

Short abstract
There has been considerable progress in finding microRNA targets computationally, but assessing success and biological significance requires a move into the 'wet' lab.
Abstract
Given that microRNAs select their targets by nucleotide base-pairing, it follows that it should be possible to find microRNA targets computationally. There has been considerable progress, but assessing success and biological significance requires a move into the 'wet' lab.
Traditional thinking regarding gene regulation was shaken by the recent discovery of microRNAs (miRNAs), an abundant class of endogenous 21-22-nucleotide RNAs that mediate post-transcriptional regulation via components of the RNA-interference (RNAi) pathway, either by directing target transcript cleavage or by translational inhibition (Figure 1a) [1,2]. Early miRNA discovery relied upon cloning and sequencing of small RNAs to find those whose corresponding genomic loci adopted an extended hairpin structure as RNA; such a structure is an obligate precursor to the mature miRNA. Effective computational methods were later developed that recognize miRNA precursors as a particular class of evolutionarily conserved RNA hairpins. Hundreds of different miRNAs have now been identified in complex eukaryotes, implying that they mediate a vast network of unappreciated regulatory interactions. Nevertheless, the in vivo functions and biologically relevant target genes are thus far known only for a few miRNAs. Given that target selection is guided by the miRNA sequence, can miRNA targets be predicted informatically?
Figure 1.
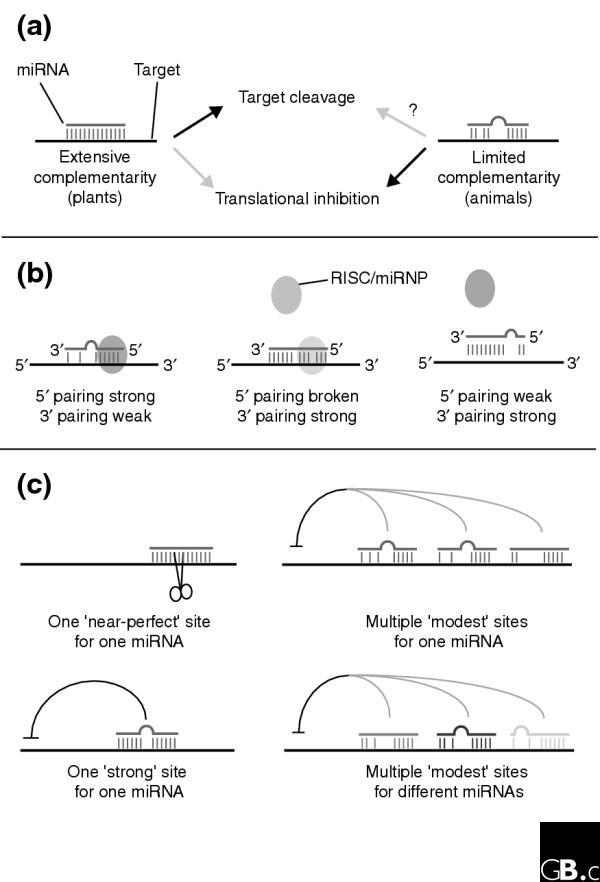
Complementarity of miRNAs and their targets. (a) Two classes of miRNA-binding site. Some miRNA-binding sites have extensive, near-perfect complementarity to miRNAs (left), whereas other miRNA-binding sites display only limited pairing to a miRNA (right). In many cases, the former leads to target cleavage while the latter causes translational inhibition (black arrows), although converse examples have also been described (gray arrows). Plant miRNAs commonly show extensive pairing to targets, whereas this is exceedingly uncommon for animal miRNAs. (b) Strong canonical base-pairing to the 5' end of a miRNA (nucleotides 2-8) is important for regulation of sites with limited complementarity. This is presumably due to specific recognition of the 5' end of the miRNA-target duplex by components of the RISC/miRNP complex (oval). It should be noted that RISC/miRNP may have physical contact along the entire miRNA:target duplex. An approximately seven-nucleotide duplex with the 5' end of a miRNA may in fact be sufficient for target recognition (left). Imperfect 5' pairing renders most sites nonfunctional, although in some cases, site functionality is 'rescued' by sufficiently extensive 3' pairing (middle). Sites that lack strong 5' pairing are nonfunctional, regardless of the degree of 3' pairing (right). (c) Examples of different configurations of miRNA-binding sites. Individual sites need to be considered within the milieu of other miRNA-binding sites present on a given transcript. Single sites can suffice for target cleavage (upper left) or strong translational inhibition (lower left), but these are typically 'near-perfect' or 'strong' sites that present extended complementarity to the miRNA. Multiple 'modest' sites that maintain 5' pairing to the miRNA can act synergistically and together confer strong regulation (upper right). Some transcripts contain multiple binding sites for different miRNAs (designated in different shades), which might also function synergistically (lower right).
Plant miRNAs have it easy
An early success in the bioinformatic hunt for miRNA target genes came in plants. In late 2002, it was reported that probable targets of most plant miRNAs were found simply by searching for highly complementary sequences in mRNA coding sequences or untranslated regions [3]; 61 putative targets were identified by looking for Arabidopsis mRNAs with three or fewer mismatches to a miRNA, with gaps and non-Watson-Crick base-pairs (G:U) not allowed. The validity of these predicted target sites was argued largely on the basis of their conservation in orthologous rice transcripts and from the observation that similar searches using randomly permuted miRNAs identified only 4.4 sites in the Arabidopsis transcriptome. When the set of possible targets with only one or two mismatches to the miRNAs was considered, 30 target sites were identified, compared to 0.2 for random 21-mers - a signal-to-noise ratio of 150:1.
It is now firmly established that highly complementary miRNA-binding sites mediate biologically relevant negative regulation in plants. Experimental evidence includes miRNA-directed cleavage of targets, down-regulation of target transcripts and phenocopy of the effects of target loss-of-function by miRNA ectopic expression, and creation of gain-of-function alleles by silent mutation of miRNA binding sites [4-7]. Experimental work also showed that limited G:U pairing, bulged nucleotides, and/or mismatches between miRNA and target are tolerated. A more comprehensive follow-up informatic study allowed for these types of mispairings, but required site conservation in rice; many new sites were thus identified and validated [8]. It is now believed that most plant miRNA targets, or at least those with extensive complementarity to miRNAs, have been identified [8,9].
Genetics identifies the first animal miRNA targets
The first miRNAs were actually characterized in nematodes in the 1990s, long before the concepts of 'miRNAs' and 'RNAi' even existed. In stark contrast to plant miRNAs, the founding miRNAs lin-4 and let-7 regulate gene expression through quite modestly complementary sites in the 3' untranslated regions (UTRs) of their target genes [10-12]. It is now abundantly clear that animal miRNAs do not generally exhibit extensive complementarity to any endogenous transcripts (Figure 1a). How were direct targets of lin-4 and let-7 then identified? Genetics was the key: their loss-of-function phenotypes showed that they regulate the timing of developmental transitions, and genetic interactions implicated a coherent set of genes as regulatory targets. This biological context enabled the identification of major developmental timers, including lin-14, lin-28, lin-41 and hbl-1, as miRNA targets [10-14]. Similar genetic analyses led to the finding that the worm miRNA lsy-6 regulates left-right neuronal asymmetry by targeting cog-1 [15], and aided the identification of the pro-apoptotic gene hid as a biologically relevant target of the Drosophila miRNA bantam [16].
In a converse set of findings, gain-of-function alleles that result in abnormal fly neuronal patterning led to the discovery of multiple families of 3' UTR motifs (Brd boxes, GY boxes and K boxes) that negatively regulate two large classes of target genes of the Notch pathway [17,18]. These motifs are six or seven nucleotides in length, lack degeneracy, and are specifically conserved in orthologous insect transcripts. Each of these motifs was subsequently appreciated to be perfectly complementary to members of three different miRNA families [19], and several of the targets have since been experimentally demonstrated to be negatively regulated by miRNAs and the RNAi pathway [20,21]. Similar thinking also helped to identify miR-273 as a negative regulator of die-1, which encodes a transcription factor that itself activates the miRNA lsy-6 during left-right asymmetric neuronal patterning in the worm [22].
The fly study [19], aside from being the first "informatic" determination of animal miRNA targets, made two additional vital observations. First, it was invariably the 5' end of the miRNA that is complementary to the 3' UTR regulatory motif, with a stretch of more than seven nucleotides of contiguous pairing in each case (Figure 1b). Second, base-pairing within this region is canonical, with no G:U base-pairs seen. The importance of the 5' miRNA end for target recognition was further suggested by the finding that, in many cases, miRNAs could be grouped according to their homologous 5' ends (for example, [19]), and that point mutations in lin-4 and let-7 loss-of-function alleles are found in the 5' region [10,11].
Algorithms to predict animal miRNA targets
Intense efforts have recently been dedicated to systematic bioinformatic identification of animal miRNA targets [20,23-27]. The common initial step is to assess and rank-order target 3' UTR complementarity to a miRNA by either duplex free energy and/or number of paired nucleotides, typically with some requirement or reward given to pairing to the 5' portion of the miRNA. The output of independent analyses of common datasets varies significantly, though, as the various algorithms perform and evaluate RNA foldings in different ways, make different allowances for bulges and loops in the duplexes, and promote 5' pairing to different extents. For example, the way that complementarity to the miRNA 5' end is treated varies considerably, from requiring a seven-nucleotide block of perfect Watson-Crick complementarity with the target [23], requiring an eight-nucleotide block but allowing G:U basepairs [20], allowing for G:U basepairs along with certain mismatches and/or bulges in the 5' region in the context of dynamically weighted pairing [24,26], or not specifically demanding or weighting 5' pairing at all [25].
Simple 'matching' of miRNAs to mRNAs in a single genome is ineffectual, since individual, genuine animal miRNA regulatory sites do not display a statistically significant amount of complementarity [20]. Confidence in a given target increases if it is conserved between species, however, because sequence preservation within UTRs signals potential functional constraint. For instance, all of the known binding sites of lin-4, let-7, bantam, miR-2, miR-7, and lsy-6 are conserved between closely related species. In particular, Lewis and colleagues [23] empirically determined that only those 7-mers that pair with the 5' end of genuine miRNAs (positions 2-8 and to a lesser extent, 1-7 or 3-9) are preferentially conserved when compared with equivalent matches in randomized miRNAs, strongly implying that these positions of the miRNA are most critical for target recognition (Figure 1b).
One disadvantage of applying the 'conservation filter' is that experimental evidence of the extent of 3' UTRs is often lacking in less-characterized species, so that for a majority of transcripts in certain species, an arbitrarily designated (and possibly incorrect) 3' UTR of a few kilobases downstream from the stop codon had to be used. In addition, a significant fraction of each predicted 3' UTR generated in this way overlaps coding sequences in some species, so that apparent conservation does not necessarily reflect regulatory constraint. Also, one should bear in mind that site divergence does not necessarily indicate that the site is nonfunctional, or that it confers quantitatively less regulation than a given conserved site.
Another strategy is to concern oneself primarily with targets that contain multiple sites, which can be factored in as a cumulative score for a given 3' UTR. While plant targets contain single miRNA-binding sites as a rule, many animal miRNA targets bear multiple binding sites. Selecting multiply-hit targets allows one to cherry-pick the 'good-looking' ones. Single sites are also known to confer regulation in vivo, however, and so should not be disregarded. Finally, the potential coordinate regulation of either paralogous genes or multiple genes in a common signaling or biochemical pathway is also a potentially useful feature for identifying compelling candidates. In Drosophila, many Notch target genes are regulated by miR-7 and probably other families of miRNAs; multiple pro-apoptotic genes are targeted by miR-2; and many enzymes involved in branched-chain amino-acid degradation contain predicted sites for binding miR-277 [19,20,24]. In general, then, these sorts of features increase one's confidence in selected candidates, but at the probable expense of discarding genuine targets.
Strategies for miRNA target validation
All miRNA target-finders return lists of candidate target genes. The vital question is, how valid is their output? Can computational approaches be validated in silico? The various methods easily recover the known targets of lin-4, let-7 and bantam when assessed at the genome level, suggesting that they do 'work'. But this evaluation is not only circular but also based on a small and potentially highly biased reference set of miRNAs with unusually strong target interactions. Few novel candidates display the number of target sites seen with, say, lin-14 (seven lin-4 sites) or hid (five bantam sites). One may also ask if more targets are predicted for genuine miRNAs than for randomly permuted miRNAs. This is indeed the case in the different published studies, and is taken to reflect an underlying biological signal. It is instructive to note, however, where the signal derives from. Four of the studies find around 1.3 times as many targets for genuine miRNAs as for randomized miRNAs when analyzing a single genome [20,24,26,27], suggesting that real miRNAs 'match' mRNAs better than might be expected by chance. On the other hand, Lewis and colleagues [23] caution against generating signal by 'matching'. They point out that sequence shuffling without strict regard for dinucleotide (or higher-order) sequence bias inherently decreases miRNA: mRNA matching, creating artifactual signal. Importantly, this is amplified with multiple observations, either as multiple sites in a given 3' UTR or as site conservation in a second genome. For example, in an extreme model where a 1.3:1 signal of single sites in a single genome is entirely artificial, this translates into a spurious (1.3)4:1 or 2.9:1 signal when conserved, two-site targets are considered. They therefore deliberately generated shuffled control miRNAs that have the same number of hits in a single genome as genuine miRNAs, and derived their signal only from asking whether predicted sites of genuine miRNAs were preferentially conserved [23].
In any case, there is modest or no overlap amongst top-predicted targets when similar genomes were analyzed (for example, groups of flies [20,24] or mammals [23,26,27]). The success of a computational approach therefore necessarily rests upon experimental validation of novel targets. From the geneticist's point of view, stringent tests might be for a given miRNA loss-of-function mutant to display corresponding misregulation of its predicted target(s), and for mutation of miRNA-binding sites in cis to at least partially phenocopy miRNA loss-of-function. This level of evidence is impractical to obtain on the genomic scale in any animal species; moreover, functional overlap between miRNAs could complicate experimental interpretation.
As an easier alternative, many have implemented tissue-culture assays using reporter gene constructs fused to target sequences. If such a construct is actively regulated by miRNAs already present in the transfected cells, one might expect it to produce lower levels of the reporter than a control construct. A more rigorous test asks if point mutation of target sites in such reporters increases their activity, which might indicate relief from endogenous miRNA-mediated regulation. Finally, for those miRNAs not normally expressed in tissue-culture cells, one can ask if reporter product levels are reduced in response to ectopically expressed miRNAs. Using the first assay, the Mourelatos/Hatzigeorgiou group [26] initially validated 3 out of 14 predicted targets, but later confirmed 7/7 top-predicted candidates from a refined list of 222 candidates. The Burge/Bartel [23] group validated 11/15 targets using the latter two assays; these 15 were selected as an unbiased subset of 451 pan-mammalian conserved targets, indicating that three-fourths of their target list should be genuine.
The Cohen group [16,20] also validated one interaction in vitro, but the strength of their studies was in testing miRNA-mediated regulation in vivo using transgenic flies. Their main assay asked if the levels of a ubiquitously expressed reporter bearing a test 3' UTR was affected by misexpression of an miRNA in a spatially delimited pattern. Six candidates (comprising pro-apoptotic genes and Notch target genes) were downregulated specifically in miRNA-expressing cells [16,20].
The different studies varied in the types, quality and context of sites that were tested. The Mourelatos/Hatzigeorgiou group [26] inserted individual 15-25 nucleotide binding sites into a completely heterologous 3' UTR, the Burge/Bartel [23] group placed 100-1,100 nucleotide 3' UTR segments typically bearing two or more predicted miRNA-binding sites into a heterologous 3' UTR, and the Cohen group [16,20] tested endogenous, complete 3' UTRs containing either single or multiple miRNA-binding sites. These different strategies have different merits, but as mentioned earlier it will probably fall to miRNA loss-of-function genetics to tell us how important any of these new miRNA:target interactions is in vivo.
Considerations for next-generation target-finding studies
Despite the recent progress, most miRNA researchers will agree that we have insufficient knowledge of how miRNAs identify their targets in vivo. This makes it a tough assignment for the informatician to assemble the desired program to predict targets. Two recent studies provide the first systematic studies of the requirements for miRNA: mRNA-target pairing [26,28]. Several unexpected insights emerge that support the notion that miRNA:target interaction is not a simple consequence of nucleic-acid hybridization. Nevertheless, the conclusions are not wholly consistent, indicating that a complete understanding of miRNA target selection is still to come.
The data [26,28] provide experimental support for the idea that pairing to the 5' region of the miRNA is a major determinant in target selection (Figure 1b). But a surprise was that the presence of G:U base-pairs in the 5' miRNA region decreased target regulation far above its thermodynamic effect on duplex formation [28]. Thus, predicted targets from schemes that allow for G:U or mismatched base-pairs in this region cannot be considered equivalent to those with perfect Watson-Crick pairing. Nevertheless, G:U base-pairs to the miRNA-targeting region cannot be totally discounted either. For example, demanding Watson-Crick pairing to the miRNA-targeting region causes one to miss the perfect complementarity between miR-196 and its bona fide target hoxb8 [29]. A further complication is that certain functional miRNA-binding sites (including one of the let-7 sites in lin-41) actually contain a bulge in the middle of the targeting region [10,26]. This phenomenon needs to be investigated further to resolve it with the so-called 5'-pairing rule. A possible resolution may be that 5' broken sites (Figure 1b) are nonfunctional unless extensive 3' pairing is present.
Another controversial point regards the general appearance of a miRNA-binding site (Figure 1b). One study concluded that miRNA-binding sites consist of an RNA duplex with a central bulge of prescribed lengths on either the miRNA or target side [26], a description that certainly fits some of the published target sites. But the other study [28] demonstrated that pairing to the 3' region of the miRNA could be entirely eliminated with minimal effects on target regulation (although strong 3' pairing became important in 5'-weak cases) [28]. This challenges common assumptions that target recognition involves an RNA duplex along the length of the miRNA and that greater complementarity indicates a better miRNA-binding site. Could it be that as little as eight nucleotides of complementarity to the 5' end of an miRNA suffices for regulation? This might be consistent with the growing appreciation of extensive 'off-target' regulation of modestly complementary transcripts by the small interfering RNAs (siRNAs) involved in RNAi [30-32]. These differing views might potentially be reconciled if miRNA-mediated regulation by multiple sites is governed primarily by the 5'-pairing rule whereas regulation by a single site might necessitate more extensive and/or specific pairing configurations (Figure 1c).
Other issues remain to be resolved. Foremost among these are the factors contributing to site insufficiency. Insertion of miRNA-binding sites into a heterologous context often suffices to bring a transcript under miRNA control, but a significant fraction of tested sites fail for unknown reasons. More importantly, some in vivo tests suggest that target regulation is not always so forgiving of context. Specifically, mutation of sequences in the lin-41 3' UTR in between two bona fide let-7 sites renders lin-41 nonresponsive to let-7 in transgenic worms [33]. Moreover, certain multimers of two let-7 or six lin-4 binding sites fail to mediate appropriate regulation in vivo [33,34]. Understanding why these site configurations do not work may improve identification of 'real' sites. For example, these failures might be due to influences of 3' UTR structure on miRNA accessibility, or the necessity for co-regulation by other factors - potentially even other miRNAs. Germane to the latter possibility is understanding the functional interactions between binding sites for the same miRNA and for different miRNAs in an individual target transcript (Figure 1c), either of which could have synergistic consequences on net regulation [28]. Another unanswered question is whether animal miRNA-binding sites can reside in coding regions or 5' UTRs, which are usually excluded from analyses. Indeed, the general possibilities that miRNAs might regulate noncoding RNAs or even DNA have been little explored [35].
Two additional biological considerations need to be included in the prediction of miRNA targets. Firstly, with the exceptions of lsy-6, let-7, mir-273, and bantam [15,16,22,36], we are generally ignorant of the spatial expression of miRNAs on a cell-by-cell basis. This means that we do not generally know that any miRNA and its predicted target are ever present in the same cell, an obvious prerequisite for a regulatory relationship. Secondly, we generally lack information on the relative levels of miRNA and target on a per-cell basis. The study by Doench and Sharp [28] showed that target regulation that can be detected when the miRNA is very abundant does not occur when the miRNA is rarer [28]. Thus, greater biological relevance to target prediction may come from incorporating data on miRNA:target coexpression and relative levels. Finally, improvements will come from having additional genomes sequenced, which will more clearly delineate functionally conserved segments of untranslated regions. The near future should see the completion of many additional drosophilid and vertebrate species, which will provide an incredible resource for all informatic studies of regulatory biology, including that of miRNAs.
Endgame
When the perfect miRNA target-finding program comes around, what will its output look like and what will it mean? There is a certain popular expectation of a series of rank-ordered lists in which the scores of the top candidates are well-separated from the rest, thus clearly defining transcripts that are strongly miRNA-regulated. But a majority of miRNAs currently lack predicted targets that stand out as obviously as do the targets of genetically studied miRNAs such as lin-4 and bantam. Does this mean that the programs do not work well enough? Or might it mean that miRNAs more often fine-tune gene expression rather than switch it off completely? In fact, it is a matter of speculation whether miRNAs generally regulate one or a few targets, or whether they have a continuum of increasingly poor targets that are nevertheless regulated to some extent. For that matter, it is not even clear if plant miRNAs regulate targets that display an animal-target-like level of complementarity.
Conversely, to what extent is miRNA-mediated regulation 'accidental'? The fact that null alleles of nearly all genes are completely recessive tells us that almost any gene can be knocked down by at least 50% without significant effect. It could be that, at any given time, a significant number of genes acquire a miRNA-binding site that places them under detectable but inconsequential regulation ('neutral targets' [37]). Are these targets less 'real' or 'interesting' than those for which loss of miRNA-mediated regulation is lethal or causes dramatic morphological or metabolic defects? In fact, it might be that genes for which miRNA-binding site acquisition is not tolerated ('anti-targets' [37]) are more interesting from the gene-regulatory perspective than are neutral targets.
One might expect that randomly acquired miRNA-binding sites are not subject to selective constraint, so that evolutionary conservation may provide one way to classify sites. This is not to say that non-conserved sites are necessarily weak; some may in fact confer quantitatively significant regulation as long as this 'knockdown' is tolerated by the animal. It is also not to say that they are not important; indeed, one idea is that they might serve as capacitors for speciation and microevolution [37]. Nevertheless, it is undoubtedly the case that the primary immediate interest is to identify essential targets - those transcripts for which loss of miRNA-mediated regulation is detrimental to the organism in some significant way. In plants, although some miRNA targets are involved in metabolism, the large majority of targets are transcription factors that regulate development [3,8,9]. This does not appear to be case for animals, since the current lists are only mildly enriched in transcription factors or transcripts involved in development [20,23,26]. Suggestions were made, however, that early Drosophila patterning and nervous system function are heavily regulated by miRNAs [24,25].
To return to the beginnings of miRNA studies, I expect that loss- and gain-of-function miRNA genetics will prove to be key in evaluating the biological relevance of the thousands of target genes predicted by informatic studies, and for evaluating the degree to which miRNA-mediated regulation of any 'validated' target actually matters to the animal or plant. This will probably necessitate detailed studies of a broad range of biological processes, and potentially the analysis of multiply-miRNA-mutant animals, or ones in which miRNA activity has been inhibited by chemical inhibitors (including 2' O-methylated oligonucleotides [38,39]). In addition, mutants of the miRNA-producing enzyme Dicer are now available in most model organisms, and these should prove useful in revealing the extent to which development, behavior and metabolism depend on miRNAs. If the first few years of the miRNA era are any indication, we may expect fast and furious progress on understanding the individual biological functions of miRNAs in the near future.
Acknowledgments
Acknowledgements
I thank Alexander Stark, Julius Brennecke, Chris Burge, Ben Lewis, Artemis Hatzigeorgiou, Zissimos Mourelatos and John Doench for enlightening discussions and critical reading of this manuscript. I am supported by a Career Development Award from the Leukemia and Lymphoma Society. I also acknowledge ongoing support from Gerald Rubin and the Howard Hughes Medical Institute.
References
- Lai EC. microRNAs: runts of the genome assert themselves. Curr Biol. 2003;13:R925–R936. doi: 10.1016/j.cub.2003.11.017. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs. Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B, Bartel DP. Prediction of plant microRNA targets. Cell. 2002;110:513–520. doi: 10.1016/S0092-8674(02)00863-2. [DOI] [PubMed] [Google Scholar]
- Kasschau KD, Xie Z, Allen E, Llave C, Chapman EJ, Krizan KA, Carrington JC. P1/HC-Pro, a viral suppressor of RNA silencing, interferes with Arabidopsis development and miRNA function. Dev Cell. 2003;4:205–217. doi: 10.1016/S1534-5807(03)00025-X. [DOI] [PubMed] [Google Scholar]
- Chen X. A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science. 2004;303:2022–2025. doi: 10.1126/science.1088060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palatnik JF, Allen E, Wu X, Schommer C, Schwab R, Carrington JC, Weigel D. Control of leaf morphogenesis by microRNAs. Nature. 2003;425:257–263. doi: 10.1038/nature01958. [DOI] [PubMed] [Google Scholar]
- Tang G, Reinhart BJ, Bartel DP, Zamore PD. A biochemical framework for RNA silencing in plants. Genes Dev. 2003;17:49–63. doi: 10.1101/gad.1048103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones-Rhoades MW, Bartel DP. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol Cell. 2004;14:787–799. doi: 10.1016/j.molcel.2004.05.027. [DOI] [PubMed] [Google Scholar]
- Bonnet E, Wuyts J, Rouze P, Van de Peer Y. Detection of 91 potential conserved plant microRNAs in Arabidopsis thaliana and Oryza sativa identifies important target genes. Proc Natl Acad Sci USA. 2004;101:11511–11516. doi: 10.1073/pnas.0404025101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart BJ, Slack F, Basson M, Pasquinelli A, Bettinger J, Rougvie A, Horvitz HR, Ruvkun G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-Y. [DOI] [PubMed] [Google Scholar]
- Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75:855–862. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- Abrahante JE, Daul AL, Li M, Volk ML, Tennessen JM, Miller EA, Rougvie AE. The Caenorhabditis elegans hunchback-like gene lin-57/hbl-1 controls developmental time and is regulated by microRNAs. Dev Cell. 2003;4:625–637. doi: 10.1016/S1534-5807(03)00127-8. [DOI] [PubMed] [Google Scholar]
- Lin SY, Johnson SM, Abraham M, Vella MC, Pasquinelli A, Gamberi C, Gottlieb E, Slack FJ. The C. elegans hunchback homolog, hbl-1, controls temporal patterning and is a probable microRNA target. Dev Cell. 2003;4:639–650. doi: 10.1016/S1534-5807(03)00124-2. [DOI] [PubMed] [Google Scholar]
- Johnston RJ, Hobert O. A microRNA controlling left/right neuronal asymmetry in Caenorhabditis elegans. Nature. 2003;426:845–849. doi: 10.1038/nature02255. [DOI] [PubMed] [Google Scholar]
- Brennecke J, Hipfner DR, Stark A, Russell RB, Cohen SM. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell. 2003;113:25–36. doi: 10.1016/S0092-8674(03)00231-9. [DOI] [PubMed] [Google Scholar]
- Lai EC, Posakony JW. The Bearded box, a novel 3' UTR sequence motif, mediates negative post-transcriptional regulation of Bearded and Enhancer of split complex gene expression. Development. 1997;124:4847–4856. doi: 10.1242/dev.124.23.4847. [DOI] [PubMed] [Google Scholar]
- Lai EC, Burks C, Posakony JW. The K box, a conserved 3' UTR sequence motif, negatively regulates accumulation of Enhancer of split complex transcripts. Development. 1998;125:4077–4088. doi: 10.1242/dev.125.20.4077. [DOI] [PubMed] [Google Scholar]
- Lai EC. Micro RNAs are complementary to 3' UTR sequence motifs that mediate negative post-transcriptional regulation. Nat Genet. 2002;30:363–364. doi: 10.1038/ng865. [DOI] [PubMed] [Google Scholar]
- Stark A, Brennecke J, Russell RB, Cohen SM. Identification of Drosophila microRNA targets. PLoS Biol. 2003;1:E60. doi: 10.1371/journal.pbio.0000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Nakahara K, Pham JW, Kim K, He Z, Sontheimer EJ, Carthew RW. Distinct roles for Drosophila Dicer-1 and Dicer-2 in the siRNA/miRNA silencing pathways. Cell. 2004;117:69–81. doi: 10.1016/S0092-8674(04)00261-2. [DOI] [PubMed] [Google Scholar]
- Chang S, Johnston RJ, Jr, Frokjaer-Jensen C, Lockery S, Hobert O. MicroRNAs act sequentially and asymmetrically to control chemosensory laterality in the nematode. Nature. 2004;430:785–789. doi: 10.1038/nature02752. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/S0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome Biol. 2003;5:R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajewsky N, Socci ND. Computational identification of microRNA targets. Dev Biol. 2004;267:529–535. doi: 10.1016/j.ydbio.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Kiriakidou M, Nelson PT, Kouranov A, Fitziev P, Bouyioukos C, Mourelatos Z, Hatzigeorgiou AG. A combined computational-experimental approach predicts human microRNA targets. Genes Dev. 2004;18:1165–1178. doi: 10.1101/gad.1184704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human microRNA targets. PLoS Biol. [DOI] [PMC free article] [PubMed]
- Doench JG, Sharp PA. Specificity of microRNA target selection in translational repression. Genes Dev. 2004;18:504–511. doi: 10.1101/gad.1184404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekta S, Shih IH, Bartel DP. MicroRNA-directed cleavage of HOXB8 mRNA. Science. 2004;304:594–596. doi: 10.1126/science.1097434. [DOI] [PubMed] [Google Scholar]
- Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, Li B, Cavet G, Linsley PS. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol. 2003;21:635–637. doi: 10.1038/nbt831. [DOI] [PubMed] [Google Scholar]
- Saxena S, Jonsson ZO, Dutta A. Small RNAs with imperfect match to endogenous mRNA repress translation. Implications for off-target activity of small inhibitory RNA in mammalian cells. J Biol Chem. 2003;278:44312–44319. doi: 10.1074/jbc.M307089200. [DOI] [PubMed] [Google Scholar]
- Scacheri PC, Rozenblatt-Rosen O, Caplen NJ, Wolfsberg TG, Umayam L, Lee JC, Hughes CM, Shanmugam KS, Bhattacharjee A, Meyerson M, Collins FS. Short interfering RNAs can induce unexpected and divergent changes in the levels of untargeted proteins in mammalian cells. Proc Natl Acad Sci USA. 2004;101:1892–1897. doi: 10.1073/pnas.0308698100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vella MC, Choi EY, Lin SY, Reinert K, Slack FJ. The C. elegans microRNA let-7 binds to imperfect let-7 complementary sites from the lin-41 3' UTR. Genes Dev. 2004;18:132–137. doi: 10.1101/gad.1165404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha I, Wightman B, Ruvkun G. A bulged lin-4/lin-14 RNA duplex is sufficient for Caenorhabditis elegans lin-14 temporal gradient formation. Genes Dev. 1996;10:3041–3050. doi: 10.1101/gad.10.23.3041. [DOI] [PubMed] [Google Scholar]
- Lai EC, Wiel C, Rubin GM. Complementary miRNA pairs suggest a regulatory role for miRNA:miRNA duplexes. RNA. 2004;10:171–175. doi: 10.1261/rna.5191904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SM, Lin SY, Slack FJ. The time of appearance of the C. elegans let-7 microRNA is transcriptionally controlled utilizing a temporal regulatory element in its promoter. Dev Biol. 2003;259:364–379. doi: 10.1016/S0012-1606(03)00202-1. [DOI] [PubMed] [Google Scholar]
- Bartel DP, Chen CZ. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet. 2004;5:396–400. doi: 10.1038/nrg1328. [DOI] [PubMed] [Google Scholar]
- Meister G, Landthaler M, Dorsett Y, Tuschl T. Sequence-specific inhibition of microRNA- and siRNA-induced RNA silencing. RNA. 2004;10:544–550. doi: 10.1261/rna.5235104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutvagner G, Simard MJ, Mello CC, Zamore PD. Sequence-specific inhibition of small RNA function. PLoS Biol. 2004;2:E98. doi: 10.1371/journal.pbio.0020098. [DOI] [PMC free article] [PubMed] [Google Scholar]