

ABSTRACT
HIV rapidly accumulates resistance mutations following exposure to subtherapeutic concentrations of antiretroviral drugs that reduces treatment efficacy. High-resolution melting analysis (HRMA) has been used to successfully identify single nucleotide polymorphisms (SNPs) and to genotype viral and bacterial species. Here, we tested the ability of HRMA incorporating short unlabeled probes to accurately assign drug susceptibilities at the 103, 181, and 184 codons of the HIV-1 reverse transcriptase gene. The analytical sensitivities of the HRMA assays were 5% of mixed species for K103N and Y181C and 20% for M184V. When applied to 153 HIV-1 patient specimens previously genotyped by Sanger population sequencing, HRMA correctly assigned drug sensitivity or resistance profiles to 80% of the samples at codon 103 (K103K/N) (Cohen's kappa coefficient [κ] > 0.6; P < 0.05), 90% at 181 (Y181Y/C) (κ > 0.74, P < 0.05), and 80% at 184 (M184M/V) (κ > 0.62; P < 0.05). The frequency of incorrect genotypes was very low (≤1 to 2%) for each assay, which in most cases was due to the higher sensitivity of the HRMA assay. Specimens for which drug resistance profiles could not be assigned (9 to 20%) often had polymorphisms in probe binding regions. Thus, HRMA is a rapid, inexpensive, and sensitive method for the determination of drug sensitivities caused by major HIV-1 drug resistance mutations and, after further development to minimize the melting effects of nontargeted polymorphisms, may be suitable for surveillance purposes.
KEYWORDS: HRMA, drug resistance, human immunodeficiency virus
INTRODUCTION
Currently, 37 million people are living with HIV, and close to 40 million people have died from AIDS-related illnesses since the beginning of the pandemic (1). Approximately 17 million individuals have access to antiretroviral therapy (ART), which inhibits different HIV enzymes at distinct stages of the virus life cycle, thereby controlling viremia and reducing mortality. However, the development of resistance to ART remains one of the greatest threats to the successful long-term chemotherapeutic treatment of HIV infection. The majority of individuals failing ART harbor at least one drug resistance mutation, typically the nucleoside/nucleotide reverse transcriptase inhibitor (NRTI) mutation M184V/I and/or the nonnucleoside reverse transcriptase inhibitor (NNRTI) mutation K103N (2–9). The NNRTI mutations Y181C and K103N are rapidly selected for in children (10, 11) and women (12, 13), respectively, following exposure to single-dose nevirapine (sd-NVP) for the prevention of mother-to-child transmission (MTCT).
Genotypic detection of known drug resistance mutations is an informative measure of drug failure and facilitates the selection of subsequent therapy options (14). While Sanger sequencing-based methodologies remain the gold standard for mutation detection, the assays are costly and resource intensive and cannot detect minority (<20%) quasispecies variants. Alternative methodologies, such as parallel and allele-specific PCR (15–17), point mutation ligation assays (LigAmp) (18, 19), oligonucleotide ligation assays (OLA) (20), line probe assays (21), and next-generation sequencing technologies (22, 23), have been implemented with various degrees of effectiveness, sensitivity, and cost.
High-resolution melting analysis (HRMA) is a closed-tube fluorescence-based single nucleotide polymorphism (SNP) detection and typing methodology (24) (Fig. 1). It allows genotyping of loci by measuring differences in DNA melting kinetics, which is a function of nucleotide sequence and length (25). Sensitive instruments and saturating double-stranded DNA (dsDNA) binding dyes allow SNP discrimination based on deviations in the melting temperature (Tm) curve and shape (26). This method has been useful for typing bacteria, viruses, and parasites associated with human disease (27) and for drug susceptibility testing in Mycobacterium tuberculosis (28), Mycoplasma pneumoniae (29), Salmonella enterica (30), and influenza A viruses (31). Incorporation of unlabeled probes has been utilized for resistance testing in hepatitis B virus (32), Cytomegalovirus (33), and M. tuberculosis (34). HRMA has not previously been used for HIV resistance testing but has been used to study HIV diversity (35), showing high concordance with population and next-generation sequencing results (36).
FIG 1.
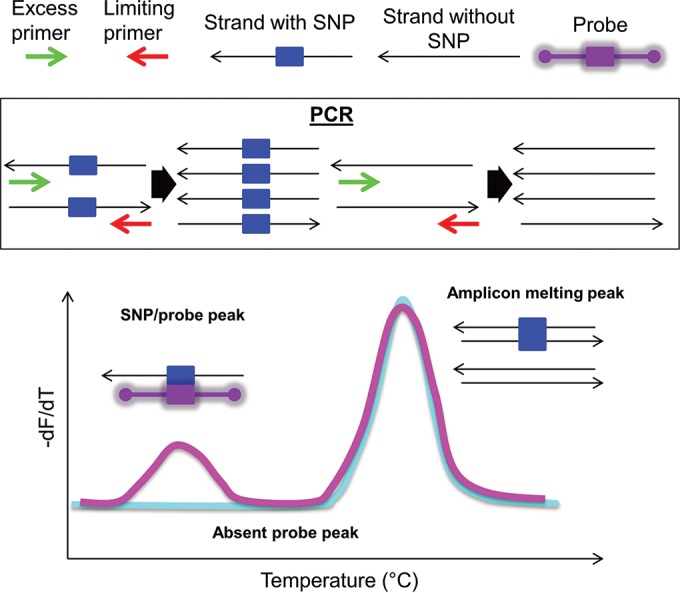
PCR and HRMA. (Top) Key to diagram. (Middle) Asymmetric PCR preferentially amplifies the sense strand. The PCR product contains the SNP of interest (left) or does not have this sequence (right). (Bottom) Normalized negative first derivative plotted against temperature visualizing the melting transitions of the probe-amplicon to the left of the amplicon-amplicon melting peak.
With the widespread use of ART, there is an urgent need to develop rapid and cheap assays to detect HIV-1 drug resistance mutations in resource-limited settings. Therefore, in this study, we set out to assess the feasibility of using a one-step, closed-tube HRMA-PCR with unlabeled oligonucleotide probes and saturating dsDNA binding dye to detect the key HIV-1 drug resistance mutations K103N, Y181C, and M184V.
RESULTS
HRMA assay validation.
HRMA PCRs and melting steps were optimized using wild-type and codon 103, 181, and 184 mutant plasmids. Four complementary probes were designed to hybridize optimally to a consensus C sequence and the 103K wild-type (AAA and AAG) and 103N mutant (AAC and AAT) sequences (Table 1). Two complementary probes were designed to genotype the 181Y wild-type (TAT) and 181C mutant (TGT) sequences. Similarly, two complementary probes were designed and applied to the 184M wild-type (ATG) and 184V mutant (GTG) plasmids. Single and distinct probe melting peaks adjacent to the amplicon melting peaks were visualized on negative-first-derivative (−dF/dT) plots. Peaks were evident for each of the wild-type (Fig. 2A and B) and mutant (Fig. 2C and D) 103 plasmids, the wild-type (Fig. 3A) and mutant (Fig. 3B and C) 181 plasmids, and the wild-type (Fig. 4A) and mutant (Fig. 4C and E) 184 plasmids.
TABLE 1.
Sequences of HRMA-PCR primers and unlabeled oligonucleotide probes
| Codon | Primer |
Probe |
||
|---|---|---|---|---|
| Name | Sequence (5′-3′) | Name | Sequence (5′-3′)a | |
| 103 | 103-Forward | AGGAATACCACACCCAGCAG | wt103A | CTGATTTTTTCTTTT |
| 103-Reverse | AAATATGCATCCCCCACATCC | wt103G | CTGATTTCTTCTTTT | |
| mt103C | CTGATTTGTTCTTTT | |||
| mt103T | CTGATTTATTCTTTT | |||
| 181 and 184 | 181/4-Forward | AGCAAAAAATCCAGAAATAGTCATC | wt181 | ATATTGATAGATGAC |
| 181/4-Reverse | CTATGTTGCCCTATTTCTAAGTC | mt181C | ATATTGACAGATGAC | |
| wt184 | ATATTGATAGATGAC | |||
| mt184V | ATATTGACAGATGAC | |||
Nucleotide base pairs with the mutant base of interest are in boldface.
FIG 2.

Application of the 103 HRMA assay to plasmids and patient samples. −dF/dT plots of the common SNP genotypes at position 103 were obtained by using either plasmids (A to D) or patient samples (E to H) and complementary probes. The presence of a probe peak to the left of the amplicon-amplicon peak indicates successful probe binding, allowing the allocation of a 103 genotype to the sample. Shown are wild-type (AAA) plasmid (A) and wild-type (AAA) patient sample (E), wild-type (AAG) plasmid (B) and wild-type (AAG) patient sample (F), mutant (AAC) plasmid (C) and mutant (AAC) patient sample (G), and mutant (AAT) plasmid (D) and mutant (AAT) patient sample (H). The large peak on the right in panel E and the small peaks on the right in panels F to H correspond to the melting of the PCR template DNA.
FIG 3.

Application of the 181 HRMA assay to plasmids and patient samples. −dF/dT plots of common SNP genotypes at position 181 were obtained by using either plasmids (A to C) or patient samples (E to G) and complementary probes or deletion probes (D and H). The smaller probe peak at a lower Tm indicates successful probe binding, allowing allocation of a 181 genotype to the sample. Shown are wild-type (TAT) plasmid (A) and wild-type (TAT) patient sample (E); mutant (TGT) plasmid (B) and mutant (TGT) patient sample (F); a mixture of wild-type (C) and mutant plasmid and patient sample (G) genotypes, indicated by the presence of multiple probe peaks; and wild-type (TAT) at position 181, obtained by using the same wild-type patient sample with complementary probes (D) and deletion probes (H).
FIG 4.

Application of the 184 HRMA assay to plasmids and patient samples. −dF/dT plots of the common SNP genotypes at position 184 were obtained by using either plasmids (A, C, and E) or patient samples (B, D, and F) and complementary probes. The presence of a probe peak to the left of the amplicon-amplicon peak indicates successful probe binding, allowing the allocation of a 184 genotype to the sample. Shown are wild-type (ATG) plasmid (A) and wild-type (ATG) patient sample (B); mutant (GTG) plasmid (C) and mutant (GTG) patient sample (D); and a mixture of wild-type (E) and mutant plasmids and patient sample (F), indicated by a peak from the wild-type and mutant probes.
To determine the analytical sensitivities and to establish cutoff thresholds for each assay, serial dilutions of mutant plasmids in wild-type plasmid were subjected to HRMA. The limit of detection for the K103N (AAC diluted in AAA) and Y181C (TGT diluted in TAT) assays were ∼5% mixed species, and for the M184V assay (GTG diluted in ATG), ∼20% mutant plasmid was detected in a background of wild-type plasmid (Fig. 5). There was 100% reproducibility and repeatability for each assay.
FIG 5.

Analytical sensitivity of HRMA assays. Shown are −dF/dT plots of dilution series. (A) Mutant AAC 103 plasmid diluted in wild-type AAA plasmid. The smallest peak that can be reliably identified is at 5%. (B) Mutant TGT 181 plasmid diluted in wild-type TAT plasmid. The lower limit of detection is 5%. (C) Mutant GTG 184 plasmid diluted in wild-type ATG plasmid. The smallest peak that can be reliably identified is at 20%.
103 HRMA assay.
When applied to 153 patient samples, HRMA assigned the correct genotype to 122 (80%; Cohen's kappa coefficient [κ] > 0.6; P < 0.05) specimens, of which 91 were drug susceptible and 31 were resistant (Table 2 and Fig. 2E to H). Further analysis of these samples revealed that 114 (93%) had identical genotypic matches to the Sanger sequence, while 8 had genotype mismatches that did not affect the drug resistance interpretation (Table 3). Only one specimen resulted in an incorrect outcome. In this case, a peak was obtained with the AAG probe (103K; susceptible), while the Sanger sequence showed AGT (103S; resistant). No probe peak was obtained (no result) for 30 specimens (95% confidence interval [CI], 13.3% to 25.9%), of which 19 were susceptible and 11 resistant, with no bias detected (χ2 = 0.88; P = 0.3).
TABLE 2.
Detection of drug-sensitive and -resistant genotypes by HRMA in 153 HIV-1-positive specimens
| Outcome | No. (%) of codons detected |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| 103 |
181 |
184 |
|||||||
| Total | Sensitive | Resistant | Total | Sensitive | Resistant | Total | Sensitive | Resistant | |
| Correcta | 122 (80) | 91 | 31 | 137 (90) | 106 | 31 | 123 (80) | 86 | 37 |
| Incorrect | 1 (1) | 0 | 1 | 2 (1) | 2 | 0 | 3 (2) | 2 | 1 |
| No result | 30 (20) | 19 | 11 | 14 (9) | 14 | 0 | 27 (18) | 22 | 5 |
| Total | 110 | 43 | 122 | 31 | 110 | 43 | |||
Includes genotype mismatches that did not alter drug resistance interpretation.
TABLE 3.
Comparison of Sanger sequences and corresponding HRMA outcomes
| Drug resistance mutation position | Sanger sequence | Amino acid translation | Drug resistance prediction | No. of samples with sequence | HRMA outcome (no.) |
|||
|---|---|---|---|---|---|---|---|---|
| Correct |
Incorrect | No result | ||||||
| Genotype match | Genotype mismatcha | |||||||
| K103 | AAA | K | Susceptible | 96 | 79 | 1 (AAR; susceptible) | 16 | |
| AAG | K | Susceptible | 8 | 6 | 2 (AAR; susceptible) | |||
| AAR | K | Susceptible | 1 | 1 | ||||
| AGA | R | Susceptible | 5 | 2 (AAA; susceptible) | 3 | |||
| AAC | N | Resistant | 23 | 19 | 1 (AAY; resistant) | 3 | ||
| AAT | N | Resistant | 6 | 3 | 2 (AAY; resistant) | 1 | ||
| AAM | K/N | Resistant | 10 | 6 | 4 | |||
| AAW | K/N | Resistant | 1 | 1 | ||||
| AAH | K/N | Resistant | 1 | 1 | ||||
| AGC | S | Resistant | 1 | 1 | ||||
| AGT | S | Resistant | 1 | 1 (AAG; susceptible) | ||||
| Total | 153 | 114 | 8 | 1 | 30 | |||
| Y181 | TAT | Y | Susceptible | 119 | 105 | 2 (TRT; resistant) | 12 | |
| TAC | Y | Susceptible | 2 | 2 | ||||
| TAY | Y | Susceptible | 1 | 1 (TAT; susceptible) | ||||
| TGT | C | Resistant | 20 | 17 | 3 (TRT; resistant) | |||
| TRT | Y/C | Resistant | 11 | 11 | ||||
| Total | 153 | 133 | 4 | 2 | 14 | |||
| M184 | ATG | M | Susceptible | 109 | 86 | 2 (RTG; resistant) | 21 | |
| GGA | G | Susceptible | 1 | 1 | ||||
| GTA | V | Resistant | 2 | 2 | ||||
| GTG | V | Resistant | 34 | 24 | 7 (RTG; resistant) | 3 | ||
| GTR | V | Resistant | 3 | 3 (GTG; resistant) | ||||
| RTG | M/V | Resistant | 3 | 3 | ||||
| RTR | M/V | Resistant | 1 | 1 (ATG; susceptible) | ||||
| Total | 153 | 113 | 10 | 3 | 27 | |||
Genotype mismatches do not alter drug resistance interpretation.
One of the specimens with a genotype mismatch to the Sanger sequence was selected for clonal analysis and sequencing. HRMA detected a mixed wild-type population (AAA and AAG; 103K), while the Sanger sequence detected only one species (103K; AAA). Analysis of over 100 clones detected an AAG population at a frequency of 3% (data not shown), suggesting that the 103 codon HRMA genotyping assay is more sensitive than Sanger sequencing. The increased sensitivity of HRMA likely explains the discrepancies in the seven additional genotype mismatches. Here, the drug sensitivity was accurately defined, but susceptible (AAR; n = 2) or resistant (AAY; n = 3) quasispecies were detected by HRMA. The final two genotype mismatches were susceptible (AGA; 103R) by Sanger sequencing and susceptible (AAA; 103K) by HRMA. While a minority AAA population might have been present in the sample, the guanine in the second position of the 103 codon likely disrupted accurate melting analysis, as the 103 assay probes were not designed for 103R.
Single nucleotide variation at either end of the probe region did not disrupt HRMA, but polymorphisms at positions 3, 7, and 10 of the probe (5′-3′) were common in specimens that showed no result (see Table S1 in the supplemental material). Polymorphism-masking probes included incorporation of ambiguous bases at these polymorphic sites, but this did not enhance probe peak formation, nor did the use of 11-bp locked nucleic acid probes (data not shown).
181 HRMA assay.
The 181 assay was applied to the patient specimens, and 137 (90%; κ > 0.74; P < 0.05) were correctly genotyped (Table 2 and Fig. 3E to G), 133 (97%) of which were genotypic matches with the Sanger sequence. Fourteen (9%) samples could not be genotyped (95% CI, 4.6% to 13.8%), as no probe melting peak was visible, and 2 (1%) were discordant with the Sanger sequence. Of the samples that were accurately genotyped, 106 were susceptible and 31 were resistant. All of the specimens that could not be genotyped were sensitive by Sanger sequencing, but this did not reach statistical significance (χ2 = 3.46; P = 0.06).
For the two discordant specimens, Sanger sequencing indicated 181Y (TAT; susceptible), while the HRMA probes detected both TAT and TGT (TRT; resistant) (Table 3). Putative mixed species were investigated by clonal analysis to assess the presence of minority resistant populations. The mutant sequence (TGT) was detected in 33% and 7% of the clones analyzed in the two samples (data not shown), suggesting that HRMA is more sensitive than Sanger sequencing for codon 181 and that the error rate for the assay is in fact zero. As a consequence of the increased sensitivity, HRMA detected quasispecies of drug-susceptible and -resistant sequences (TRT) in three samples that were completely resistant (TGT) by Sanger sequencing. The final genotype mismatch may have occurred because the HRMA was not designed to detect the TAC (susceptible) genotype. Therefore, for the specimen that harbored TAT and TAC (TAY; susceptible), the HRMA indicated only TAT (susceptible). Two samples that contained TAC at codon 181 generated no result, as expected.
Sequence variation in the probe binding region was examined in the 14 specimens for which no result was obtained (see Table S1 in the supplemental material). While most polymorphisms toward the probe ends did not disrupt HRMA, a guanine at position 12 of the probe (5′-3′) may have disrupted probe binding in 5 specimens (see Table S1, gray shading, in the supplemental material). A probe lacking the mismatched guanine was tested and successfully genotyped 4 of the 5 specimens (Fig. 3H), while probes incorporating universal bases at this position did result in probe peak formation.
184 HRMA assay.
The 184 HRMA assay correctly predicted genotypes for 123 of 153 (80%; κ> 0.62; P < 0.05) patient samples; 86 (56%) were drug susceptible, and 37 (24%) were resistant, with 113 (92%) genotypic matches (Table 2 and Fig. 4B, D, and F) to the Sanger sequence. Twenty-seven samples (18%) could not be genotyped (95% CI, 11.6% to 23.6%) by HRMA, 22 of which were susceptible and 5 resistant, with no bias detected (χ2 = 1.07; P = 0.3).
Two specimens exhibited major discordances, where the HRMA predicted a mixed susceptible/resistant (RTG) sequence but susceptibility (ATG) was present in the population sequence (Table 3). Clonal analysis of these two samples showed that M184V (GTG) was present in 25% and 20% of the specimen clones, respectively (data not shown), indicating the superior sensitivity of HRMA over Sanger sequencing.
The increased sensitivity of HRMA over Sanger sequencing likely accounts for the minority quasispecies detected by HRMA in 7 samples that were mixed susceptible and resistant (RTG) by HRMA and completely resistant (GTG) by Sanger sequencing. The final genotype mismatch arose because the sequence GTG (resistant) was accurately identified by HRMA but the GTA (susceptible) population, for which the probes were not designed, was not detected in the GTR (resistant) sequences. One sample with a sensitive and resistant quasispecies (RTR), as determined by Sanger sequencing, was genotyped as sensitive (ATG) by HRMA and constitutes the real error of the 184 assay. RTR indicates a diverse population (ATA/GTG/ATG/GTA), among which the M184V HRMA assay was designed to genotype ATG and GTG.
Analysis of the sequences corresponding to the probe binding region indicated that sequence variation directly adjacent to the mutation site of interest (n = 13; see Table S1 in the supplemental material) was associated with no melting peak formation, whereas variation at the end of the probe did not prevent melting peak formation. Repositioning of the HRMA primers to directly flank the 184 locus to mask the adjacent variation did not improve the outcome.
HRMA on longitudinal samples.
Thirteen patients had specimens collected at two time points as part of longitudinal studies to assess the emergence of HIV-1 drug resistance following exposure to sd-NVP for the prevention of MTCT of HIV-1 (10, 37). The first sample was taken at study enrollment, and no drug resistance mutations were detected by Sanger sequencing. The second sample was collected 6 weeks post-sd-NVP exposure when resistance mutations, including K103N (n = 10), Y181C (n = 4), and M184V (n = 1), had developed.
When HRMA was applied to the 103 locus, six of the patients were found to be wild type (AAA or AAG) (Fig. 6A1) at the first time point and mutant (AAC or AAT) at the second time point (Fig. 6A2). Two patients were wild type at both time points. The remaining samples generated no genotype at both time points (n = 1) or at the second time point (n = 4), possibly due to the evolution of polymorphism in the probe binding region. Four patients developed Y181C (TGT) (Fig. 6B1 and B2) at the second time point, as indicated by the 181 HRMA assay, only three of which were identified by population sequencing. Eight patients remained wild type (TAT) at both time points, and one patient could not be genotyped by HRMA. One patient developed M184V (GTG) at the second time point, which was not detected by Sanger sequencing. Eleven remained wild type (ATG) at the 184 locus, and one could not be genotyped by HRMA.
FIG 6.

Application of the HRMA assay to patients sampled at two time points. −dF/dT plots of the SNP genotypes at positions 103 (A) and 181 (B) were obtained with patient samples. Two time points were available for each of the samples. The first time point consisted of drug-naive wild-type virus (A1 and B1), and the second time point consisted of drug-resistant virus (A2 and B2) 6 weeks after single-dose nevirapine exposure. Sample B2 generated a wild-type and a mutant probe peak.
DISCUSSION
HIV drug resistance develops rapidly under suboptimal antiretroviral drug levels as a consequence of viral evolution that disrupts binding between the antiviral compound and viral proteins. Common drug resistance mutations include K103N (AAA/G→AAC/T) and Y181C (TAT→TGT), which confer high-level resistance to NVP and efavirenz (EFV), while M184V (ATG→GTG) causes resistance to lamivudine (3TC) and emtricitabine (FTC) (38, 39) and is usually detected by Sanger population sequencing following regimen failure.
The biggest obstacles to successful drug resistance surveillance and testing in resource-limited settings are cost, access to equipment, and expertise. While Sanger population sequencing is the gold standard genotyping methodology due to its accuracy and long read lengths, the equipment and reagent costs remain prohibitive for many in low-resource settings, prompting research into simple and cost-effective detection systems. HRMA is a rapid and relatively simple DNA-typing and mutation-scanning methodology that genotypes by melting curve analysis (24). Thus, we sought to utilize the technology to develop HRMA-PCRs to rapidly genotype key HIV-1 drug resistance codons in samples consisting of predominately subtype C viruses. These assays were shown to correctly genotype 80%, 90%, and 80% of specimens at the 103, 181, and 184 codons, respectively (κ > 0.6; P < 0.05). A minority of the HRMA genotype predictions were discordant with the Sanger population sequence (1%, 1%, and 2% per assay). The remaining specimens, 9% to 20% (95% CI, 4.6% to 25.9%), could not be genotyped by HRMA due to nontargeted polymorphisms that prevented probe binding. The agreement between the Sanger sequence and specimens that HRMA was able to genotype was very high for all 3 assays (κ > 0.94; P < 0.05). The 103 and 184 assays displayed no bias in genotyping susceptible or resistant samples (P = 0.3), while there was possible preferential genotyping of sensitive samples in the 181 assay, although it was not statistically significant (P = 0.06). The limits of detection of the 103, 181, and 184 assays were 5%, 5%, and 20% of quasispecies, respectively, and they were consistently more sensitive than Sanger population sequencing, as confirmed by clonal analysis. Although this study used a particular HRM instrument and dyes, the primers and probes developed in the study could be used with other platforms.
Considering the increased sensitivity of HRMA, only two specimens were found to be truly discordant between HRMA and population sequencing. In one patient sample, the 103 HRMA-PCR indicated drug susceptibility (AAG; 103K) at the codon, while the Sanger sequence was resistant (AGT; 103S). The second incorrect prediction occurred with the 184 HRMA-PCR detecting sensitivity (ATG; 184M) while the Sanger sequence was mixed susceptible/resistant (RTR; 184M/V). In both cases, the discordance was likely a result of the nontargeted polymorphism position adjacent to the variable bases of interest. The melting effects of nontargeted polymorphism can be minimized with polymorphism-masking probes consisting of unlabeled oligonucleotide probes incorporating universal nucleotides, mismatched nucleotides, or deletions of mismatched nucleotides (40, 41). Incorporation of probes integrating inosine into the 103, 181, and 184 assays, however, did not allow genotyping of additional specimens. Limited success was achieved in genotyping additional samples with probes incorporating a deletion within the probe of the nontargeted polymorphism that displaced the complementary nucleotide by bypassing melting effects of the polymorphic site.
The inability of the HRMA to genotype up to 20% of the patient samples was partially due to the presence of known nontargeted polymorphisms, which are infrequent and can confer drug resistance (e.g., K103S and M184I) or function as susceptible transitional mutations (e.g., K103R and M184G). Similar to the incorrectly genotyped specimens, these polymorphisms are adjacent to the variable bases targeted by the probes. Additionally, polymorphisms proximal to the codons of interest were often observed in specimens that could not be genotyped. As probe modifications can bypass polymorphisms, a large panel of probes would be required to increase the ability of the assays to genotype the majority of patient samples. HRMA-PCR assays were developed and tested for additional drug resistance mutations (M41L, K65R, D67N, K70R, V106M, Q151M, G190A, T215F/Y, and P225H) with limited or no success, highlighting the challenge the genetic variation of HIV presents. Thus, in areas where specific drug resistance mutations, such as 103, 181, and 184, are present at high frequency, HRMA assays may be useful to rapidly genotype patient samples on a population basis, such as for surveillance purposes, with high specificity and sensitivity. These assays may also be useful in selected patients to study the emergence of resistance over time.
HRMA genotyping is suited to relatively invariant genetic material that presents a challenge to the genotyping of RNA viruses, such as HIV. Nevertheless, we have shown that it is possible to genotype 80 to 90% of samples with high accuracy and sensitivity. Expanding the panel of drug resistance codons to incorporate additional prevalent antiretroviral therapy drug resistance mutations would likely require the integration of polymorphism-masking probes to allow more accurate genotyping of a larger proportion of patient samples. Additionally, combining viral quantification by real-time PCR prior to melting analysis for genotyping is valuable for research, diagnostic, and treatment formulation purposes. Thus, despite limitations due to inherent genetic polymorphisms, HRMA with unlabeled probes is a viable methodology to cheaply and rapidly genotype some of the most common HIV-1 drug resistance mutations.
MATERIALS AND METHODS
HRMA-PCR assay design.
Asymmetric PCR was adopted to minimize the amplicon melting peak magnitude and to maximize the probe signal following preferential amplification of the sense strand, to which the probes annealed (42). Primers and probes were designed to hybridize to a consensus C sequence compiled from 648 subtype C sequences downloaded from the Los Alamos National Laboratory HIV database. The 103 assay primers (20 to 25 bp) amplified nucleotides 2391 to 2421, and the 181 and 184 assay primers amplified nucleotides 2636 to 2668 of HIV-1 reverse transcriptase (Table 1) (HXB2; GenBank accession number K03455). The unlabeled 3′ phosphorylation-blocked oligonucleotide probes (15 bp) were designed to identify the wild-type and mutant sequences at the 103, 181, and 184 positions (Table 1).
PCR was performed in a final volume of 10 μl and contained 2.5 mM MgCl2, 10 μM forward primer, 2 μM probe (one probe per well), ∼3 ng of template DNA, and 1× LightCycler HRM Master (Roche Applied Science, GmbH, Germany). The concentrations of the limiting reverse primers were 0.3 μM 100R (103 codon assay) and 0.6 μM 181R (181 and 184 codon assays). PCR (and subsequent HRMA) was performed on the LightCycler 480 real-time (RT) PCR instrument (Roche Applied Science, GmbH, Germany). The PCR conditions were 94°C for 5 min and 35 cycles of 95°C for 10 s, 50°C for 15 s, and 72°C for 15 s. The HRMA conditions were 95°C for 1 min and 40°C for 1 min, followed by temperature increases from 40°C to 95°C in increments of 1°C/s. Fluorescence data were obtained in 25 data acquisitions per degree to ensure the melting transitions were recorded. The melting data were analyzed using the LightCycler 480 Gene Scanning software (version 1.2.0.0625; Roche Applied Science, GmbH, Germany) and plotted as fluorescence (y axis) relative to temperature (x axis). Maximum and minimum fluorescence for each PCR were normalized to 100% and 0%, respectively, on the same axis. Software-generated −dF/dT plots of the normalized data allowed the probe-amplicon and amplicon-amplicon duplexes to be easily discriminated as separate peaks on a melting curve (Fig. 1). A clear separation between the probe and amplicon melting peaks, adequate peak height above background, and the predicted melting temperature were considered when assigning a genotype or no result. The melting curves corresponding to the probes allowed the nucleotide sequence to be determined, and the translated amino acid was used to infer a drug-susceptible or -resistant phenotype. The presence of quasispecies was inferred by the detection of multiple probe melting peaks.
The wild-type and mutant plasmids were used to validate the HRMA assays. Each reaction was repeated five times on one PCR plate and again on different days to assess HRMA-PCR repeatability and reproducibility, respectively. Samples were run singly, and the runs were repeated on different days to assess agreement between runs. Serial dilutions consisting of 100%, 50%, 20%, 10%, 5%, 1%, and 0% mutant mixed with wild-type plasmids were utilized to determine genotype assignment cutoffs for each mutation.
Specimen selection.
Stored plasma or PCR products from 153 specimens previously genotyped by population sequencing were selected for analysis, 26 of which were collected at 2 different time points from 13 patients (5, 10, 11, 37). HIV-1 RNA extraction and RT-PCR were performed as previously described (43). Of the 153 specimens tested, 150 were subtype C and 3 were subtype B. Thirty-five specimens were from drug-naive patients and 118 from drug-experienced patients. By population sequencing, 43 (28%) specimens contained the K103N mutation, 2 K103S (1%), 5 (3%) K103R, 31 (20%) Y181C, 43 (28%) M184V, 1 (0.7%) M184G, and 1 (0.7%) M184I.
Outcome definitions.
Agreement between the amino acids at the 103, 181, and 184 codons with the Sanger population sequence, as determined by HRMA, was considered a correct result and in most cases also had identical genotypic matches. However, in some cases, there was a difference in the genotypes between the two methods that did not affect the drug resistance interpretation, which we refer to as genotype mismatches. Cases where genotypic mismatches resulted in an altered drug resistance interpretation were referred to as incorrect results. If no probe melting peak was visible, no genotype could be assigned, and no result was recorded.
Clonal analysis.
Clonal analysis was performed on 5 selected nested-PCR products to assess the presence of minority populations. The TOPO TA cloning kit with the pCR 2.1-TOPO vector and TOP10 cells (Invitrogen, USA) was utilized according to the manufacturer's protocol. Over 100 colonies from each of five samples were amplified by an in-house nested RT-PCR (43) and then genotyped by HRMA.
Statistical analysis.
Cohen's kappa coefficient (κ) was utilized to measure agreement between the HRMA and Sanger population sequences. A chi-square test with the n − 1 correction for small sample sizes was applied to test if HRMA preferentially genotyped sensitive or resistant samples. Statistics were calculated with Stata version 13 (StataCorp, College Station, TX).
Supplementary Material
ACKNOWLEDGMENTS
David Sacks received a Poliomyelitis Research Foundation bursary. Gillian M. Hunt received funding for consumables from the National Health Laboratory Service Research Trust.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JCM.01291-16.
REFERENCES
- 1.Joint United Nations Programme on HIV/ AIDS. 2016. Global AIDS Update 2016. Joint United Nations Programme on HIV/AIDS, Geneva, Switzerland. [Google Scholar]
- 2.Marconi VC, Sunpath H, Lu Z, Gordon M, Koranteng-Apeagyei K, Hampton J, Carpenter S, Giddy J, Ross D, Holst H, Losina E, Walker BD, Kuritzkes DR, South Africa Resistance Cohort Study Team. 2008. Prevalence of HIV-1 drug resistance after failure of a first highly active antiretroviral therapy regimen in KwaZulu Natal, South Africa. Clin Infect Dis 46:1589–1597. doi: 10.1086/587109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoffmann CJ, Charalambous S, Sim J, Ledwaba J, Schwikkard G, Chaisson RE, Fielding KL, Churchyard GJ, Morris L, Grant AD. 2009. Viremia, resuppression, and time to resistance in human immunodeficiency virus (HIV) subtype C during first-line antiretroviral therapy in South Africa. Clin Infect Dis 49:1928–1935. doi: 10.1086/648444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orrell C, Walensky RP, Losina E, Pitt J, Freedberg KA, Wood R. 2009. HIV type-1 clade C resistance genotypes in treatment-naive patients and after first virological failure in a large community antiretroviral therapy programme. Antivir Ther 14:523–531. [PMC free article] [PubMed] [Google Scholar]
- 5.El-Khatib Z, Ekstrom AM, Ledwaba J, Mohapi L, Laher F, Karstaedt A, Charalambous S, Petzold M, Katzenstein D, Morris L. 2010. Viremia and drug resistance among HIV-1 patients on antiretroviral treatment: a cross-sectional study in Soweto, South Africa. AIDS 24:1679–1687. doi: 10.1097/QAD.0b013e32833a097b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wallis CL, Mellors JW, Venter WD, Sanne I, Stevens W. 2010. Varied patterns of HIV-1 drug resistance on failing first-line antiretroviral therapy in South Africa. J Acquir Immune Defic Syndr 53:480–484. doi: 10.1097/QAI.0b013e3181bc478b. [DOI] [PubMed] [Google Scholar]
- 7.Murphy RA, Sunpath H, Lu Z, Chelin N, Losina E, Gordon M, Ross D, Ewusi AD, Matthews LT, Kuritzkes DR, Marconi VC, South Africa Resistance Cohort Study Team. 2010. Outcomes after virologic failure of first-line ART in South Africa. AIDS 24:1007–1012. doi: 10.1097/QAD.0b013e3283333639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sigaloff KC, Ramatsebe T, Viana R, de Wit TF, Wallis CL, Stevens WS. 2012. Accumulation of HIV drug resistance mutations in patients failing first-line antiretroviral treatment in South Africa. AIDS Res Hum Retroviruses 28:171–175. doi: 10.1089/aid.2011.0136. [DOI] [PubMed] [Google Scholar]
- 9.van Zyl GU, Liu TF, Claassen M, Engelbrecht S, de Oliveira T, Preiser W, Wood NT, Travers S, Shafer RW. 2013. Trends in genotypic HIV-1 antiretroviral resistance between 2006 and 2012 in South African patients receiving first- and second-line antiretroviral treatment regimens. PLoS One 8:e67188. doi: 10.1371/journal.pone.0067188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinson NA, Morris L, Gray G, Moodley D, Pillay V, Cohen S, Dhlamini P, Puren A, Bhayroo S, Steyn J, McIntyre JA. 2007. Selection and persistence of viral resistance in HIV-infected children after exposure to single-dose nevirapine. J Acquir Immune Defic Syndr 44:148–153. doi: 10.1097/QAI.0b013e31802b920e. [DOI] [PubMed] [Google Scholar]
- 11.Hunt GM, Coovadia A, Abrams EJ, Sherman G, Meyers T, Morris L, Kuhn L. 2011. HIV-1 drug resistance at antiretroviral treatment initiation in children previously exposed to single-dose nevirapine. AIDS 25:1461–1469. doi: 10.1097/QAD.0b013e3283492180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson JA, Li JF, Morris L, Martinson N, Gray G, McIntyre J, Heneine W. 2005. Emergence of drug-resistant HIV-1 after intrapartum administration of single-dose nevirapine is substantially underestimated. J Infect Dis 192:16–23. doi: 10.1086/430741. [DOI] [PubMed] [Google Scholar]
- 13.Coovadia A, Hunt G, Abrams EJ, Sherman G, Meyers T, Barry G, Malan E, Marais B, Stehlau R, Ledwaba J, Hammer SM, Morris L, Kuhn L. 2009. Persistent minority K103N mutations among women exposed to single-dose nevirapine and virologic response to nonnucleoside reverse-transcriptase inhibitor-based therapy. Clin Infect Dis 48:462–472. doi: 10.1086/596486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanna GJ, D'Aquila RT. 2001. Clinical use of genotypic and phenotypic drug resistance testing to monitor antiretroviral chemotherapy. Clin Infect Dis 32:774–782. doi: 10.1086/319231. [DOI] [PubMed] [Google Scholar]
- 15.Cai F, Chen H, Hicks CB, Bartlett JA, Zhu J, Gao F. 2007. Detection of minor drug-resistant populations by parallel allele-specific sequencing. Nat Methods 4:123–125. doi: 10.1038/nmeth995. [DOI] [PubMed] [Google Scholar]
- 16.Paredes R, Marconi VC, Campbell TB, Kuritzkes DR. 2007. Systematic evaluation of allele-specific real-time PCR for the detection of minor HIV-1 variants with pol and env resistance mutations. J Virol Methods 146:136–146. doi: 10.1016/j.jviromet.2007.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rowley CF, Boutwell CL, Lee EJ, MacLeod IJ, Ribaudo HJ, Essex M, Lockman S. 2010. Ultrasensitive detection of minor drug-resistant variants for HIV after nevirapine exposure using allele-specific PCR: clinical significance. AIDS Res Hum Retroviruses 26:293–300. doi: 10.1089/aid.2009.0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi C, Eshleman SH, Jones D, Fukushima N, Hua L, Parker AR, Yeo CJ, Hruban RH, Goggins MG, Eshleman JR. 2004. LigAmp for sensitive detection of single-nucleotide differences. Nat Methods 1:141–147. doi: 10.1038/nmeth713. [DOI] [PubMed] [Google Scholar]
- 19.Church JD, Towler WI, Hoover DR, Hudelson SE, Kumwenda N, Taha TE, Eshleman JR, Eshleman SH. 2008. Comparison of Ligamp and an ASPCR assay for detection and quantification of K103N-containing HIV variants. AIDS Res Hum Retroviruses 24:595–605. doi: 10.1089/aid.2007.0224. [DOI] [PubMed] [Google Scholar]
- 20.Troyer RM, Lalonde MS, Fraundorf E, Demers KR, Kyeyune F, Mugyenyi P, Syed A, Whalen CC, Bajunirwe F, Arts EJ. 2008. A radiolabeled oligonucleotide ligation assay demonstrates the high frequency of nevirapine resistance mutations in HIV type 1 quasispecies of NVP-treated and untreated mother-infant pairs from Uganda. AIDS Res Hum Retroviruses 24:235–250. doi: 10.1089/aid.2007.0138. [DOI] [PubMed] [Google Scholar]
- 21.Stuyver L, Wyseur A, Rombout A, Louwagie J, Scarcez T, Verhofstede C, Rimland D, Schinazi RF, Rossau R. 1997. Line probe assay for rapid detection of drug-selected mutations in the human immunodeficiency virus type 1 reverse transcriptase gene. Antimicrob Agents Chemother 41:284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fisher RG, Smith DM, Murrell B, Slabbert R, Kirby BM, Edson C, Cotton MF, Haubrich RH, Kosakovsky Pond SL, Van Zyl GU. 2015. Next generation sequencing improves detection of drug resistance mutations in infants after PMTCT failure. J Clin Virol 62:48–53. doi: 10.1016/j.jcv.2014.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bushman FD, Hoffmann C, Ronen K, Malani N, Minkah N, Rose HM, Tebas P, Wang GP. 2008. Massively parallel pyrosequencing in HIV research. AIDS 22:1411–1415. doi: 10.1097/QAD.0b013e3282fc972e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wittwer CT, Reed GH, Gundry CN, Vandersteen JG, Pryor RJ. 2003. High-resolution genotyping by amplicon melting analysis using LCGreen. Clin Chem 49:853–860. doi: 10.1373/49.6.853. [DOI] [PubMed] [Google Scholar]
- 25.Ririe KM, Rasmussen RP, Wittwer CT. 1997. Product differentiation by analysis of DNA melting curves during the polymerase chain reaction. Anal Biochem 245:154–160. doi: 10.1006/abio.1996.9916. [DOI] [PubMed] [Google Scholar]
- 26.Herrmann MG, Durtschi JD, Wittwer CT, Voelkerding KV. 2007. Expanded instrument comparison of amplicon DNA melting analysis for mutation scanning and genotyping. Clin Chem 53:1544–1548. doi: 10.1373/clinchem.2007.088120. [DOI] [PubMed] [Google Scholar]
- 27.Montgomery JL, Sanford LN, Wittwer CT. 2010. High-resolution DNA melting analysis in clinical research and diagnostics. Expert Rev Mol Diagn 10:219–240. doi: 10.1586/erm.09.84. [DOI] [PubMed] [Google Scholar]
- 28.Pietzka AT, Indra A, Stöger A, Zeinzinger J, Konrad M, Hasenberger P, Allerberger F, Ruppitsch W. 2009. Rapid identification of multidrug-resistant Mycobacterium tuberculosis isolates by rpoB gene scanning using high-resolution melting curve PCR analysis. J Antimicrob Chemother 63:1121–1127. doi: 10.1093/jac/dkp124. [DOI] [PubMed] [Google Scholar]
- 29.Wolff B, Thacker W, Schwartz S, Winchell J. 2008. Detection of macrolide resistance in Mycoplasma pneumoniae by real-time PCR and high-resolution melt analysis. Antimicrob Agents Chemother 52:3542–3549. doi: 10.1128/AAC.00582-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Slinger R, Bellfoy D, Desjardins M, Chan F. 2007. High-resolution melting assay for the detection of Gyra mutations causing quinolone resistance in Salmonella enterica serovars Typhi and Paratyphi. Diagn Microbiol Infect Dis 57:455–458. doi: 10.1016/j.diagmicrobio.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 31.Arvia R, Corcioli F, Azzi A. 2013. High resolution melting analysis as a tool to detect molecular markers of antiviral resistance in influenza A viruses. J Virol Methods 189:265–270. doi: 10.1016/j.jviromet.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 32.Hsiao CC, Chang J, Wu JY, Liu WH, Han SY, Chen PJ, Yeh SH. 2012. High-resolution melting and real-time PCR for quantification and detection of drug-resistant HBV mutants in a single amplicon. Antivir Ther 17:291–303. doi: 10.3851/IMP2022. [DOI] [PubMed] [Google Scholar]
- 33.Zhao XT, Zhou DQ, Wu S, Chen YW, Shao Y, Zhang J, Xia CS, Wang KP, Yang H, Wan J, Yu B, Zhang Z, Zhang W. 2012. Genotyping cytomegalovirus UL97 mutations by high-resolution melting analysis with unlabeled probe. Arch Virol 157:475–481. doi: 10.1007/s00705-011-1173-y. [DOI] [PubMed] [Google Scholar]
- 34.Ramirez MV, Cowart KC, Campbell PJ, Morlock GP, Sikes D, Winchell JM, Posey JE. 2010. Rapid detection of multidrug-resistant Mycobacterium tuberculosis by use of real-time PCR and high-resolution melt analysis. J Clin Microbiol 48:4003–4009. doi: 10.1128/JCM.00812-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Towler WI, James MM, Ray SC, Wang L, Donnell D, Mwatha A, Guay L, Nakabiito C, Musoke P, Jackson JB, Eshleman SH. 2010. Analysis of HIV diversity using a high-resolution melting assay. AIDS Res Hum Retroviruses 26:913–918. doi: 10.1089/aid.2009.0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cousins MM, Ou SS, Wawer MJ, Munshaw S, Swan D, Magaret CA, Mullis CE, Serwadda D, Porcella SF, Gray RH, Quinn TC, Donnell D, Eshleman SH, Redd AD. 2012. Comparison of a high-resolution melting assay to next-generation sequencing for analysis of HIV diversity. J Clin Microbiol 50:3054–3059. doi: 10.1128/JCM.01460-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martinson NA, Morris L, Johnson J, Gray GE, Pillay V, Ledwaba J, Dhlamini P, Cohen S, Puren A, Steyn J, Heneine W, McIntyre JA. 2009. Women exposed to single-dose nevirapine in successive pregnancies: effectiveness and nonnucleoside reverse transcriptase inhibitor resistance. AIDS 23:809–816. [DOI] [PubMed] [Google Scholar]
- 38.WHO. 2013. Consolidated guidelines on the use of antiretroviral drugs for treating and preventing HIV infection: recommendations for a public health approach. WHO, Geneva, Switzerland. [PubMed] [Google Scholar]
- 39.Wensing AM, Calvez V, Günthard HF, Johnson VA, Paredes R, Pillay D, Shafer RW, Richman DD. 2014. Update of the drug resistance mutations in HIV-1. Top Antivir Med 22:642–650. [PMC free article] [PubMed] [Google Scholar]
- 40.Margraf RL, Mao R, Wittwer CT. 2006. Masking selected sequence variation by incorporating mismatches into melting analysis probes. Hum Mutat 27:269–278. doi: 10.1002/humu.20290. [DOI] [PubMed] [Google Scholar]
- 41.Margraf RL, Mao R, Highsmith WE, Holtegaard LM, Wittwer CT. 2007. RET proto-oncogene genotyping using unlabeled probes, the masking technique, and amplicon high-resolution melting analysis. J Mol Diagn 9:184–196. doi: 10.2353/jmoldx.2007.060091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou L, Myers AN, Vandersteen JG, Wang L, Wittwer CT. 2004. Closed-tube genotyping with unlabeled oligonucleotide probes and a saturating DNA dye. Clin Chem 50:1328–1335. doi: 10.1373/clinchem.2004.034322. [DOI] [PubMed] [Google Scholar]
- 43.Loubser S, Balfe P, Sherman G, Hammer S, Kuhn L, Morris L. 2006. Decay of K103N mutants in cellular DNA and plasma RNA after single-dose nevirapine to reduce mother-to-child HIV transmission. AIDS 20:995–1002. doi: 10.1097/01.aids.0000222071.60620.1d. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.