

Abstract
Gemcitabine is a commonly used chemotherapy drug in pancreatic cancer. The function of activator protein 1 (AP-1) is cell-specific, and its function depends on the expression of other complex members. In the present study, we added gemcitabine to the media of Panc-1 and SW1990 cells at clinically achieved concentrations (10 µM). Compared with constitutive c-Fos expression, c-Jun expression increased in a dose-dependent manner upon gemcitabine treatment. c-Jun overexpression increased gemcitabine-induced apoptosis through Bim activation, while cell apoptosis and Bim expression decreased following c-Jun knockdown. Furthermore, gemcitabine-induced apoptosis and Bim levels decreased when c-Jun phosphorylation was blocked by SP600125. Our findings suggest that c-Jun, which is a member of the AP-1 complex, functions in gemcitabine-induced apoptosis by regulating its downstream target Bim in pancreatic cancer cells.
Keywords: activator protein 1, gemcitabine, c-Jun, Bim, apoptosis
Introduction
As one of the most aggressive and lethal diseases, pancreatic ductal adenocarcinoma is the fourth leading cause of cancer-related mortality worldwide, despite accounting for only 2.2% of all cancers (1,2). The estimated 5-year survival rate is less than 5%, and the overall median survival time is less than 1 year following diagnosis (3,4). Gemcitabine monotherapy was initially approved by the US Food and Drug Administration (FDA) in 1996, and it has been the standard of care for patients with metastatic pancreatic cancer for several decades (5). Although various combination therapies have been developed, most demonstrate a minimal or no significant change in overall survival compared with treatment of gemcitabine alone (6), highlighting the requirement for further mechanistic studies. Numerous factors have been reported to be involved in gemcitabine's effects, including various genes, proteins, signalling pathways and microRNAs (7–10). Apoptosis is a core signalling pathway in human pancreatic cancer, and a detailed understanding of apoptosis is essential for the development of more effective or ‘targeted’ therapies (11–13).
The transcription factor activator protein 1 (AP-1) is a dimeric complex comprised of the Jun, Fos, activating transcription factor and musculoaponeurotic fibrosarcoma protein families (14), and it is involved in cellular proliferation, transformation and death (15). The AP-1 complex forms various combinations of heterodimers and homodimers, and this combination determines the genes that are regulated by AP-1 (16). c-Jun and c-Fos function differentially modulates their target genes. It has been reported that c-Jun−/− fibroblasts are resistant to alkylating agent-induced apoptosis, which may be mediated by Fas ligand induction (17). Additionally, rhomboid domain-containing 1 inhibits UV-induced cell apoptosis by activating and upregulating c-Jun and its downstream target B-cell lymphoma 3 (Bcl-3) (18). c-Fos downregulation in MCF-7/ADR cells resulted in enhanced apoptosis, and altered expression of apoptosis-associated proteins, including Bax, Bcl-2, p53 and PUMA (19).
In this study, we investigated the biological effects of AP-1 on gemcitabine-induced apoptosis in pancreatic cancer cells. Our results indicate that endogenous c-Jun expression, but not c-Fos expression, increased following gemcitabine treatment. Furthermore, c-Jun functioned as a pro-apoptotic protein by regulating the downstream AP-1 target Bim. These results are likely to provide further insight into the molecular mechanisms of chemotherapy in pancreatic cancer.
Materials and methods
Cell culture, plasmids and transfection
Cell lines were obtained from the Cell Resource Centre at Peking Union Medical College (PUMC), China. Panc-1 cells were cultured in Dulbecco's modified Eagle's medium (HyClone; GE Healthcare Life Sciences, Logan, UT, USA) with 10% foetal bovine serum (FBS), and SW1990 cells were cultured in RPMI-1640 (HyClone) with 10% FBS in 5% CO2 at 37°C. Adherent cells were passaged every 2–3 days with 0.5 mg/ml trypsin (1:250) and 0.53 mM ethylenediaminetetraacetic acid. c-Jun expression plasmids were cloned into pIRES-puro2 with a C-terminal Myc tag. siRNA oligonucleotides were designed against c-Jun as follows: GAU GGA AAC GAC CUU CUAU. The plasmids were constructed according to standard cloning techniques. Cells were transfected using Lipofectamine™ 2000 (Invitrogen Life Technologies, Carlsbad, CA, USA).
Antibodies
The Bcl-3 and Bim antibodies were purchased from Santa Cruz Biotechnology. The antibody against c-Jun was purchased from BD Pharmingen (San Diego, CA, USA). Antibodies against phospho-c-Jun (Ser73), Bax, PARP, caspase-7 and GAPDH were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). Horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology, Inc. (Dallas, MA, USA).
Western blot analysis
Proteins were extracted with sodium dodecyl sulphate (SDS) lysis buffer [50 mM Tris-HCl (pH 6.8), 10% glycerol and 2% SDS] and quantified using the bicinchoninic acid protein assay reagent (Thermo Fisher Scientific, Waltham, MA, USA). Extracts were separated on a 12% SDS-polyacrylamide gel and electrophoretically transferred to polyvinylidene fluoride membrane (GE Healthcare Life Sciences). The membrane was blocked in 5% skimmed milk for 1 h at room temperature and then incubated overnight with the indicated antibodies at 4°C. The membrane was incubated with an anti-rabbit or an anti-mouse IgG-HRP (Santa Cruz Biotechnology, Inc.) for 1 h at room temperature. Chemiluminescence was detected using an enhanced chemiluminescence blot detection system (Santa Cruz Biotechnology, Inc.).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA from cells was extracted with TRIzol reagent (Invitrogen Life Technologies) and 1 µg isolated total RNA was converted to cDNA using a First-Strand cDNA synthesis kit (Takara Biotechnology Co., Ltd., Dalian, China). Power SYBR-Green master mix (Applied Biosystems, Foster City, CA, USA) was added to cDNA samples that were then subjected to RT-qPCR using the StepOne Real-Time PCR system (Applied Biosystems). Relative mRNA levels were normalised against the housekeeping gene GAPDH. The primers for RT-qPCR were as follows: c-Jun sense, 5′-TCCAAGTGCCGAAAAAGGAAG-3′ and antisense, 5′-CGAGTTCTGAGCTTTCAAGGT-3′; c-Fos sense, 5′-GGGGCAAGGTGGAACAGTTAT-3′ and antisense, 5′-CCGCTTGGAGTGTATCAGTCA-3′; GAPDH sense, 5′-TGAGTACGTCGTGGAGTCCA-3′, and antisense, 5′-TAGACTCCACGACATACTCA-3′.
Cell proliferation assay
Cell proliferation was assessed by the Cell Counting Kit-8 (CCK-8) assay (Dojindo Molecular Technologies, Inc., Shanghai, China). Cells were seeded at a density of 2,000 cells/well in 96-well plates. A total of 10 µl CCK-8 solution was added to each well containing 100 µl culture medium and incubated for 2 h at 37°C. Absorbance was measured at 450 nm using a multiwell spectrophotometer (BioTek, Winooski, VT, USA).
Flow cytometric analysis
Cells were harvested by trypsinisation and collected by centrifugation, and then a fluorescein isothiocyanate (FITC)-Annexin V kit (NeoBioscience, Shenzhen, China) was used to stain cells according to the manufacturer's instructions. Apoptotic cells were analysed with a BD Accuri® C6 flow cytometer and corresponding CellFIT software (both from BD Biosciences, San Diego, CA, USA).
Terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL)
Cells were fixed with 4% paraformaldehyde solution for 30 min at room temperature. After rinsing with phosphate-buffered saline (PBS), the samples were incubated with a TUNEL reaction mixture containing terminal deoxynucleotidyl transferase and FITC-dUTP (Roche Applied Science, Indianapolis, IN, USA) for 1 h at 37 °C using an apoptosis detection kit (Roche Applied Science). These cells were then stained with 4,6-diamidino-2-phenylindole (DAPI) to detect the cell nucleus.
Statistical analyses
Statistical analyses were performed using Student's t-test in Microsoft Excel software (Redmont, WA, USA). The results were presented as the means ± standard deviation of triplicates of each experiment. All experiments were performed three times, unless stated otherwise.
Results
Gemcitabine induces apoptosis in human pancreatic cancer cells
To determine the physiologically relevant dose of gemcitabine that induces apoptosis in Panc-1 and SW1990 cells, we first determined gemcitabine's cytotoxic effects and used 293A cells as a control. We treated cells with increasing doses of gemcitabine (from 0.1 to 1 mM) or PBS for control cells. The results identified the 50% inhibitory concentration (IC50) value in Panc-1 and SW1990 cells (Fig. 1A and B) while 293A cells did not respond to gemcitabine (Fig. 1C). Based on the data and previous studies reporting that micromolar gemcitabine concentrations may be clinically achieved (20), we used 10 µM gemcitabine for all subsequent experiments. Flow cytometric analysis revealed that 10 µM gemcitabine significantly increased apoptosis in Panc-1 and SW1990 cells (Fig. 1D and E), but not in 293A cells (Fig. 1F).
Figure 1.

Gemcitabine induces apoptosis in human pancreatic cancer cells. (A-C) Cytotoxicity of gemcitabine in Panc-1 cells (A), SW1990 cells (B) and 293A cells (C). Cells were treated with various concentrations of gemcitabine for 48 h and cell survival was determined by Cell Counting Kit-8 assay. Cells treated with phosphate-buffered saline (PBS) were used as a control, with survival set at 100%. (D-F) Gemcitabine (GEM) induced apoptosis in Panc-1 cells (D), SW1990 cells (E) and 293A cells (F). Annexin V/propidium iodide (PI) fluorescence-activated cell sorting assay was used to analyse apoptosis in Panc-1 cells following gemcitabine treatment for 24 h. Cells treated with PBS were used as a control. The percentages of apoptotic cells, including early apoptotic cells (Annexin V+, PI−) and late apoptotic cells (Annexin V−, PI+), are shown as bar graphs. All data are representative of three different experiments, and the error bars represent the standard deviations of triplicate samples.
Gemcitabine treatment induces c-Jun expression in a dose-dependent manner
c-Jun and c-Fos are extensively studied components of the AP-1 complex, which is involved in numerous cell activities, including proliferation, apoptosis, survival, tumourigenesis and tissue morphogenesis (21). To further define the AP-1 mechanism in gemcitabine-induced pancreatic cancer cell apoptosis, we examined the expression of c-Jun and c-Fos following exposure to 10 µM gemcitabine at the indicated time points through western blot analysis and RT-qPCR. As shown in Fig. 2A and B, gemcitabine had little effect on c-Fos expression but significantly increased c-Jun expression. These results suggest that gemcitabine specifically activates the AP-1 pathway through c-Jun but not c-Fos. In addition, gemcitabine increased c-Jun expression in a concentration-dependent manner (Fig. 2C and D).
Figure 2.

Gemcitabine treatment induces c-Jun expression in a dose-dependent manner. (A and B) Gemcitabine increased c-Jun expression in a time-dependent manner. The cells were exposed to 10 µM gemcitabine for the indicated time periods, and extracted for reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and western blot analysis using the indicated antibodies. (C and D) Gemcitabine increased c-Jun expression in a concentration-dependent manner. The cells were treated with different amounts of gemcitabine for 24 h, and extracted for RT-qPCR and western blot analysis using the indicated antibodies.
c-Jun promotes gemcitabine-induced apoptosis by upregulating Bim
Based on the experimental data above, we hypothesised that c-Jun regulates gemcitabine-induced apoptosis. To confirm this hypothesis, we transfected c-Jun (c-Jun-Myc) or a control vector into Panc-1 and SW1990 cells for 24 h. We then exposed the cells to gemcitabine and performed fluorescence-activated cell sorting (FACS) analysis to measure apoptosis at the indicated time points. The results revealed that gemcitabine-induced apoptosis increased upon c-Jun overexpression (Fig. 3A and B). Western blot analysis also suggested that PARP and cleaved caspase 7 levels increased, indicative of the increased apoptosis. AP-1 likely affects apoptosis through the differential regulation of pro-apoptotic and anti-apoptotic downstream factors (22,23). Therefore, we analysed Bcl-3, Bax and Bim expression. As shown in Fig. 4A and B, Bcl-3 and Bax expression did not change upon c-Jun overexpression, whereas Bim expression increased. Therefore, c-Jun overexpression promotes gemcitabine-induced apoptosis though Bim upregulation.
Figure 3.
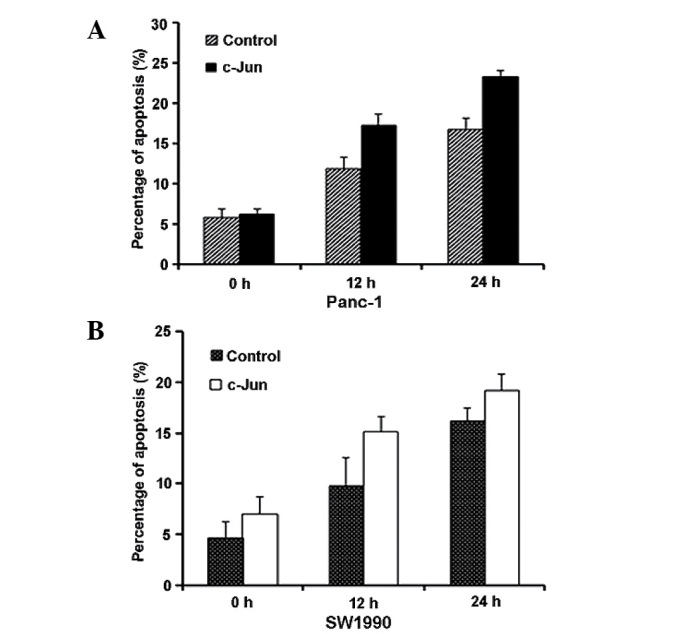
c-Jun promotes gemcitabine-induced apoptosis by upregulating Bim. Gemcitabine-induced apoptosis increased upon c-Jun overexpression in Panc-1 cells (A) and SW1990 cells (B). We transfected c-Jun (c-Jun-Myc) or a control vector into Panc-1 and SW1990 cells for 24 h, and then exposed the cells to gemcitabine and performed Annexin V/propidium iodide fluorescence-activated cell sorting assay to measure apoptosis at the indicated time points. The data are representative of three different experiments, and the error bars represent the standard deviations of triplicate samples.
Figure 4.
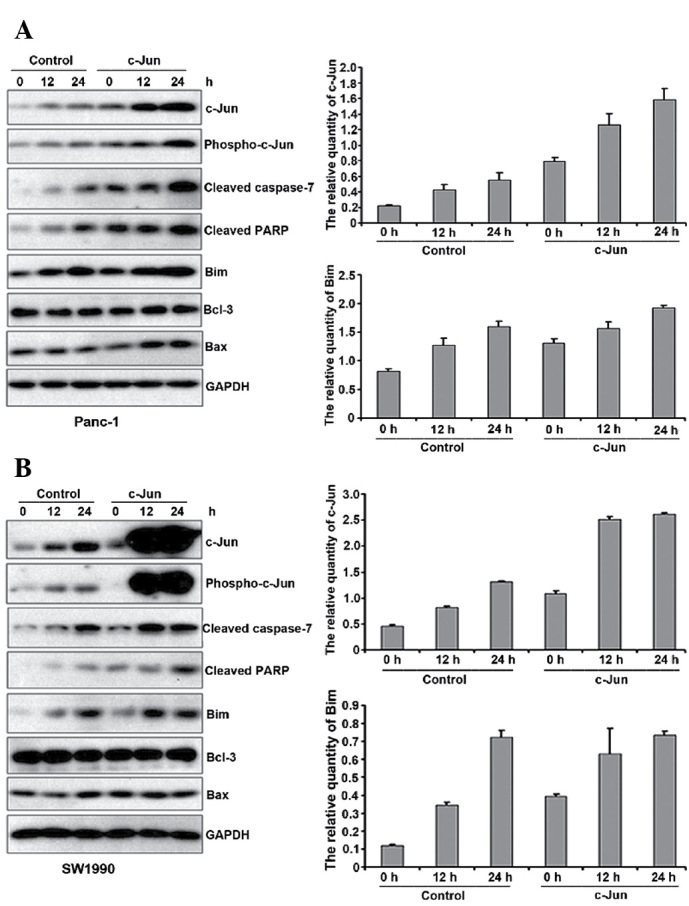
c-Jun promotes gemcitabine-induced apoptosis by upregulating Bim. c-Jun overexpression promotes gemcitabine-induced apoptosis though Bim upregulation in Panc-1 cells (A) and SW1990 cells (B). Following gemcitabine treatment, the cells were extracted at the indicated time points for western blot analysis. The right panel shows the quantitative intensity of bands boxed in the left panel.
Endogenous c-Jun suppression inhibits gemcitabine-induced apoptosis by reducing Bim
To further determine the pro-apoptotic properties of c-Jun in gemcitabine resistance, we suppressed endogenous c-Jun expression in Panc-1 cells by transfecting cells with siRNA against c-Jun. FACS and western blot analysis indicated that c-Jun knockdown inhibited gemcitabine-induced apoptosis (Fig. 5A). We also observed that Bim expression decreased following gemcitabine treatment in c-Jun knockdown cells. However, c-Jun did not affect Bax and Bcl-3 expression under the same conditions (Fig. 5B). Taken together, our findings suggest that c-Jun exerts pro-apoptotic effects on gemcitabine-treated cells by regulating its downstream target Bim in pancreatic cancer cells.
Figure 5.
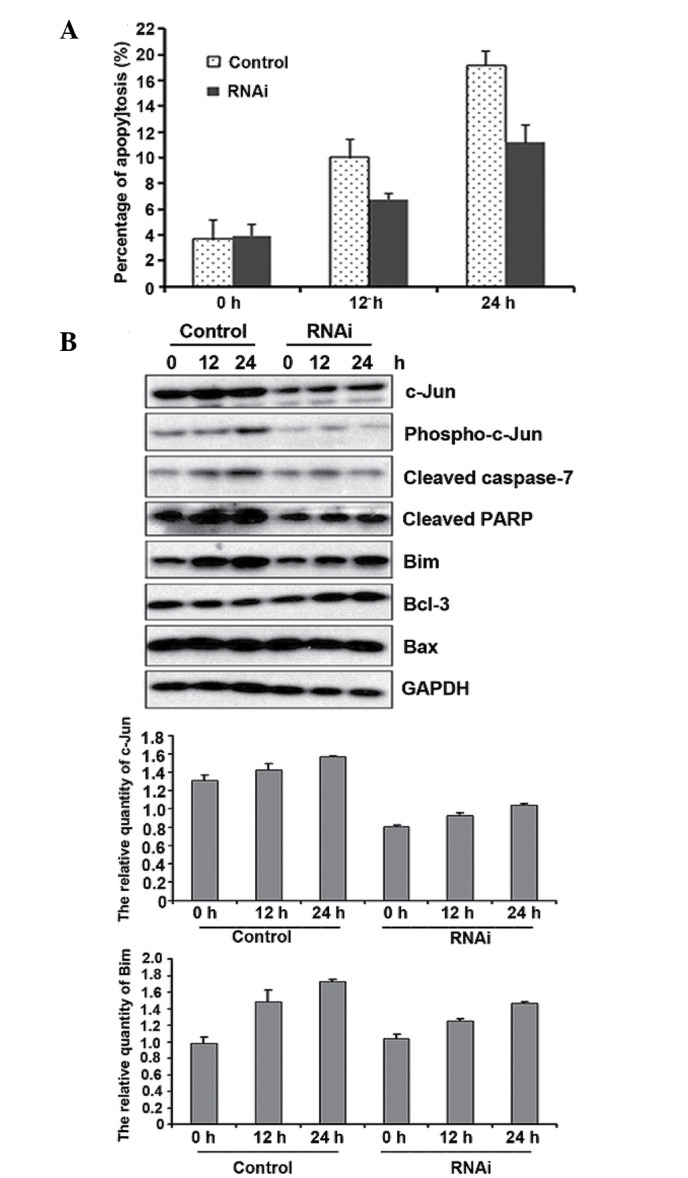
Endogenous c-Jun suppression inhibits gemcitabine-induced apoptosis by reducing Bim expression. (A) c-Jun knockdown inhibited gemcitabine-induced apoptosis. c-Jun siRNA or non-targeting siRNA were transfected into Panc-1 cells for 24 h. The cells were exposed to 10 µM gemcitabine, and Annexin V/propidium iodide fluorescence-activated cell sorting assay was performed at the indicated time points. (B) c-Jun knockdown inhibited gemcitabine-induced apoptosis by reducing Bim expression. The cells were treated as described in (A), and were harvested for western blot analysis with the corresponding antibodies. The bottom panels show the quantitative intensity of bands boxed in the above panel.
Reduced c-Jun activity inhibits gemcitabine-induced apoptosis by decreasing Bim expression
In addition to the expression level, the activity of c-Jun is also critical for its function. Therefore, we treated cells with SP600125, which blocks c-Jun phosphorylation. We exposed Panc-1 cells to gemcitabine for 24 h in the presence or absence of 20 µM (24) SP600125 and examined apoptosis by FACS analysis. The results revealed that SP600125 alone had little effect on Panc-1 cells, but SP600125 pre-treatment partially decreased gemcitabine-mediated apoptosis (Fig. 6A). TUNEL staining was used to further confirm the apoptosis results and the data also indicated that SP600125 pre-treatment partially decreased gemcitabine-mediated apoptosis (Fig. 6B). SP600125 attenuated PARP and cleaved caspase 7 levels (Fig. 6C). These results suggest that c-Jun activation is required for gemcitabine-induced apoptosis. Moreover, compared with the uniform Bcl-3 and Bax expression, a gemcitabine-induced increase in Bim expression was attenuated upon SP600125 treatment.
Figure 6.

The inactivity of c-Jun inhibits gemcitabine-mediated apoptosis by decreasing the expression of Bim. SP600125 pre-treatment partially decreased gemcitabine-mediated apoptosis. (A and B) Panc-1 cells were treated with 10 µM gemcitabine (GEM) in the presence or absence of SP600125 (SP) for 24 h, then Annexin V/propidium iodide fluorescence-activated cell sorting assay (A) and terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) (B) were applied to measure the apoptosis. Data represent the means ± standard deviation from three independent experiments. (C) The inactivity of c-Jun inhibited gemcitabine-induced apoptosis by reducing Bim expression. The cells were treated as described in (A), and were harvested for western blot analysis with the corresponding antibodies. The right panel shows the quantitative intensity of bands boxed in the left panel.
Discussion
Pancreatic cancer is one of the most aggressive diseases due to the difficulties in early detection and the low resection rate (7,25). At present, ~40% of patients have metastatic disease, and these patients are primarily treated with palliative therapy (26). The current chemotherapeutic agent of choice for pancreatic cancer is gemcitabine, which was approved by the FDA in 1996. Gemcitabine confers a median survival advantage of 6 months (27), and 5-fluorouracil contributes to an improvement of only 1 month over gemcitabine. Erlotinib targets epidermal growth factor receptor and adds only two additional weeks to the average overall survival time (28). Since gemcitabine remains the first line of chemotherapy, an understanding of its molecular mechanism is essential in developing new therapeutic approaches.
Chemotherapy resistance occurs primarily due to cellular evasion of apoptosis, one of the characteristics of cancer. Apoptosis is a genetically controlled physiological process characterised by its morphology and biochemical events, including cellular shrinkage, chromatin condensation and apoptotic body formation (29). In the development and regulation of multicellular organisms, apoptosis is an active and well-defined programmed cell death. However, the balance between proliferation and apoptosis is disrupted at certain stages in tumour development. The imbalance leads to deregulated cell proliferation and subsequent tumour formation (30). Therefore, induction of tumour cell apoptosis with limited or minimal toxicity to surrounding normal cells has been recognised as an effective cancer chemotherapy target. In the present study, we screened various gemcitabine concentrations in two pancreatic cancer cell lines and calculated the IC50 value. Considering the gemcitabine concentrations used in the clinic, we used 10 µM gemcitabine in subsequent experiments to induce various degrees of apoptosis throughout the time course.
Gemcitabine sensitivity has been reported to correlate with the activation of the p38 (31), c-Jun N-terminal kinase (32) and ERK (33) signalling pathways. Significantly, the AP-1 transcription factor is at the hub of the signalling network. Jun proteins preferentially regulate genes involved in proliferation and apoptosis, including Bim (34), Bcl-3 (35) and cyclin D1 (36), whereas Fos proteins, including DNA (cytosine-5)-methyltransferase 1 (37) and matrix metalloproteinase 1, are often required for angiogenesis and invasion by malignant tumours (38). To clarify the factors involved in gemcitabine-induced apoptosis, we analysed c-Jun and c-Fos. Western blot analysis demonstrated that gemcitabine increased c-Jun expression, but had little effect on c-Fos. Flow cytometric analysis and the alterations in cleaved PARP and caspase 7 expression revealed that c-Jun promotes gemcitabine-induced apoptosis.
Bcl-2 protein family members regulate apoptotic mitochondrial events (39). c-Jun has been demonstrated to lead to induction of pro-apoptotic molecules, including Bim. However, c-Jun functions as an anti-apoptotic or pro-apoptotic factor depending on the downstream targets. Immunoblotting assays indicated that c-Jun overexpression enhanced Bim activity, and the increase in Bim was inhibited when c-Jun expression decreased. However, other pro-apoptotic and anti-apoptotic factors, including Bcl-3 and Bax, were not affected by c-Jun (21). We also used SP600125 to reduce c-Jun activity and compared its effect with that of gemcitabine treatment. The results demonstrated that SP600125 treatment inhibited gemcitabine-induced apoptosis and attenuated the increase in Bim expression. Gemcitabine is a nucleoside analogue that causes cytotoxicity by inducing DNA replication blocks (40), and Bim suppression was reported to reduce cyclins and cyclin-dependent kinases, which would control DNA replication (41). Taken together, c-Jun regulates gemcitabine-induced apoptosis in pancreatic cancer by activating its downstream target Bim.
In summary, our results indicate that c-Jun is a pro-apoptotic protein that promotes gemcitabine-induced cell apoptosis by upregulating Bim activity and expression. Our study is the first to implicate the AP-1 pathway in gemcitabine-induced apoptosis, and we suggest that it plays the most significant role since c-Jun is at the crossroads with several signalling pathways. The effect of c-Jun in regulating gemcitabine-induced apoptosis is recognised for its potential value in treating pancreatic cancer.
Acknowledgements
This study was supported by a grant for the youth from the National Natural Science Foundation of China (81201734), the Research Special Fund for the Public Welfare Industry of Health (201402001), and the Research Fund for the Doctoral Program of Higher Education (20131106110008 and 20121106120048).
References
- 1.Jones OP, Melling JD, Ghaneh P. Adjuvant therapy in pancreatic cancer. World J Gastroenterol. 2014;20:14733–14746. doi: 10.3748/wjg.v20.i40.14733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 3.Silvestris N, Gnoni A, Brunetti AE, Vincenti L, Santini D, Tonini G, Merchionne F, Maiello E, Lorusso V, Nardulli P, et al. Target therapies in pancreatic carcinoma. Curr Med Chem. 2014;21:948–965. doi: 10.2174/09298673113209990238. [DOI] [PubMed] [Google Scholar]
- 4.Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardière C, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 5.Thota R, Pauff JM, Berlin JD. Treatment of metastatic pancreatic adenocarcinoma: a review. Oncology (Williston Park) 2014;28:70–74. [PubMed] [Google Scholar]
- 6.Lima CM Rocha, Green MR, Rotche R, Miller WH, Jr, Jeffrey GM, Cisar LA, Morganti A, Orlando N, Gruia G, Miller LL. Irinotecan plus gemcitabine results in no survival advantage compared with gemcitabine monotherapy in patients with locally advanced or metastatic pancreatic cancer despite increased tumor response rate. J Clin Oncol. 2004;22:3776–3783. doi: 10.1200/JCO.2004.12.082. [DOI] [PubMed] [Google Scholar]
- 7.Xu J, Wang T, Cao Z, Huang H, Li J, Liu W, Liu S, You L, Zhou L, Zhang T, Zhao Y. MiR-497 downregulation contributes to the malignancy of pancreatic cancer and associates with a poor prognosis. Oncotarget. 2014;5:6983–6993. doi: 10.18632/oncotarget.2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Al-Hajeili M, Azmi AS, Choi M. Nab-paclitaxel: potential for the treatment of advanced pancreatic cancer. Onco Targets Ther. 2014;7:187–192. doi: 10.2147/OTT.S40705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiorean EG, Von Hoff DD: Taxanes. impact on pancreatic cancer. Anticancer Drugs. 2014;25:584–592. doi: 10.1097/CAD.0000000000000073. [DOI] [PubMed] [Google Scholar]
- 10.Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, Collisson EA, Connor F, Van Dyke T, Kozlov S, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159:80–93. doi: 10.1016/j.cell.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Z, Li D, Zheng X, Wang E, Wang J. Selective induction of apoptosis: promising therapy in pancreatic cancer. Curr Pharm Des. 2013;19:2259–2268. doi: 10.2174/1381612811319120013. [DOI] [PubMed] [Google Scholar]
- 12.Bianco R, Melisi D, Ciardiello F, Tortora G. Key cancer cell signal transduction pathways as therapeutic targets. Eur J Cancer. 2006;42:290–294. doi: 10.1016/j.ejca.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 13.Nicholson DW. From bench to clinic with apoptosis-based therapeutic agents. Nature. 2000;407:810–816. doi: 10.1038/35037747. [DOI] [PubMed] [Google Scholar]
- 14.Zhao C, Qiao Y, Jonsson P, Wang J, Xu L, Rouhi P, Sinha I, Cao Y, Williams C, Dahlman-Wright K. Genome-wide profiling of AP-1-regulated transcription provides insights into the invasiveness of triple-negative breast cancer. Cancer Res. 2014;74:3983–3994. doi: 10.1158/0008-5472.CAN-13-3396. [DOI] [PubMed] [Google Scholar]
- 15.Shaulian E. AP-1 - the Jun proteins: oncogenes or tumor suppressors in disguise? Cell Signal. 2010;22:894–899. doi: 10.1016/j.cellsig.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 16.Chinenov Y, Kerppol TK. Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene. 2001;20:2438–2452. doi: 10.1038/sj.onc.1204385. [DOI] [PubMed] [Google Scholar]
- 17.Kolbus A, Herr I, Schreiber M, Debatin KM, Wagner EF, Angel P. c-Jun-dependent CD95-L expression is a rate-limiting step in the induction of apoptosis by alkylating agents. Mol Cell Biol. 2000;20:575–582. doi: 10.1128/MCB.20.2.575-582.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ren X, Song W, Liu W, Guan X, Miao F, Miao S, Wang L. Rhomboid domain containing 1 inhibits cell apoptosis by upregulating AP-1 activity and its downstream target Bcl-3. FEBS Lett. 2013;587:1793–1798. doi: 10.1016/j.febslet.2013.04.033. [DOI] [PubMed] [Google Scholar]
- 19.Shi R, Peng H, Yuan X, Zhang X, Zhang Y, Fan D, Liu X, Xiong D. Down-regulation of c-fos by shRNA sensitizes adriamycin-resistant MCF-7/ADR cells to chemotherapeutic agents via P-glycoprotein inhibition and apoptosis augmentation. J Cell Biochem. 2013;114:1890–1900. doi: 10.1002/jcb.24533. [DOI] [PubMed] [Google Scholar]
- 20.Leijen S, Veltkamp SA, Huitema AD, van Werkhoven E, Beijnen JH, Schellens JH. Phase I dose-escalation study and population pharmacokinetic analysis of fixed dose rate gemcitabine plus carboplatin as second-line therapy in patients with ovarian cancer. Gynecol Oncol. 2013;130:511–517. doi: 10.1016/j.ygyno.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 21.Meng Q, Xia Y. c-Jun, at the crossroad of the signaling network. Protein Cell. 2011;2:889–898. doi: 10.1007/s13238-011-1113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Passegué E, Jochum W, Schorpp-Kistner M, Möhle-Steinlein U, Wagner EF. Chronic myeloid leukemia with increased granulocyte progenitors in mice lacking junB expression in the myeloid lineage. Cell. 2001;104:21–32. doi: 10.1016/S0092-8674(01)00188-X. [DOI] [PubMed] [Google Scholar]
- 23.Kashatus D, Cogswell P, Baldwin AS. Expression of the Bcl-3 proto-oncogene suppresses p53 activation. Genes Dev. 2006;20:225–235. doi: 10.1101/gad.1352206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li J, Liang X, Yang X. Ursolic acid inhibits growth and induces apoptosis in gemcitabine-resistant human pancreatic cancer via the JNK and PI3K/Akt/NF-κB pathways. Oncol Rep. 2012;28:501–510. doi: 10.3892/or.2012.1827. [DOI] [PubMed] [Google Scholar]
- 25.Dai MH, Liu SL, Chen NG, Zhang TP, You L, Q Zhang F, Chou TC, Szalay AA, Fong Y, Zhao YP. Oncolytic vaccinia virus in combination with radiation shows synergistic antitumor efficacy in pancreatic cancer. Cancer Lett. 2014;344:282–290. doi: 10.1016/j.canlet.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 26.Chugh R, Sangwan V, Patil SP, Dudeja V, Dawra RK, Banerjee S, Schumacher RJ, Blazar BR, Georg GI, Vickers SM, Saluja AK. A preclinical evaluation of Minnelide as a therapeutic agent against pancreatic cancer. Sci Transl Med. 2012;4:156ra139. doi: 10.1126/scitranslmed.3004334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trouilloud I, Dubreuil O, Boussaha T, Lepère C, Landi B, Zaanan A, Bachet JB, Taieb J. Medical treatment of pancreatic cancer: new hopes after 10 years of gemcitabine. Clin Res Hepatol Gastroenterol. 2011;35:364–374. doi: 10.1016/j.clinre.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 28.Lee JG, Wu R. Erlotinib-cisplatin combination inhibits growth and angiogenesis through c-MYC and HIF-1α in EGFR-mutated lung cancer in vitro and in vivo. Neoplasia. 2015;17:190–200. doi: 10.1016/j.neo.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Evans GL, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- 31.Bu HQ, Luo J, Chen H, Zhang JH, Li HH, Guo HC, Wang ZH, Lin SZ. Oridonin enhances antitumor activity of gemcitabine in pancreatic cancer through MAPK-p38 signaling pathway. Int J Oncol. 2012;41:949–958. doi: 10.3892/ijo.2012.1519. [DOI] [PubMed] [Google Scholar]
- 32.Teraishi F, Zhang L, Guo W, Dong F, Davis JJ, Lin A, Fang B. Activation of c-Jun NH2-terminal kinase is required for gemcitabine's cytotoxic effect in human lung cancer H1299 cells. FEBS Lett. 2005;579:6681–6687. doi: 10.1016/j.febslet.2005.10.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang XL, Lin FJ, Guo YJ, Shao ZM, Ou ZL. Gemcitabine resistance in breast cancer cells regulated by PI3K/AKT-mediated cellular proliferation exerts negative feedback via the MEK/MAPK and mTOR pathways. Onco Targets Ther. 2014;7:1033–1042. doi: 10.2147/OTT.S63145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang H, Yang YB, Shen HM, Gu J, Li T, Li XM. ABT-737 induces Bim expression via JNK signaling pathway and its effect on the radiation sensitivity of HeLa cells. PLoS One. 2012;7:e52483. doi: 10.1371/journal.pone.0052483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rebollo A, Dumoutier L, Renauld JC, Zaballos A, Ayllón V, Martínez-A C. Bcl-3 expression promotes cell survival following interleukin-4 deprivation and is controlled by AP1 and AP1-like transcription factors. Mol Cell Biol. 2000;20:3407–3416. doi: 10.1128/MCB.20.10.3407-3416.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang W, Du Z, Yan J, Ma D, Shi M, Zhang M, Peng C, Li H. Mesenchymal stem cells promote liver regeneration and prolong survival in small-for-size liver grafts: involvement of C-Jun N-terminal kinase, cyclin D1, and NF-κB. PLoS One. 2014;9:e112532. doi: 10.1371/journal.pone.0112532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Niu Q, Liu H, Guan Z, Zeng Q, Guo S, He P, Guo L, Gao P, Xu B, Xu Z, et al. The effect of c-Fos demethylation on sodium fluoride-induced apoptosis in L-02 cells. Biol Trace Elem Res. 2012;149:102–109. doi: 10.1007/s12011-012-9392-y. [DOI] [PubMed] [Google Scholar]
- 38.Renaud SJ, Kubota K, Rumi MA, Soares MJ. The FOS transcription factor family differentially controls trophoblast migration and invasion. J Biol Chem. 2014;289:5025–5039. doi: 10.1074/jbc.M113.523746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ganesan V, Colombini M. Regulation of ceramide channels by Bcl-2 family proteins. FEBS Lett. 2010;584:2128–2134. doi: 10.1016/j.febslet.2010.02.032. [DOI] [PubMed] [Google Scholar]
- 40.Smith SC, Petrova AV, Madden MZ, Wang H, Pan Y, Warren MD, Hardy CW, Liang D, Liu EA, Robinson MH, et al. A gemcitabine sensitivity screen identifies a role for NEK9 in the replication stress response. Nucleic Acids Res. 2014;42:11517–11527. doi: 10.1093/nar/gku840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gautam S, Kirschnek S, Wiesmeier M, Vier J, Häcker G. Roscovitine-induced apoptosis in neutrophils and neutrophil progenitors is regulated by the Bcl-2-family members Bim, Puma, Noxa and Mcl-1. PLoS One. 2013;8:e79352. doi: 10.1371/journal.pone.0079352. [DOI] [PMC free article] [PubMed] [Google Scholar]