

Abstract
Acquired hemophilia A (AHA) is a relatively rare and life-threatening bleeding disorder whose pathogenesis is not completely understood. The present study reports a rare case of immunogubulin (IgG)4-related AHA with multisystemic involvement. A 55-year old male patient presented with symptoms of bronchial asthma and multiple subdermal hematomas. Chest computed tomography showed multiple diffuse nodular lesions with thickening of bronchovascular bundles, and scattered high-density spots in both lung lobes. Laboratory investigations showed increased activated partial prothrombin time (120.0 sec), a markedly decreased factor VIII (FVIII) activity (0.5%), a high-titer of FVIII inhibitor (27.2 Bethesda units/ml) and a marked increase in serum IgG4 (>4.03 g/l) level. Left inguinal lymph node biopsy revealed capsular thickening with marked lymphoplasmacytic infiltration, occlusive phlebitis and irregular fibrosis. Immunostaining revealed numerous IgG4-positive plasma cells (>100 cells/human plasma fibronectin) in the nodular lesions, with an IgG4/IgG ratio of >40%. The symptoms were markedly alleviated following corticosteroid therapy. The current study presents the first reported case of a rare IgG4-related AHA that presented with unusual clinical features and multisystemic involvement. The patient responded well to corticosteroid therapy. Documentation of such rare cases will help in characterizing the pathogenesis, and prompt recognition and timely treatment of this rare disorder.
Keywords: immunoglobulin 4-related disease, acquired hemophilia, bronchial asthma, lymph node, factor VIII, corticosteroid
Introduction
Acquired hemophilia A (AHA) is a relatively rare and life-threatening bleeding disorder caused by spontaneous development of autoantibodies against factor VIII (FVIII). The reported annual incidence of AHA is of the order of 1.5 individuals per million (1). The disease most commonly presents as spontaneous excessive hemorrhage in muscles, skin or soft tissues, or uncontrolled bleeding during surgery. To date, the pathogenesis of AHA remains unclear. There is no apparent underlying cause in ~50% of the reported cases; other cases are typically associated with autoimmune disorders, malignancy, adverse drug reactions and various skin diseases (2).
In 1993, a 78-year old male patient developed generalized lymphadenopathy accompanied with a disproportionately elevated serum IgG4 level. The IgG4-related disease has attracted increasing attention ever since (3). In addition to the elevated IgG4 levels, the disease is characterized by lymph node involvement due to lymphoplasmacytic infiltration with IgG4-positive plasma cells, marked interstitial fibrosis, eosinophilic infiltration and obliterative phlebitis of the terminal venules (4).
IgG4-related disease is a fibroinflammatory systemic disease which affects multiple organs, including the biliary system, salivary glands, lymph nodes, pancreas, retroperitoneum, periorbital tissue, lungs, meninges, aorta, breast, prostate, thyroid gland, pericardium, skin and kidney (3,5–12). Owing to multisystemic involvement, clinical manifestation of IgG4-related disease varies widely, and depends on the severity of the affected organs. An extensive literature search revealed only two documented cases of AHA with co-existing IgG4 related disease (13,14). Herein, a rare case of IgG4 -related AHA with multisystemic involvement is described that presented with a myriad of clinical characteristics. The clinical relevance of AHA has been discussed to provide a better understanding of this rare disorder.
Case report
A 55-year-old Chinese male, who provided written informed consent, presented at Shanxi DAYI Hospital of Shanxi Medical University (Shanxi, China) in November 2014 with a history of chronic cough since half a year ago with no obvious cause. The cough tended to aggravate at night. In the previous 1 month there was progressive aggravation of cough, which could be induced by cooking fumes or pungent odor. Six days prior to admission, the patient caught a cold with further aggravation of cough, but without fever, hemoptysis, chest pain, palpitations and sweating. The patient did not respond to antibiotic therapy prescribed at a local health clinic. On examination, the patient was not febrile (36.5°C), and his vitals were stable with a systolic blood pressure of systolic 141 mmHg and diastolic 94 mmHg. There were no signs of inflammation of pharyngeal mucosa or tonsillar enlargement. His systemic examination was unremarkable except for the presence of a small palpable submandibular lymph node and an enlarged left inguinal lymph node measuring 3×1 cm. His abdomen was soft, non-tender with non-palpable liver and spleen. The result of bronchial provocation test was positive and the patient was diagnosed as having bronchial asthma.
The patient had a significant past medical history, including hospitalization for autoimmune hepatitis with serum alanine aminotransferase (ALT) 421 IU/ml and aspartate aminotransferase (AST) 600 IU/ml. Anti-smooth muscle antibody was positive (titre, 1:100). Abdominal computed tomography (CT) showed multiple enlarged lymph nodes in the ligament of liver and stomach, around the abdominal aorta and bilateral inguinal regions, with the left inguinal region being more affected. The upper pancreatic bile duct was widened, indicating a possibility of cholecystitis or cholangitis. Furthermore, there was thickening and consolidation in the wall of left ureter, indicating a possibility of a space occupying lesion (SOL) of the ureter, accompanied with an inflammatory response in the renal pelvis and the upper ureter.
Magnetic resonance cholangiopancreatography revealed multiple gallbladder and bile duct calculi, intra- and extrahepatic bile duct dilation, and abnormal signals in caput pancreatis accompanied with multiple enlarged small lymph nodes around the abdominal artery. Based on these findings, the patient was diagnosed with gallbladder and cystic duct calculi, acute cholecystitis and acute pancreatitis. The symptoms of the patient improved with anti-inflammatory drugs and supportive treatment, with restoration of normal serum ALT (25.2 IU/ml) and AST (19.4 IU/ml) levels.
To rule out the possibility of SOL in the left ureter, the patient underwent cystoscopic examination with double J tube placement. However, there was no evidence of SOL. The patient also had urticaria, diabetes and low triiodothyronine (T3) levels.
Positron emission tomography (PET)/CT performed three months prior to admission showed abnormal hypermetabolic signals in multiple organ systems (including bone marrow, multiple lymph nodes, parotid gland, lung, gallbladder, bile duct, pancreas, prostate and testis), left hydronephrosis and dilatation of the upper left ureter. Assays for serum lupus anticoagulant combination, antinuclear antibody, antineutrophil cytoplasmic antibodies, immunoglobulin and complement levels were all negative. Three days after admission, the patient developed subcutaneous hemorrhages and pain in the right hip (Fig. 1). Ultrasound examination revealed a subdermal hematoma over the right hip measuring 9×3x5 cm. On detailed enquiry, the patient revealed a past history of spontaneous bleeding in bilateral upper extremities 2 months prior to admission. Chest CT showed multiple diffuse nodular lesions with thickening of bronchovascular bundles, and multiple scattered high density spots in both lung lobes. In addition, there was multiple lymph node enlargement in the mediastinum and pulmonary hila (Fig. 2).
Figure 1.
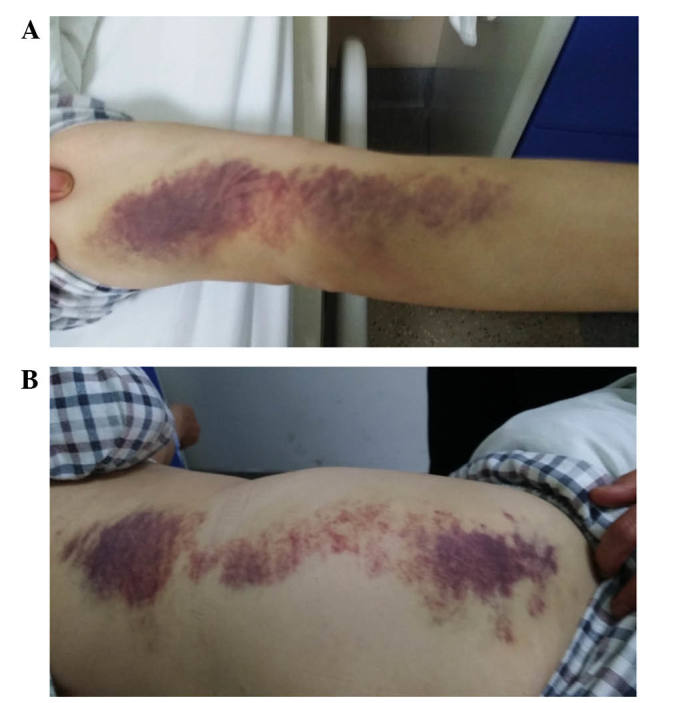
Subdermal hematoma in immunoglubulin 4-related acquired hemophilia. Large diffuse subdermal hematoma on the patient's (A) arm and (B) right waist and hip.
Figure 2.

Chest CT showing multiple nodular lesions and thickening of bronchovascular bundles in bilateral lung lobes. (A) Chest CT 3 months before admission showing multiple diffuse nodular lesions with thickening of bronchovascular bundles and scattered high-density spots in both lung lobes. (B) Chest CT at admission showing more diffuse nodular lesions with thickening of bronchovascular bundles and scattered high-density spots in both lung lobes compared with at 3 months before admission. (C) Chest CT one month after admission showing markedly diminished nodular lesions and scattered high-density spots in both lung lobes. CT, computed tomography.
Laboratory investigations at admission showed activated partial prothrombin time (APTT) of 120.0 sec (normal range, 24.0–40.0 sec), but with normal prothrombin time, thrombin time and fibrinogen levels. The platelet count was also normal. Coagulation factor assay revealed a markedly decreased factor VIII (FVIII) activity at 0.5% (normal range, 60–150%), and a high-titer of FVIII inhibitor at 27.2 Bethesda units/ml (BU/ml) (normal range, 0–0.6 BU/ml). In addition, serum IgG4 level was markedly increased (>4.03 g/l).
Left inguinal lymph node biopsy revealed capsular thickening, marked lymphoplasmacytic infiltration with irregular fibrosis and obliterative phlebitis (Fig. 3). Further positive immunostaining for human herpes virus type 8, Lambda and Kappa light chains showed reactive lymphoid hyperplasia in the nodular lesion (Fig. 4). In addition, immunohistochemical staining for IgG4 revealed numerous IgG4-positive plasma cells [>100 cells/human plasma fibronectin (HPF)] in the nodular lesion, with an increased IgG4/IgG ratio of >40% (Fig. 5). A diagnosis of IgG4-related AHA was rendered.
Figure 3.

Histological examination of the left inguinal lymph node biopsy specimen before treatment. (A) Hematoxylin and eosin staining showing a marked diffuse proliferation of large atypical lymphoid cells and lymphoplasmacytic infiltration with irregular fibrosis. (B) Marked thickening of lymph node capsule. (C) Obliterating phlebitis. (D) Reduced area of germinal centers. Magnification, ×100.
Figure 4.

Immunostaining for the HHV, Lambda and Kappa light chains in a biopsy specimen from the left inguinal lymph node before treatment. (A) Negative immunostaining for HHV. Magnification, ×100. (B) Kappa and (C) lambda positive immunostaining for Lambda and Kappa showing similar proportions of positive cells, indicating a reactive lymphoid hyperplasia. Magnification, ×200. HHV, human herpes virus.
Figure 5.

Immunostaining for IgG and IgG4 of the biopsy specimen of the left inguinal lymph node before treatment. (A) Immunostaining for IgG showing numerous IgG positive plasma cells. Magnification, ×200. (B) Immunostaining for IgG4 showing numerous IgG4-positive plasma cells in the nodular lesion, with an increased IgG4/IgG ratio (>40%). Magnification, ×100. IgG, immunoglobulin.
IgG4-related disease is responsive to corticosteroid therapy. The patient was treated with oral prednisone (initial dose, 40 mg/day; maintenance dose, 15 mg/day; cat. no. H33021207; Zhejiang Xianju Pharmaceutical Co., Ltd., Taizhou, China). After one week of treatment, the patient's symptoms were relieved. After 2 weeks of treatment, the activated partial thromboplastin time was within the normal range. Furthermore, one month after treatment, chest CT scan showed clearing of the nodular lesions and lymph nodes. There was no instance of relapse during follow-up (Table I) after one and three months.
Table I.
Key laboratory parameters on follow up in IgG4-related acquired hemophilia.
| Follow-up | CRP (mg/l) | ESR (mm/h) | IgG (g/l) | IgG4 (g/l) | APTT (sec) | FVIII (%) | FVIII inhibitor (BU/ml) |
|---|---|---|---|---|---|---|---|
| Normal range | 0–8 | 0–20 | 7.51–15.6 | 0.03–2.01 | 25.1–36.5 | 50–150 | 0–0.6 |
| Nov 2014 | <2.5 | 66 | 21.40 | >4.030 | 120.0 | 0.5 | 27.2 |
| Dec 2014 | <2.5 | 8 | 14.80 | >4.030 | 31.7 | 50.0 | 8.0 |
| Mar 2015 | <2.5 | 6 | 12.0 | >4.040 | 30.0 | 70.8 | <0.6 |
CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; IgG, immunoglobulin; APTT, activated partial thromboplastin time; FVIII, factor VIII.
Discussion
Since the first reported case of IgG4-related disease in 1993 (3), there have been numerous reports of complicated diseases overlapping this clinical entity (14,15). Although the understanding of IgG4-related disease has rapidly increased, its etiology remains elusive. According to the clinicopathological characteristics of IgG4-related disease, its proposed comprehensive diagnostic criteria consists of the following (14,16): i) Diffuse or localized swelling or masses in single or multiple organs on clinical examination; ii) elevated serum IgG4 level (≥135 mg/dl); and iii) marked lymphocyte and plasmacyte infiltration, fibrosis and infiltration of IgG4-positive plasma cells with a ratio of IgG4/IgG positive cells >40%, and >10 IgG4-positive plasma cells/HPF on histopathological examination.
AHA is a rare but life-threatening hemorrhagic disorder caused by the presence of spontaneous antibodies against FVIII (17). Although IgG4-related disease can potentially affect any organ besides co-existence with other complicated diseases; it is extremely rare for this condition to overlap AHA disease. Only two documented cases of AHA overlapping with IgG4 related disease have been identified in the literature (13,14). In the present case, the patient was hospitalized because of developing bronchial asthma. Subsequently, multiple subcutaneous hemorrhages occurred. The patient was diagnosed with AHA based on the laboratory findings that included markedly increased APTT, markedly decreased factor VIII (FVIII) activity, and a high-titer of FVIII inhibitor. Notably, the clinicopathological characteristics of the patient fulfilled all the diagnostic criteria of IgG4-related disease described above. A review of the patient's past history revealed multiple organ diseases, including gallbladder and cystic duct calculi, acute cholecystitis, acute pancreatitis, urticaria, diabetes and low T3 syndrome. Considering that IgG-4-related disease could affect any organ and can present with myriad manifestations, it is suggested that the multiple organ disease identified in this patient was caused by IgG4-related AHA. The specific treatment of IgG4-related AHA resulted in the following response: i) The manifestations of IgG4-related AHA, such as cough, nausea, abdominal pain and subcutaneous hemorrhage, were completely resolved; ii) blood glucose level and thyroid function were restored to normal range and remained stable, unlike that in IgG4-related disease only, which frequently involves the pancreas and thyroid gland, causing diabetes and low T3 syndrome; iii) the specific serological markers of IgG4-related AHA, such as IgG, APTT, FVIII and FVIII inhibitor were within normal range; iv) the abnormal hypermetabolic signals in multiple organ systems identified by PET/CT were alleviated.
IgG4-related disease is a glucocorticoid-responsive disorder (18), and this was well manifested in the present case. The initial findings of multiple diffuse nodular lesions with thickening of bronchovascular bundles and scattered high-density spots were markedly diminished in both lung lobes, and there was a decrease in the lymph nodes. The patient's symptoms further improved and there was no relapse on follow-up. The efficacy of glucocorticoid treatment in multiple organ diseases in this patient was consistent with the diagnosis of IgG4-related AHA.
However, the serum IgG4 level remained high even after glucocorticoid treatment, which was not consistent with most of the other documented cases. Although IgG4-related disease typically demonstrates high serum IgG4 levels, approximately 20% of patients with biopsy-proven IgG4-related disease may have normal serum IgG4 level (15,19,20). A review of the literature revealed a case report wherein a patient was diagnosed with IgG4-related sclerosing disease, despite serum IgG4 levels remaining normal. The patient responded to corticosteroid treatment (prednisolone) with alleviation of pulmonary lesions and improved renal function; however, the serum IgG4 level increased (6). A plausible explanation for the seronegativity of the patient may be that active synthesis blocked IgG4 secretion, and then corticosteroid treatment suppressed the synthesis, which restored IgG4 secretion from the plasma cells. Thus, IgG4-related disease may have various pathological features, and is likely to be associated with a variable response to treatment. The current patient is currently being followed-up with monitoring of serological markers, including IgG4.
In conclusion, the present study describes the first reported case of IgG4-related AHA in a 55-year-old male who presented with unusual clinical features and systemic manifestations. Awareness of such an entity is necessary as it is a curable disease, and timely treatment could be life-saving. Corticosteroids remain the mainstay of the treatment. Documentation of such rare cases will help in further characterizing the pathogenesis of this rare disorder.
Acknowledgements
The authors thank Medjaden Bioscience Limited for scientific editing of the manuscript.
References
- 1.Collins P, Macartney N, Davies R, Lees S, Giddings J, Majer R. A population based, unselected, consecutive cohort of patients with acquired haemophilia A. Br J Haematol. 2004;124:86–90. doi: 10.1046/j.1365-2141.2003.04731.x. [DOI] [PubMed] [Google Scholar]
- 2.Aljasser MI, Sladden C, Crawford RI, Au S. Bullous pemphigoid associated with acquired hemophilia a: A rare association of autoimmune disease. J Cutan Med Surg. 2014;18:123–126. doi: 10.2310/7750.2013.13060. [DOI] [PubMed] [Google Scholar]
- 3.Suzuki S, Kida S, Ohira Y, Ohba T, Miyata M, Nishimaki T, Morito T, Kasukawa R, Hojyo H, Wakasa H. A case of Sjögren's syndrome accompanied by lymphadenopathy and IgG4 hypergammaglobulinemia. Ryumachi. 1993;33:249–254. [PubMed] [Google Scholar]
- 4.Zen Y, Inoue D, Kitao A, Onodera M, Abo H, Miyayama S, Gabata T, Matsui O, Nakanuma Y. IgG4-related lung and pleural disease: A clinicopathologic study of 21 cases. Am J Surg Pathol. 2009;33:1886–1893. doi: 10.1097/PAS.0b013e3181bd535b. [DOI] [PubMed] [Google Scholar]
- 5.Ryu JH, Sekiguchi H, Yi ES. Pulmonary manifestations of immunoglobulin G4-related sclerosing disease. Eur Respir J. 2012;39:180–186. doi: 10.1183/09031936.00025211. [DOI] [PubMed] [Google Scholar]
- 6.Khosroshahi A, Stone JH. A clinical overview of IgG4-related systemic disease. Curr Opin Rheumatol. 2011;23:57–66. doi: 10.1097/BOR.0b013e328341a229. [DOI] [PubMed] [Google Scholar]
- 7.Raissian Y, Nasr SH, Larsen CP, Colvin RB, Smyrk TC, Takahashi N, Bhalodia A, Sohani AR, Zhang L, Chari S, et al. Diagnosis of IgG4-related tubulointerstitial nephritis. J Am Soc Nephrol. 2011;22:1343–1352. doi: 10.1681/ASN.2011010062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khosroshahi A, Deshpande V, Stone JH. The clinical and pathological features of IgG (4)-related disease. Curr Rheumatol Rep. 2011;13:473–481. doi: 10.1007/s11926-011-0213-7. [DOI] [PubMed] [Google Scholar]
- 9.Zen Y, Nakanuma Y. IgG4-related disease: A cross-sectional study of 114 cases. Am J Surg Pathol. 2010;34:1812–1819. doi: 10.1097/PAS.0b013e3181f7266b. [DOI] [PubMed] [Google Scholar]
- 10.Kamisawa T, Okamoto A. Autoimmune pancreatitis: Proposal of IgG4-related sclerosing disease. J Gastroenterol. 2006;41:613–625. doi: 10.1007/s00535-006-1862-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamano H, Kawa S, Ochi Y, Unno H, Shiba N, Wajiki M, Nakazawa K, Shimojo H, Kiyosawa K. Hydronephrosis associated with retroperitoneal fibrosis and sclerosing pancreatitis. Lancet. 2002;359:1403–1404. doi: 10.1016/S0140-6736(02)08359-9. [DOI] [PubMed] [Google Scholar]
- 12.Takeda S, Haratake J, Kasai T, Takaeda C, Takazakura E. IgG4-associated idiopathic tubulointerstitial nephritis complicating autoimmune pancreatitis. Nephrol Dial Transplant. 2004;19:474–476. doi: 10.1093/ndt/gfg477. [DOI] [PubMed] [Google Scholar]
- 13.Nagao Y, Yamanaka H, Harada H. A patient with hypereosinophilic syndrome that manifested with acquired hemophilia and elevated IgG4: A case report. J Med Case Rep. 2012;6:63. doi: 10.1186/1752-1947-6-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sugino K, Gocho K, Ishida F, Kikuchi N, Hirota N, Sato K, Sano G, Isobe K, Sakamoto S, Takai Y, et al. Acquired hemophilia A associated with IgG4-related lung disease in a patient with autoimmune pancreatitis. Intern Med. 2012;51:3151–3154. doi: 10.2169/internalmedicine.51.8133. [DOI] [PubMed] [Google Scholar]
- 15.Sekiguchi H, Horie R, Aksamit TR, Yi ES, Ryu JH. Immunoglobulin G4-related disease mimicking asthma. Can Respir J. 2013;20:87–89. doi: 10.1155/2013/619453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Umehara H, Okazaki K, Masaki Y, Kawano M, Yamamoto M, Saeki T, Matsui S, Yoshino T, Nakamura S, Kawa S, et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol. 2012;22:21–30. doi: 10.3109/s10165-011-0571-z. [DOI] [PubMed] [Google Scholar]
- 17.Kessler CM. Acquired factor VIII autoantibody inhibitors: Current concepts and potential therapeutic strategies for the future. Haematologica. 2000;85:S57–S61. (10 Suppl) discussion 61–63. [PubMed] [Google Scholar]
- 18.Umehara H, Okazaki K, Masaki Y, Kawano M, Yamamoto M, Saeki T, Matsui S, Sumida T, Mimori T, Tanaka Y, et al. A novel clinical entity, IgG4-related disease (IgG4RD): General concept and details. Mod Rheumatol. 2012;22:1–14. doi: 10.3109/s10165-011-0508-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamisawa T, Takuma K, Tabata T, Inaba Y, Egawa N, Tsuruta K, Hishima T, Sasaki T, Itoi T. Serum IgG4-negative autoimmune pancreatitis. J Gastroenterol. 2011;46:108–116. doi: 10.1007/s00535-010-0317-2. [DOI] [PubMed] [Google Scholar]
- 20.Ebbo M, Daniel L, Pavic M, Sève P, Hamidou M, Andres E, Burtey S, Chiche L, Serratrice J, Longy-Boursier M, et al. IgG4-related systemic disease: Features and treatment response in a French cohort: Results of a multicenter registry. Medicine (Baltimore) 2012;91:49–56. doi: 10.1097/MD.0b013e3182433d77. [DOI] [PubMed] [Google Scholar]