

Abstract
Transforming growth factor β (TGFβ) ligands, including TGFβ1, are multifunctional cytokines known as key regulators of cell growth, differentiation and inflammation. Dysregulated TGFβ signaling is common in numerous solid tumors, including head and neck squamous cell carcinoma (HNSCC). Previously, TGFβ ligands were also reported to be associated with an enhancement of stemness in glioma stem-like cells. However, their role in HNSCC cancer stem cells (CSCs) has not been explored. The present study demonstrated that TGFβ1 enriches the properties of HNSCC CSCs. TGFβ1 promoted sphere formation and increased stemness-associated gene expression (Oct4 and Sox2) of primary HNSCC CSCs. Additionally, the population of aldehyde dehydrogenase (ALDH)-positive cells was increased subsequent to exogenous treatment of cells with TGFβ1. In addition, following stimulation with TGFβ1, the cells exhibited more resistance to cisplatin and elevated expression of Twist, Snail and Slug. Mechanistically, TGFβ1 acts as an upstream stimulator of Wnt/β-catenin signaling. Collectively, the present findings provide insights toward the development of TGFβ1 signaling inhibition strategies for treating HNSCC CSCs.
Keywords: head and neck squamous cell carcinoma, cancer stem cells, TGFβ1
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer worldwide and has a poor prognosis (1). The overall survival rate has not changed over the past 30 years, with >400,000 patients diagnosed and >200,000 mortalities annually (2). Despite numerous advances in treatment modalities and techniques, including surgery, chemotherapy, radiation therapy and combinations of these, only 50–60% of patients survive for >5 years (3). The static survival rate is considered to be the result of a high incidence of treatment resistance, resulting in local/regional recurrences and frequent distant metastases (4). Thus, to improve the survival of patients with HNSCC, an increased understanding of the pathophysiology and tumorigenesis of HNSCC is urgently required.
A previous study revealed that a small subpopulation of tumor cells, cancer stem cells (CSCs), can initiate and maintain tumors and metastasize (5). As conventional therapies usually target differentiated cancer cells, CSCs are considered to be responsible for treatment failure (6). Thus, a considerable understanding and identification of the promoters and regulators that control expression of HNSCC CSCs may lead to methods to increase the prognosis of HNSCC against treatment failure.
Transforming growth factor β (TGFβ) is a well-known and potent regulator of the extracellular matrix (7). In human cancer, TGFβ shows biphasic roles in tumor development and progression (8). TGFβ functions as a tumor suppressor by cellular proliferation inhibition or by promoting differentiation of normal cells (9). Mutations of type II receptors in the TGFβ signaling pathway have been demonstrated for head and neck cancer (10). However, once tumor cells become resistant to the proliferation-inhibiting function of TGFβ, they increase their production of TGFβ. As a result, the increase in TGFβ leads to TGFβ-mediated angiogenesis stimulation, immune suppression and cell motility stimulation (11). Consequently, an increased TGFβ level results in increased invasiveness of tumor cells. Previously, it was reported that TGFβ is able to increase the proportion of cluster of differentiation (CD)133+ (a putative marker of CSCs) cells in liver cancer cells, suggesting a positive role for TGFβ in the ‘stemness’ of cancer cells (12). However, whether TGFβ stimulates the key features of this cell subpopulation in HNSCC has not been reported.
In the present study, the effect of an increased TGFβ level on the stemness of HNSCC CSCs was investigated. The association with the Wnt/β-catenin signaling pathway was also examined to elucidate the downstream effect of TGFβ on stemness regulation in HNSCC CSCs.
Materials and methods
Cell culture and reagents
The primary sphere K3 cell line was used. This cell was isolated from a surgical specimen from a HNSCC patient, and the CSC properties of the cell line have been validated using a number of functional assays, testing self-renewal capability, stem cell marker expression, chemoresistance and in vivo tumorigenicity, as previously reported (5). Cells were cultured in serum-free Dulbecco's modified Eagle's medium (DMEM)-Ham's F-12 (F12) medium supplemented with human recombinant basic fibroblast growth factor (bFGF; 10 ng/ml; R&D Systems, Minneapolis, MN, USA), N2 supplement (1 ml per 500 ml medium; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA), and epidermal growth factor (EGF; 10 ng/ml; R&D Systems). The primary β-catenin antibody was purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA), and the secondary anti-mouse IgG antibody was obtained from Jackson ImmunoResearch Laboratories (West Grove, PA, USA).
Tumorsphere formation assay
To assess self-renewal in vitro, the cells were dissociated into a single-cell suspension, seeded in a 24-well plate at a density of 500 cells per well, and cultured in serum-free medium, with EGF and bFGF added every other day. Untreated cells (cells not treated with EGF or bFGF) were used as a control. Tumorspheres were allowed to grow for 7 days, and spheres with a diameter >30 µm were counted.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and western blotting
RT-qPCR and western blotting experiments were performed as previously described (5). RT-qPCR analysis of Wnt signaling target genes (β-catenin, c-myc and cyclin D) was subsequently performed on an iCycler IQ real-time detection system (Bio-Rad Laboratories, Hercules, CA, USA), using IQ Supermix with SYBR-Green (Bio-Rad Laboratories). The sequences of the human-specific primers used were as follows: Oct4 forward, 5′-GCAATTTGCCAAGCTCCTGAA-3′ and reverse, 5′-GCAGATGGTCGTTTGGCTGA-3′; Sox2 forward, 5′-CCTCCGGGACATGATCAG-3′ and reverse, 5′-TTCTCCCCCCTCCAGTTC-3′; Twist forward, 5′-CAGTCTTACGAGGAGCTGCA-3′ and reverse, 5′-TCTTGCTCAGCTTGTCCGAG-3′; Snail forward, 5′-CACCTCCAGACCCACTCAGAT-3′ and reverse, 5′-CCTGAGTGGGGTGGGAGCTTC-3′; Slug forward, 5′-CTCCATGCCTGTCATACCAC-3′ and reverse, 5′-GGAAAGAGGAGAGAGGCCAT-3′; β-catenin forward, 5′-GGCAGTGCGTTTAGCTGGTG-3′ and reverse, 5′-AGCTTGGGGTCCACCACTAG-3′; c-myc forward, 5′-GTATGTGGACGGCTTCTCGC-3′ and reverse, 5′-GCTGCTGTCGTTGAGAGGGT-3′; and cyclin D forward, 5′-GGAGCTGCTGCAAATGGAGC-3′ and reverse, 5′-CAGAGGGCAACGAAGGTCTG-3′.
Aldehyde dehydrogenase (ALDH) activity assay
ALDH activity was analyzed using an Aldefluor assay kit (Stem Cell Technologies, Vancouver, BC, Canada). Briefly, cells were suspended in Aldefluor assay buffer containing the ALDH substrate (1 µmol/l per 1×106 cells). A sample of cells was stained with the specific ALDH inhibitor diethylaminobenzaldehyde (DEAB) as a negative control. Fluorescent ALDH+ cells were detected in the green fluorescence channel, and samples treated with the specific ALDH inhibitor DEAB were used as the control to set the gates defining the ALDH+ region. Flow cytometric sorting was conducted using a FACSAria flow cytometer (Becton Dickinson, San Jose, CA, USA).
Chemosensitivity assay
Cells were dissociated into a single-cell suspension and then plated in a 24-well plate at a density of 1×105 cells per well under serum-free culture conditions. The cells were treated with cisplatin at the indicated concentrations and then cultured at 37°C in a humidified 5% CO2 atmosphere. Subsequently, 24 h later, 50 µl of MTT solution (5 mg/ml in PBS) was added to each well, and the plate was incubated at room temperature for 2 h. Absorbance was measured on a SpectraMax 190 device (Molecular Devices, LLC, Sunnyvale, CA, USA) at a wavelength of 570 nm.
Optimal Tcf-binding site (TOP)/far-from-optimal Tcf-binding site (FOP) luciferase assay
Cells were seeded in 24-well plates at a density of 1×105 cells per well. Cells were transiently transfected using Polyexpress® (Excellgen, Rockville, MD, USA). For the luciferase assay, cells were transfected with 0.5 µg of the lymphoid enhancer factor-reporter pTOPFLASH/pFOPFLASH along with 50 ng of pRL-SV40. TGFβ1 treatment was initiated at 24 h post-transfection and performed for 48 h. Luciferase activity was monitored using the Dual-Glo luciferase assay system (Promega Corporation, Madison, WI, USA). The transfection efficiency was normalized to the cotransfected Renilla luciferase activity according to the manufacturer's protocol.
Statistical analysis
Experimental data were statistically assessed using either 2-tailed Student's t-test or analysis of variance with Scheffe's post-hoc analysis. P<0.05 was considered to indicate a statistically significant difference.
Results
TGFβ1 increases the self-renewal capacity of cancer stem cells
The capacity for self-renewal is one of the putative traits of CSCs (5). Thus, it was evaluated whether TGFβ1 treatment enhances the self-renewal capacity of HNSCC CSCs by performing a sphere-forming assay. TGFβ1 (2 ng/ml) treatment significantly increased the sphere formation ability of HNSCC CSCs with a diameter of >30 µm compared to the control treatment group (Fig. 1).
Figure 1.

TGFβ1 treatment increases the sphere-forming ability of HNSCC K3 CSCs. (A) Photomicrographs of representative spheres after exogenous TGFβ1 or control (dimethyl sulfoxide) treatment in HNSCC CSCs, (B) with quantification. Scale bar, 30 µM. **P<0.01. TGFβ1, transforming growth factor β; HNSCC, head and neck squamous cell carcinoma; CSCs, cancer stem cells.
TGFβ1 increases the levels of stemness markers of CSCs
Oct4 and Sox2 are critical regulators of pluripotency in the mammalian embryo, and deregulated expression of these genes can be found in HNSCC CSCs (13). Thus, whether TGFβ1 treatment increases the transcriptional expression of Oct4 and Sox2 was examined. The results showed that mRNA expression levels of Oct4 and Sox2 were significantly increased in the TGFβ1 treatment group compared to the control treatment group (Fig. 2A). ALDH is considered to be a marker of HNSCC CSCs (14). Therefore, ALDH activity was examined in TGFβ1- and control-treated HNSCC CSCs. Fig. 2B shows that the proportion of ALDH+ cells was significantly increased in the TGFβ1 treatment group compared with the control treatment group (7.24 vs. 19.28%).
Figure 2.

TGFβ1 treatment increases stemness-associated marker expression in HNSCC CSCs. (A) Transcript levels of Oct4 and Sox2 in TGFβ1- or control (DMSO)-treated HNSCC CSCs. (B) ALDH activity subsequent to TGFβ1 or control (DMSO) treatment of HNSCC CSCs. TGFβ1, tumor growth factor β; HNSCC, head and neck squamous cell carcinoma; CSCs, cancer stem cells; DMSO, dimethyl sulfoxide; DEAB, diethylaminobenzaldehyde.
TGFβ1 increases chemoresistance to cisplatin for HNSCC CSCs by overexpression of ATP-binding cassette sub-family G member 2 (ABCG2)
HNSCC CSCs possess a chemoresistance ability against anticancer drugs, and the ABCG2 transporter is responsible for this resistance phenotype (5). Therefore, chemoresistance levels were compared using MTT assay subsequent to cisplatin administration at various concentrations with the TGFβ1 and control treatment groups. The results showed that TGFβ1-treated HNSCC CSCs had significantly increased cisplatin resistance compared with control-treated HNSCC CSCs (Fig. 3A). Furthermore, an increased ABCG2 expression level was identified in TGFβ1-treated HNSCC CSCs compared to control-treated HNSCC CSCs, indicating that TGFβ1 may enhance the chemoresistance of HNSCC CSCs through increased expression of ABCG2 (Fig. 3B).
Figure 3.
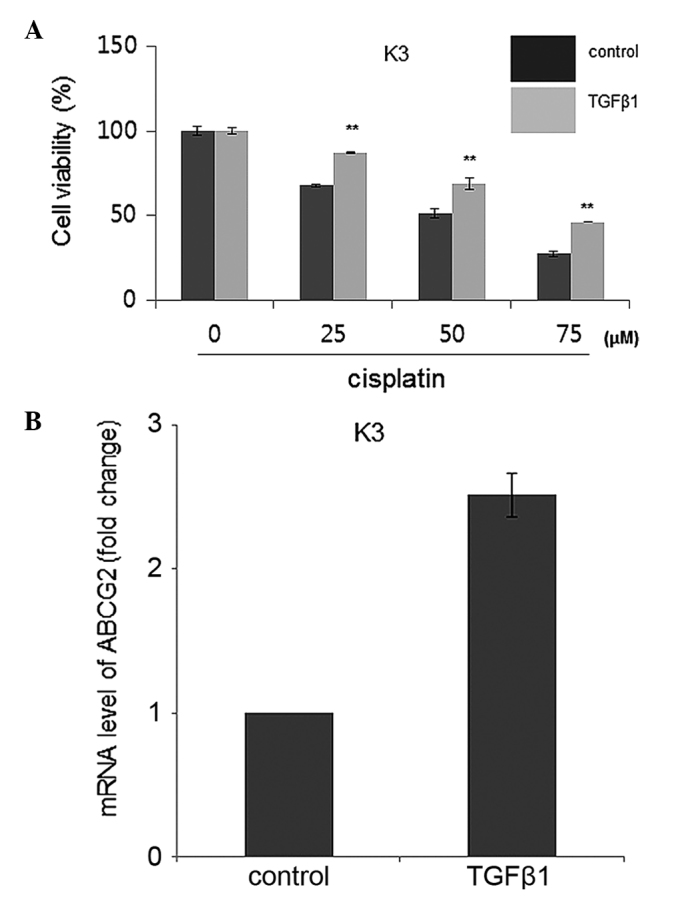
TGFβ1 treatment enhances cisplatin resistance of HNSCC K3 CSCs. (A) Cell viability subsequent to cisplatin administration at various concentrations of TGFβ1- or control (DMSO)-treated HNSCC CSCs. (B) mRNA levels of ABCG2 in TGFβ1- or control (DMSO)-treated HNSCC CSCs. **P<0.01.TGFβ1, transforming growth factor β; HNSCC, head and neck squamous cell carcinoma; CSCs, cancer stem cells; DMSO, dimethyl sulfoxide; ABCG2, ATP-binding cassette sub-family G member 2
TGFβ1 increases the expression of epithelial-mesenchymal transition (EMT) regulators
EMT is a key process in tumor invasion and metastasis (15). Previously, it was reported that the EMT process can generate cancer cells with a stem cell phenotype (15). Thus, whether TGFβ1 can increase central regulators of the EMT process, such as Twist, Snail and Slug, was investigated. As shown in Fig. 4, mRNA expression levels of all three EMT regulators were increased in TGFβ1-treated HNSCC CSCs compared with control-treated HNSCC CSCs.
Figure 4.
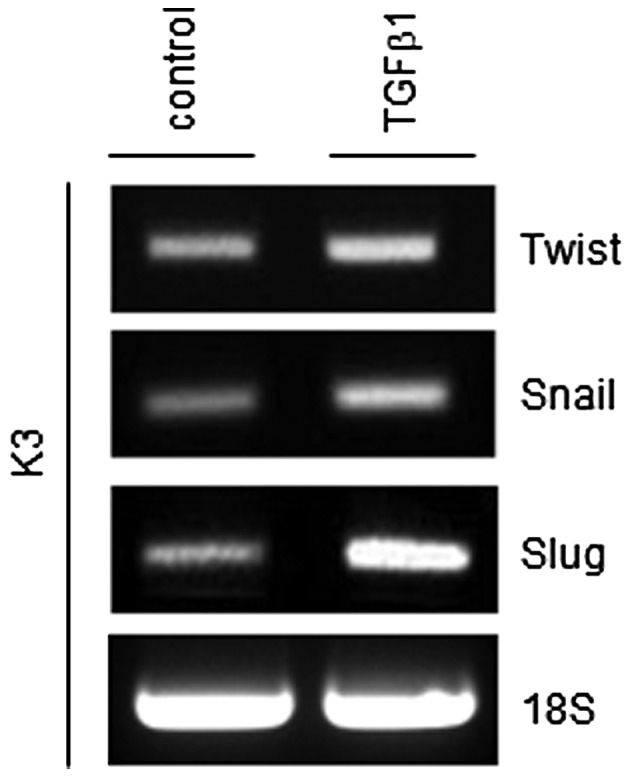
TGFβ1 treatment increases transcript levels of the epithelial-mesenchymal transition mediators Twist, Snail and Slug. TGFβ1, transforming growth factor β.
TGFβ1 activates canonical Wnt/β-catenin signaling
It was previously suggested that Wnt/β-catenin contributes to the stemness of HNSCC cells (16). Thus, whether TGFβ1 acts as an upstream stimulator of Wnt/β-catenin signaling was investigated. Administration of TGFβ1 increased the expression of β-catenin, which is an effector of the Wnt pathway (Fig. 5A). Also, TGFβ1 treatment increased the activity of a Wnt/β-catenin-dependent reporter as a functionally more relevant indicator (Fig. 5B). Furthermore, the transcript levels of Wnt/β-catenin signaling target genes, such as the c-myc and cyclin D1 genes, were increased in TGFβ1-treated HNSCC CSCs (Fig. 5C). Overall, these data suggest that TGFβ1-induced stemness may be associated with Wnt pathway upregulation in HNSCC CSCs.
Figure 5.
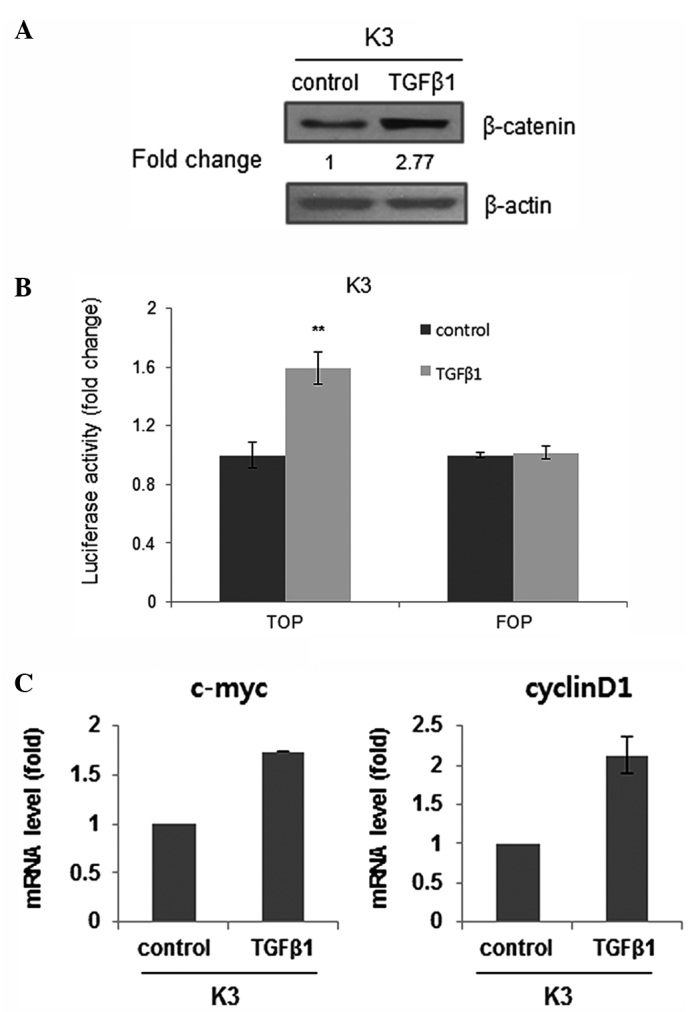
TGFβ1 treatment activates the canonical Wnt signaling pathway. (A) Protein level of β-catenin in TGFβ1- or control (DMSO)-treated HNSCC CSCs. (B) TOP/FOP luciferase activity in TGFβ1- or control (DMSO)-treated HNSCC CSCs. **P<0.01. (C) Transcript levels of β-catenin target genes (c-Myc and cyclin D1) in TGFβ1- or control (DMSO)-treated HNSCC CSCs. TGFβ1, transforming growth factor β; HNSCC, head and neck squamous cell carcinoma; CSCs, cancer stem cells; DMSO, dimethyl sulfoxide; TOP, optimal Tcf-binding site; FOP, far-from-optimal Tcf-binding site
Discussion
Numerous previous studies have indicated that a rare population of cells within the entire tumor bulk, termed CSCs or cancer-initiating cells, possess capabilities for tumor initiation and progression, and in certain cases, metastasis, which are not found in the majority of cells (5,14,17). In clinical settings, these cells exhibit an intrinsic resistance to popular chemotherapeutic agents, preventing complete eradication of the tumor cells following treatment. Therefore, an understanding of the molecular mechanism of CSCs as a treatment target is critical for the design and development of effective anticancer therapies against tumor relapse.
Several signal transmission pathways regulating CSC pathobiology have been suggested, including the Wnt, Notch and Hh pathways (18). Previously, members of the TGFβ cytokine family, such as TGFβs, bone morphogenetic proteins (BMPs), Nodal and activins, were also shown to be involved in the control of the CSC phenotype, particularly in glioblastoma multiforme (GM) (19,20). Notably, BMP and TGFβ have conflicting effects on GM CSC proliferation, although they are included in the same family (TGFβ) (19,20). BMP has been shown to inhibit tumor growth and to induce differentiation of GM CSCs (19). By contrast, TGFβ promotes self-renewal and prevents differentiation, enhancing the oncogenic ability of GM CSCs (21).
TGFβ1 expression is observed in ~80% of cases of human HNSCC and correlates with more advanced disease and reduced survival (11). In addition, TGFβ1 overexpression promotes tumorigenesis in a paracrine manner, leading to increased inflammation and angiogenesis (11). However, different TGFβ signaling disruptions also enhance epithelial carcinogenesis through various mechanisms and are common in HNSCC, suggesting biphasic roles for this signaling pathway in tumorigenesis of HNSCC (22). Also, there are extremely few previous studies regarding the effects of TGFβ ligands on HNSCC CSCs. Thus, the present study aimed to clarify the role of TGFβ1 in HNSCC CSC biology.
In the present study, it was shown that TGFβ1 treatment enhances self-renewal and stemness-associated genes (Oct4 and Sox2) expression in HNSCC CSCs. In addition, this treatment was shown to result in enrichment of the ALDH+ cell population, expressing a putative marker of HNSCC CSCs. Furthermore, TGFβ1 treatment results in an increase of stem cell-like traits, such as resistance to conventional chemotherapy (cisplatin) for treatment of HNSCC patients. Collectively, the present data suggest that TGFβ1 may be involved in the regulation of cells with stem cell-like traits in HNSCC.
Mechanistically, it has been shown that the regulation of GM CSC self-renewal by TGFβ is mediated by leukemia inhibitory factor (LIF), a member of the interleukin 6 (IL6) family of cytokines, and by signaling through a heterodimeric receptor complex composed of the glycoprotein 130 and the LIF receptor inducing the Janus kinase-signal transducers and activators of transcription pathway (21). In addition, IL6 may also be a mediator of the induction of GM CSC self-renewal by TGFβ (23). Notably, the present study showed that TGFβ administration increased the expression of β-catenin, a major effector of the canonical Wnt signal, although a detailed mechanism is not presented here. Serra et al observed that Wnt5a, a Wnt ligand, was directly regulated by TGFβ in primary mammary gland cells, and they identified Smad binding sites in the Wnt5a promoter (24). Thus, crosstalk between TGFβ signaling and Wnt signaling may be significant in HNSCC CSC biology. Additional studies are required to show how TGFβ signaling interacts with Wnt signaling.
Based on these data, the present findings demonstrate that TGFβ1 may have an important role in HNSCC CSC genesis and that its inhibitors may be valuable for eradicating HNSCC CSCs.
Acknowledgements
This study was supported by Konkuk University Hospital (grant no., 201403).
References
- 1.Jemal A, Murray T, Samuels A, Ghafoor A, Ward E, Thun MJ. Cancer statistics, 2003. CA Cancer J Clin. 2003;53:5–26. doi: 10.3322/canjclin.53.1.5. [DOI] [PubMed] [Google Scholar]
- 2.Forastiere A, Koch W, Trotti A, Sidransky D. Head and neck cancer. N Engl J Med. 2001;345:1890–1900. doi: 10.1056/NEJMra001375. [DOI] [PubMed] [Google Scholar]
- 3.Yamano Y, Uzawa K, Saito K, Nakashima D, Kasamatsu A, Koike H, Kouzu Y, Shinozuka K, Nakatani K, Negoro K, et al. Identification of cisplatin-resistance related genes in head and neck squamous cell carcinoma. Int J Cancer. 2010;126:437–449. doi: 10.1002/ijc.24704. [DOI] [PubMed] [Google Scholar]
- 4.Haddad RI, Shin DM. Recent advances in head and neck cancer. N Engl J Med. 2008;359:1143–1154. doi: 10.1056/NEJMra0707975. [DOI] [PubMed] [Google Scholar]
- 5.Lim YC, Oh SY, Cha YY, Kim SH, Jin X, Kim H. Cancer stem cell traits in squamospheres derived from primary head and neck squamous cell carcinomas. Oral Oncol. 2011;47:83–91. doi: 10.1016/j.oraloncology.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 6.Mannelli G, Gallo O. Cancer stem cells hypothesis and stem cells in head and neck cancers. Cancer Treat Rev. 2012;38:515–539. doi: 10.1016/j.ctrv.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 7.Liu L, Jiao J, Wang Y, Wu J, Huang D, Teng W, Chen L. TGF-beta1 gene polymorphism in association with diabetic retinopathy susceptibility: A systematic review and meta-analysis. PLoS One. 2014;9:e94160. doi: 10.1371/journal.pone.0094160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White RA, Malkoski SP, Wang XJ. TGFβ signaling in head and neck squamous cell carcinoma. Oncogene. 2010;29:5437–5446. doi: 10.1038/onc.2010.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siegel PM, Massagué J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3:807–821. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- 10.Lu SL, Herrington H, Reh D, Weber S, Bornstein S, Wang D, Li AG, Tang CF, Siddiqui Y, Nord J, et al. Loss of transforming growth factor-beta type II receptor promotes metastatic head-and-neck squamous cell carcinoma. Genes Dev. 2006;20:1331–1342. doi: 10.1101/gad.1413306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu SL, Reh D, Li AG, Woods J, Corless CL, Kulesz-Martin M, Wang XJ. Overexpression of transforming growth factor beta1 in head and neck epithelia results in inflammation, angiogenesis, and epithelial hyperproliferation. Cancer Res. 2004;64:4405–4410. doi: 10.1158/0008-5472.CAN-04-1032. [DOI] [PubMed] [Google Scholar]
- 12.You H, Ding W, Rountree CB. Epigenetic regulation of cancer stem cell marker CD133 by transforming growth factor-beta. Hepatology. 2010;51:1635–1644. doi: 10.1002/hep.23544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kashyap V, Rezende NC, Scotland KB, Shaffer SM, Persson JL, Gudas LJ, Mongan NP. Regulation of stem cell pluripotency and differentiation involves a mutual regulatory circuit of the NANOG, OCT4, and SOX2 pluripotency transcription factors with polycomb repressive complexes and stem cell microRNAs. Stem Cells Dev. 2009;18:1093–1108. doi: 10.1089/scd.2009.0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zou B, Sun S, Qi X, Ji P. Aldehyde dehydrogenase activity is a cancer stem cell marker of tongue squamous cell carcinoma. Mol Med Rep. 2012;5:1116–1120. doi: 10.3892/mmr.2012.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee SH, Koo BS, Kim JM, Huang S, Rho YS, Bae WJ, Kang HJ, Kim YS, Moon JH, Lim YC. Wnt/β-catenin signalling maintains self-renewal and tumourigenicity of head and neck squamous cell carcinoma stem-like cells by activating Oct4. J Pathol. 2014;234:99–107. doi: 10.1002/path.4383. [DOI] [PubMed] [Google Scholar]
- 17.Dalerba P, Clarke MF. Cancer stem cells and tumor metastasis: First steps into uncharted territory. Cell Stem Cell. 2007;1:241–242. doi: 10.1016/j.stem.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 18.Campelo MR Garcia, Curbera G Alonso, Gallego G Aparicio, Pulido E Grande, Antón Aparicio LM. Stem cell and lung cancer development: Blaming the Wnt, Hh and Notch signalling pathway. Clin Transl Oncol. 2011;13:77–83. doi: 10.1007/s12094-011-0622-0. [DOI] [PubMed] [Google Scholar]
- 19.Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, Brem H, Olivi A, Dimeco F, Vescovi AL. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444:761–765. doi: 10.1038/nature05349. [DOI] [PubMed] [Google Scholar]
- 20.Lee J, Son MJ, Woolard K, Donin NM, Li A, Cheng CH, Kotliarova S, Kotliarov Y, Walling J, Ahn S, et al. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell. 2008;13:69–80. doi: 10.1016/j.ccr.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seoane J. TGFbeta and cancer initiating cells. Cell Cycle. 2009;8:3787–3788. doi: 10.4161/cc.8.23.10054. [DOI] [PubMed] [Google Scholar]
- 22.Levy L, Hill CS. Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006;17:41–58. doi: 10.1016/j.cytogfr.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 23.Wang H, Lathia JD, Wu Q, Wang J, Li Z, Heddleston JM, Eyler CE, Elderbroom J, Gallagher J, Schuschu J, et al. Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells. 2009;27:2393–2404. doi: 10.1002/stem.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Serra R, Easter SL, Jiang W, Baxley SE. Wnt5a as an effector of TGFβ in mammary development and cancer. J Mammary Gland Biol Neoplasia. 2011;16:157–167. doi: 10.1007/s10911-011-9205-5. [DOI] [PMC free article] [PubMed] [Google Scholar]