apoptosis, autophagy and inflammasome activation: impact on the aging process. Ageing Res. Rev. 2013;12(2):520–534. doi: 10.1016/j.arr.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 326.Qi L., Zhang X.D. Role of chaperone-mediated autophagy in degrading Huntington's disease-associated huntingtin protein. Acta Biochim. Biophys. Sin. (Shanghai) 2014;46(2):83–91. doi: 10.1093/abbs/gmt133. [DOI] [PubMed] [Google Scholar]
- 327.Kang Y.A. Autophagy driven by a master regulator of hematopoiesis. Mol. Cell Biol. 2012;32(1):226–239. doi: 10.1128/MCB.06166-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Pernodet N., Dong K., Pelle E. Autophagy in human skin fibroblasts: comparison between young and aged cells and evaluation of its cellular rhythm and response to Ultraviolet A radiation. J. Cosmet. Sci. 2016;67(1):13–20. [PubMed] [Google Scholar]
- 329.Pike L.R. Transcriptional up-regulation of ULK1 by ATF4 contributes to cancer cell survival. Biochem. J. 2013;449(2):389–400. doi: 10.1042/BJ20120972. [DOI] [PubMed] [Google Scholar]
- 330.Kwon J. Assurance of mitochondrial integrity and mammalian longevity by the p62-Keap1-Nrf2-Nqo1 cascade. EMBO Rep. 2012;13(2):150–156. doi: 10.1038/embor.2011.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Kuusisto E., Salminen A., Alafuzoff I. Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport. 2001;12(10):2085–2090. doi: 10.1097/00001756-200107200-00009. [DOI] [PubMed] [Google Scholar]
- 332.Mizuno Y. Immunoreactivities of p62, an ubiqutin-binding protein, in the spinal anterior horn cells of patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 2006;249(1):13–18. doi: 10.1016/j.jns.2006.05.060. [DOI] [PubMed] [Google Scholar]
- 333.Bitto A. P62/SQSTM1 at the interface of aging, autophagy, and disease. Age (Dordr.) 2014;36(3):9626. doi: 10.1007/s11357-014-9626-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Puissant A., Fenouille N., Auberger P. When autophagy meets cancer through p62/SQSTM1. Am. J. Cancer Res. 2012;2(4):397–413. [PMC free article] [PubMed] [Google Scholar]
- 335.Lee J.H., Bodmer R., Bier E., Karin M. Sestrins at the crossroad between stress and aging. Aging (Albany NY) 2010;2(6):369–374. doi: 10.18632/aging.100157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Lee J.H. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science. 2010;327(5970):1223–1228. doi: 10.1126/science.1182228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Gamerdinger M. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. 2009;28(7):889–901. doi: 10.1038/emboj.2009.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Gamerdinger M., Kaya A.M., Wolfrum U., Clement A.M., Behl C. BAG3 mediates chaperone-based aggresome-targeting and selective autophagy of misfolded proteins. EMBO Rep. 2011;12(2):149–156. doi: 10.1038/embor.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Franceschelli S. Bag3 gene expression is regulated by heat shock factor 1. J. Cell Physiol. 2008;215(3):575–577. doi: 10.1002/jcp.21397. [DOI] [PubMed] [Google Scholar]
- 340.Voellmy R., Boellmann F. Chaperone regulation of the heat shock protein response. Adv. Exp. Med. Biol. 2007;594:89–99. doi: 10.1007/978-0-387-39975-1_9. [DOI] [PubMed] [Google Scholar]
- 341.Sheng Z., Ma L., Sun J.E., Zhu L.J., Green M.R. BCR-ABL suppresses autophagy through ATF5-mediated regulation of mTOR transcription. Blood. 2011;118(10):2840–2848. doi: 10.1182/blood-2010-12-322537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Murata H. NRF2 regulates PINK1 expression under OxidativeStress conditions. PLoS One. 2015;10(11):e0142438. doi: 10.1371/journal.pone.0142438. [DOI] [PMC free article] [PubMed] [Google Scholar]