

Summary
Antibiotic resistance is a rapidly evolving health concern that requires a sustained effort to understand mechanisms of resistance and develop new agents that overcome those mechanisms. The dihydrofolate reductase (DHFR) inhibitor, trimethoprim (TMP), remains one of the most important orally administered antibiotics. However, resistance through chromosomal mutations and mobile, plasmid-encoded insensitive DHFRs threatens the continued use of this agent. We are pursuing the development of new propargyl-linked antifolate (PLA) DHFR inhibitors designed to evade these mechanisms. While analyzing contemporary TMP-resistant clinical isolates of methicillin-resistant and sensitive Staphylococcus aureus, we discovered two mobile resistance elements, dfrG and dfrK. This is the first identification of these resistance mechanisms in the United States. These resistant organisms were isolated from a variety of infection sites, show clonal diversity and each contain distinct resistance genotypes for common antibiotics. Several PLAs showed significant activity against these resistant strains by direct inhibition of the TMP resistance elements.
eTOC Blurb
Reeve et al. identified two plasmid-borne genes that confer high-level resistance to trimethoprim for the first time in MRSA isolates from US hospitals. A series of charged propargyl-linked antifolates overcome this resistance via direct inhibition of the acquired resistance elements.
Introduction
Methicillin-resistant Staphylococcus aureus (MRSA) continues to spread globally causing moderate to life-threatening infections. The organism can be acquired through nosocomial (HA-MRSA) or community (CA-MRSA) routes; CA-MRSA and HA-MRSA are genetically distinct and have differing patterns of antibiotic resistance, however CA-MRSA is now epidemic within health care systems (Lodise et al., 2007; Bush et al., 2015). Trimethoprim-sulfamethoxazole (TMP-SMX), branded as Bactrim and Septra, is a first-line treatment for MRSA infections in the community setting (Frei et al., 2010; Gorowitz et al., 2006; Nathwani et al., 2008; Liu et al., 2011), owing to its oral bioavailability, low cost and general tolerability. In fact, prescriptions of TMP-SMX numbered more than 21 million in 2013, putting it in the group of top ten oral antibiotics prescribed in 2013 (CDC, 2013). This combination therapy employs two drugs that synergistically inhibit the folate biosynthetic pathway, which is essential for the production of deoxythymidine monophosphate, purine nucleotides and some amino acids. Trimethoprim (TMP) is an inhibitor of dihydrofolate reductase (DHFR) and sulfamethoxazole inhibits dihydropteroate synthase.
Resistance to TMP-SMX in S. aureus began to arise in the 1980s (Dale et al., 1995, 1997; Huovinen et al, 1995). Reports of contemporary TMP-SMX resistance vary regionally: 21 % of resistance is reported in travel clinics in Europe (Nurjadi et al., 2015), whereas ~3 % has been reported in a group of US hospitals (Sader et al., 2015). TMP-SMX resistance is on the rise according to a survey of 2193 isolates showing that TMP-SMX resistance rose from 3.4 % in 2007 to 6 % in 2012 (Pate et al., 2015). The mechanisms of resistance are temporally segregated. In the 1990s, point mutations were observed in the chromosomal gene, dfrB in S. aureus; these modifications of the enzyme were responsible for increases in MIC values to 256 μg/mL (~800-fold). Noteworthy in this group was the F98Y mutation that conferred ~400-fold decrease in affinity between TMP and DHFR (Dale et al., 1997). Compensatory double mutations (F98Y/H149R and F98Y/H30N) arose that increased the fitness of the mutated enzyme. Horizontally transferred, plasmid-encoded resistant DHFR enzymes also appeared. The translated protein of the dfrA gene, often called S1 (but will be called DfrA here for clarity), was observed to confer 338-fold resistance to TMP at the enzyme level (Dale et al., 1995). DfrA has three mutations in comparison to DHFR from the TMP-sensitive S. epidermidis: V31I, G43A and F98Y; F98Y is the major determinant of loss of affinity. The resistance gene, dfrG, was first reported in isolates from Thailand (Sekiguchi et al., 2005) and was later reported as abundant in sub-Saharan Africa (Nurjadi et al. 2014) with subsequent isolation from European travelers who had visited Africa. This later study reported that 54 % of 598 isolates were resistant to TMP and 19 % to the combination of TMP-SMX. The gene dfrK was originally identified in swine in multi-drug resistant MRSA ST398 where it caused MIC values to rise to 256 μg/mL (Kadlec et al. 2010), but began to be observed in farmers with MRSA infections. In 2012, a MRSA isolate from a patient in Spain was identified as linezolid-resistant, carrying the ERGB plasmid that links resistance genes cfr (oxazolidinones, phenicols, lincosamides, pleuromutilins, and streptogramin A), ant(4′)-Ia (tobramycin), tet(L) (tetracycline) and dfrK (trimethoprim) (de Gopegui et al., 2012). DfrG and DfrK, like DfrA, are innately resistant enzymes carried on plasmids and incorporated into the chromosome via horizontal gene transfer. DfrG and DfrK share 89 % sequence similarity to each other, however, they are less similar to DfrA (38 and 39 %) and DfrB (41 and 42 %). The origins of DfrG and DfrK are unknown. No recent survey of MRSA isolates from US hospitals has identified common molecular mechanisms of TMP-SMX resistance.
Over the past decade, we have focused on the development of next generation propargyl-linked antifolates (PLAs) that maintain activity against many of the important pathogenic bacterial DHFR enzymes while expanding coverage to include both mutant and naturally TMP-insensitive DHFR enzymes that give rise to TMP resistance within S. aureus (Frey et al., 2009, 2010a, 2012; Keshipeddy et al., 2015; Lamb et al., 2014; Lombardo et al., 2016). The occurrence of a common F98Y mutation shared by resistant S. aureus mutants and plasmid-encoded DHFRs provided rationale that new antifolates that are effective against this mutant could expand coverage for these resistant enzymes. We anticipated that optimizing the PLAs to overcome the central F98Y resistance mechanism would lead to robust inhibitors capable of targeting multiple enzymes that possess this substitution. Recently we reported a series of PLAs that potently inhibit the F98Y mutant enzyme as well as S. aureus strains harboring the F98Y mutation (Keshipeddy et al., 2015).
As antibiotic resistance is a naturally evolving phenomenon, it is critical to map compound design to contemporary resistance profiles found in clinical strains of bacteria in order to properly target the prevailing molecular mechanisms during lead optimization. In the work described here, we present a recent investigation of the molecular mechanisms of TMP resistance for MRSA and MSSA strains isolated in the state of Connecticut, the results of which have guided compound development to yield potent inhibitors of an emerging group of TMP-resistant strains. We collected clinical isolates of TMP-resistant MRSA over a two-year period in order to determine which mechanisms are currently predominant. Using whole genome sequencing and PCR, we identified the plasmid-encoded dfrG, dfrA and dfrK genes in these isolates. It is noteworthy that this is the first report of the resistance-conferring dfrG and dfrK genes in strains of MRSA and MSSA in the United States. Interestingly, we did not identify any of the well-characterized mutations to the chromosomal gene, dfrB. Excitingly, several of the PLAs potently inhibit the growth of the clinical isolates that possess these plasmid-encoded dfrA, dfrG and dfrK genes as well as inhibit the purified enzymes at nanomolar levels of potency.
Results
Clinical bacterial isolates of MRSA and MSSA harbor dfrA, dfrG and dfrK
Over the past two years, we obtained several TMP-resistant strains from the clinical microbiology laboratories at UConn Health/John Dempsey Hospital (UCH) and Hartford Hospital (HH) isolated in the course of routine clinical care. The strains were originally submitted for routine susceptibility testing. TMP/SMX resistance rates at UCH and HH are reported at ≤1 % and 2% for MSSA and 2–3% and 5% for MRSA isolates, respectively. Seven of the obtained strains were classified as MRSA and one was MSSA. These strains were collected from different patients and from a variety of sources including blood, skin and soft tissue (SSTI), sputum and the sinus cavity. The blood sample was derived from a hospitalized patient; the other isolates were derived from outpatients. Clearly, each of the strains is highly resistant to TMP and SMX, with MIC values between 250 and >1,000 μg/mL or 16–500 μg/mL, respectively (Table 1). The strains show varying resistance profiles to a wide range of commonly used antibiotics including erythromycin, fluoroquinolones, aminoglycosides and tetracycline (Table S1).
Table 1.
Characterization of TMPR Clinical Isolates
| Strain Designation | Infection Source | TMP Resistant Gene | dfrB | TMP MIC (μg/mL) | SMX MIC (μg/mL) | Doubling Time (min) |
|---|---|---|---|---|---|---|
| UCH MRSA 1 | Blood | dfrG | WT | >1,000 | >500 | 40.33 |
| UCH MRSA 115 | SSTI | dfrA | WT | 250 | 500 | 35.19 |
| UCH MRSA 121 | Sputum | dfrG | WT | >1,000 | >500 | 28.17 |
| UCH MRSA 127 | SSTI | dfrG | WT | >1000 | 16 | 38.61 |
| HH MRSA 714 | SSTI | dfrG | WT | >1000 | >500 | 27.39 |
| HH MRSA 1144 | SSTI | dfrA | WT | 250 | >500 | 32.54 |
| HH MRSA 1184 | Sputum | dfrK | WT | >1000 | 32 | 29.38 |
| UCH MSSA 1 | Sinus Cavity | dfrG | WT | >1000 | >500 | 36.49 |
| Sa43300 | ATCC | None | WT | 0.3125 | 8 | 38.16 |
To determine the molecular basis of TMP resistance, we initially probed for mutations in the chromosomal dfrB gene. Somewhat surprisingly, no mutations were observed in any of the strains. In order to investigate further, we initially conducted whole genome sequencing on a single strain, UCH MRSA 1. Genomic analysis revealed the presence of a second DHFR enzyme encoded by the gene, dfrG that had been integrated into the chromosome. Although this gene has been observed in S. aureus in Asia (Sekiguchi et al., 2005), Africa and European travelers (Nurjadi et al., 2014) and in Streptococcus pyogenes in India (Bergmann et al., 2014), to our knowledge, this is the first report of dfrG in North America. We then used PCR to evaluate the remaining strains for the presence of dfrG as well as two other plasmid-encoded genes, dfrA and dfrK, known to occur in S. aureus. Surprisingly, we found that all of the clinical isolates carried one of the plasmid-encoded genes, with the dfrG gene predominating (Table 1). We also identified, for the first time in the United States, the presence of the dfrK gene. The better characterized dfrA appeared in the remaining two strains (Table 1, Figure S1). One report (de Gopegui et al., 2012) showed that the presence of dfrK was associated with cfr, a gene that confers linezolid non-susceptibility. We probed the strains for the cfr gene, but no strains, even HH1184 that contains dfrK, positively identified the gene. This result correlates with the observed linezolid susceptibility of all of the strains (Table S1). Interestingly, the acquisition of these dfr genes did not appear to be associated with obvious negative fitness costs as the doubling times were generally shorter than the ATCC 43300 strain (Table 1).
Given the limited number of strains presented here, the occurrence of these diverse resistance elements is striking. To better understand the possible relationships of these TMP-resistant strains, the genetic diversity was determined through sequencing of the spaA gene (Petersson et al., 2010). The analysis showed that five of the strains (UCH MRSA115, UCH MRSA121, UCH MSSA1, HH MRSA714 and HH MRSA1144), including strains from both hospitals and the MSSA strain, were clonally indistinguishable. However, UCH MRSA127, HH MRSA1184 and UCH MRSA1 are clonally distinct isolates. Importantly, within the group of five clonal isolates, dfrA and dfrG are represented and dfrK is found the distinct yet related strain, HH MRSA 1184, Figure 1. The appearance of the different dfr isoforms within the closely related cluster as well as more genetically distinct strains suggests that they are on potentially highly mobile resistance elements.
Figure 1.
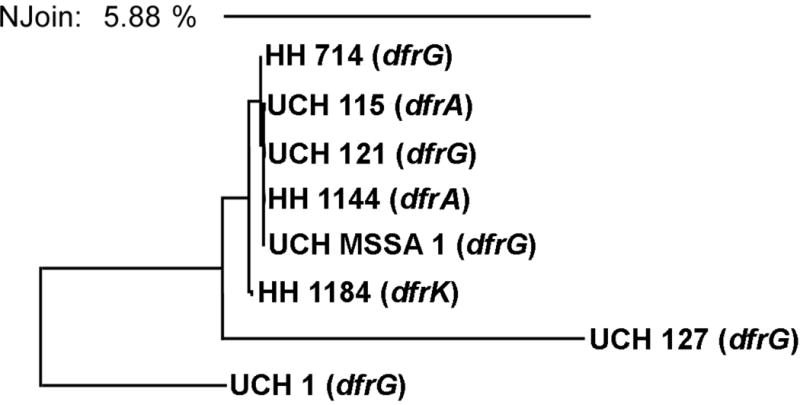
Clonal characterization of S. aureus strains by spaA sequencing
Clinical Isolates exhibit a range of antibiotic susceptibilities
The phenotypes to commonly prescribed non-TMP antibiotics were determined for the isolates revealing diverse variability in susceptibilities. All strains were susceptible to vancomycin, linezolid, rifampin and daptomycin and have varied susceptibilities to sulfamethoxazole, erythromycin, clindamycin, tetracycline, gentamicin and a variety of fluoroquinolones, based on breakpoint MICs (Table S1). To better understand the diversity of the isolates, the remaining seven genomes were sequenced and the molecular mechanisms of resistances were identified. Interestingly, the only common resistance mechanism found within the clonal group of strains was five previously reported folP mutations (F17L, T28S, T59S, L64M, E205K) conferring high-level SMX resistance. UCH 1 contains thirteen folP mutations (F17L, V30I, T31N, M37I, I58V, T59S, V60L, L64M, I110M, V117I, V126I, E208K, F226L) while HH1184 contains nine (V30I, I58V, T59S, V60L, L64M, I100M, V117I, V126I, F226L) increasing the MIC to near its clinical resistance breakpoint (Hampele et al., 1997). TetM, a ribosome protection protein, is identified as the tetracycline resistance determinant observed in UCH115 and HH1144 (Trzcinski et al., 2000). Gentamicin resistance in UCH115 and HH1144 is conferred by a plasmid borne AAC(6′)-APH(2″) aminoglycoside resistance enzyme(Daigle et al., 1999). Tetracycline and aminoglycoside resistance was only identified in strains containing dfrA, however these strains differ both in fluoroquinolone resistance mechanisms and macrolide susceptibility.
Resistance to fluoroquinolone in all strains except HH MRSA 1184 is conferred through a variety of mutations in the quinolone resistance-determining regions (QRDRs) of DNA Gyrase subunit A (gyrA) and Topoisomerase IV, subunits A and B (grlA and grlB). The combinations of mutations vary from single gyrA mutations to the accumulation of four mutations between gyrA, grlA, and grlB were observed (Schmitz et al., 1998; Pan et al., 2002). NorA efflux activity in fluoroquinolone resistance was determined by MIC in the presence of reserpine, a NorA inhibitor. Minimal shifts in MIC for levofloxacin and up to 8-fold decrease in ciprofloxacin MIC indicate that NorA has minimal influence on fluoroquinolone resistance (Table S3) (Kaatz and Seo, 1995; Aeschlimann et al., 1999). mphC, a 2′-phosphotransferase which directly inactivates the macrolides via phosphorylation determines selective macrolide resistance (erythromycin) in UCH 121, UCH 127 and HH1184, all clonally distinct isolates(Matsuoka et al., 2003; Juda et al., 2016). mphC is not commonly reported to confer macrolide resistance in human S. aureus isolates, instead it is more frequently reported in agricultural studies (Li et al., 2015). ermC, a 23s rRNA methyl transferase, found in UCH MRSA 1, HH MRSA 1144 and UCH MSSA 1 confers resistance to both macrolides and lincosamines (clindamycin) (Khan et al., 1999). HH MRSA 1184 is the only strain to contain the Panton-Valenitine leukocidin, a virulence factor that produces a cytotoxin associated with tissue necrosis and leukocyte damage (Lina et al., 1999). All strains clinically classified as ‘methicillin resistant’ via susceptibility are mecA positive. A full list of target mutations, efflux activity and resistance determinants are summarized in Tables S2 and S3.
Propargyl-linked antifolates potently inhibit clinical isolates
Whole genome sequencing of the clinical strains showed notable variation in the molecular basis of TMP resistance as well as several common antibiotics. This provided a useful panel of clinically relevant strains as an important tool for lead optimization. Therefore, we screened a variety of previously developed PLA lead compounds against the panel of TMP-resistant strains to identify candidates with broad activity. Excitingly, several of the compounds (UCP1039, UCP1164, UCP1172, UCP1173 and UCP1175; Figure 2, Table 2 and Table 3) showed very potent activity against these highly TMP-resistant strains. Although we observed activity with earlier generation inhibitors that contained a pyridyl C-ring (eg. UCP1039, Figure 2 and Table 2), the most potent activity was observed with a recently disclosed charged/zwitterionic series possessing an ionizable carboxylic acid on the distal C-ring (UCP1164, UCP1172, UCP1173 and UCP1175). Overall, the PLAs were most potent against strains carrying dfrG and dfrK with MIC values as low as 0.1563 μg/mL, two-fold lower than the MIC for TMP against wild-type S. aureus. Compound UCP1173 showed the most potent activity against strains possessing dfrA with MIC values of 1.25 and 2.5 μg/mL. Interestingly, compound UCP1172 is the antipode of UCP1173 but does not significantly inhibit dfrA-possessing strains.
Figure 2.
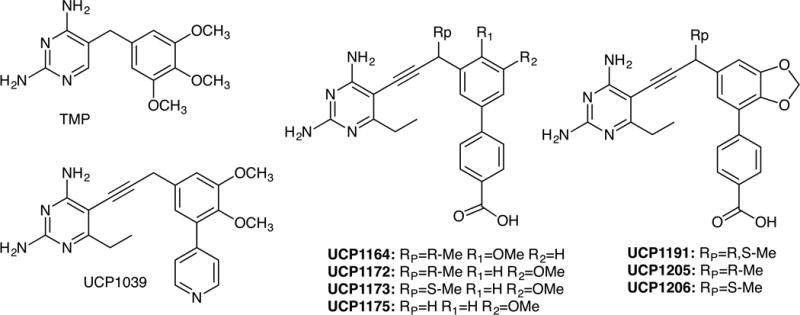
Structure of propargyl-linked antifolates in this study
Table 2.
PLA Antibacterial Activity against Clinical Isolates (MICs in μg/mL)
| Inhibitor | UCH MRSA 1 (dfrG) | UCH MRSA 115 (dfrA) | UCH MRSA 121 (dfrG) | UCH MRSA 127 (dfrG) | HH MRSA 714 (dfrG) | HH MRSA 1144 (dfrA) | HH MRSA 1184 (dfrK) | UCH MSSA 1 (dfrG) | ATCC 43300 |
|---|---|---|---|---|---|---|---|---|---|
| UCP1039 | 1.25 | >20 | 0.625 | 0.3125 | 1.25 | >10 | 1.25 | 0.625 | 0.0391 |
| UCP1164 | 2.5 | 10 | 5 | 2.5 | 5 | 5 | 0.625 | 5 | 0.0391 |
| UCP1172 | 0.625 | 5 | 0.625 | 0.3125 | 0.625 | 5 | 0.3125 | 0.3125 | 0.0098 |
| UCP1173 | 5 | 2.5 | 5 | 2.5 | 5 | 1.25 | 2.5 | 2.5 | 0.0098 |
| UCP1175 | 2.5 | >20 | 10 | 5 | 10 | >20 | 5 | 10 | 0.0195 |
| UCP1191 | 0.625 | 20 | 0.625 | 0.1563 | 0.625 | 10 | 0.1563 | 0.3125 | 0.0195 |
| UCP1205 | 0.625 | >10 | 0.625 | 0.3125 | 0.625 | >10 | 0.3125 | 0.3125 | 0.0195 |
| UCP1206 | 2.5 | >10 | 2.5 | 1.25 | 2.5 | >10 | 1.25 | 1.25 | 0.0098 |
| UCP1191 +SMXa | 0.0391 | 00.625 | 0.0781 | 0.3125 | 0.0391 | 0.625 | ≤ 0.0098 | 0.0098 | ≤ 0.0048 |
MIC values with 100 μg/mL Sulfamethoxazole (SMX), ATCC 43300 and UCH 127 at 1 μg/mL SMX
Table 3.
Enzyme Inhibition (IC50 values shown in μM)
| Inhibitor | DfrB (wt SaDHFR) | DfrG | DfrA | DfrK |
|---|---|---|---|---|
| TMP | 0.023 ± 0.002 | 380 ± 12 | 15.1 ± 0.7 | 43 ± 2 |
| UCP1039 | 0.014 ± 0.001 | 0.45 ± 0.02 | 0.36 ± 0.02 | 0.022 ± 0.003 |
| UCP1164 | 0.037 ± 0.002 | 1.4 ± 0.1 | 8.8 ± 0.9 | 0.073 ± 0.002 |
| UCP1172 | 0.0089 ± 0.0007 | 0.22 ± 0.02 | 0.41 ± 0.01 | 0.030 ± 0.001 |
| UCP1173 | 0.014 ± 0.001 | 0.19 ± 0.01 | 0.27 ± 0.02 | 0.091 ± 0.008 |
| UCP1175 | 0.0110 ± 0.0006 | 1.4 ± 0.2 | 0.98 ± 0.008 | 0.17 ± 0.01 |
| UCP1191 | 0.010 ± 0.0002 | 0.087 ± 0.005 | 0.32 ± 0.03 | 0.041 ± 0.006 |
| UCP1205 | 0.018 ± 0.003 | 0.159 ± 0.007 | 0.34 ± 0.01 | 0.10 ± 0.01 |
| UCP1206 | 0.017 ± 0.002 | 0.19 ± 0.01 | 0.18 ± 0.03 | 0.054 ± 0.005 |
It was noted that C3′, C4′ dioxygenation with a pyridyl C-ring (UCP1039) afforded some of the strongest activity in this series and as such, we investigated combining this pattern of functionality with the preference for a C-ring carboxylic acid. To explore this design, a dioxalane ring was chosen as a convenient isostere as it afforded antibacterial activity against strains that possessed a DHFR with the F98Y mutation (Keshipeddy et al., 2015). Using routes previously described (Keshipeddy et al., 2015), we synthesized racemic inhibitor UCP1191 and the individual enantiomers, UCP1205 and UCP1206. We were delighted to see a significant increase in activity against both dfrG- and dfrK-possessing strains with MIC values of 0.1563−0.625 μg/mL. Interestingly, despite these strains also being SMX-resistant, an apparently strong synergistic interaction between the PLAs and SMX was observed (Table 2 and Table S4).
Further validation that antibacterial activity of the PLAs in these resistant organisms was directly related to their ability to inhibit the resistance-conferring enzymes was provided by cloning, expressing, purifying and evaluating enzyme inhibition. The three genes, dfrA, dfrG and dfrK were cloned into expression vectors and the resulting proteins purified to homogeneity. The PLAs were evaluated for enzyme inhibition using standard assays that measure the oxidation of the NAPDH cofactor (Table 3) (Frey et al., 2009, 2010a, 2010b, 2012; Keshipeddy et al., 2015; Reeve et al. 2014, 2016). As expected, TMP exhibits high inhibition concentration 50 % (IC50) values for all three TMP-resistant DHFRs. Despite the fact that there has been no directed optimization of the PLAs against these TMP-resistant proteins, the PLAs showed relatively potent inhibition with the majority of IC50 values less than 100 nM, highlighting the value of our approach to use structure-based targeting of common resistance mechanisms in DHFRs (Keshipeddy et al., 2015; Reeve et al., 2015). Remarkably, this is an approximately 4400-, 55-, or 2000-fold increase in potency over TMP for the DfrG, DfrA and DfrK proteins, respectively. Although a variety of factors beyond target inhibition contribute to the overall antibacterial activity, there is a correlation between PLA activity against the TMP-resistant enzymes and the MICs against the corresponding strains. Additional validation that the PLAs exert their antibiotic effect through blockade of the folate pathway was provided by rescue experiments whereby the culture media was supplemented with thymidine and MIC values rose by at least 8-fold (data not shown). The potent inhibitors described above, specifically those with zwitterionic character, are promising drug leads that show strong antibiotic activity, low mammalian cell toxicity and good metabolic stability (Scocchera et al., 2016).
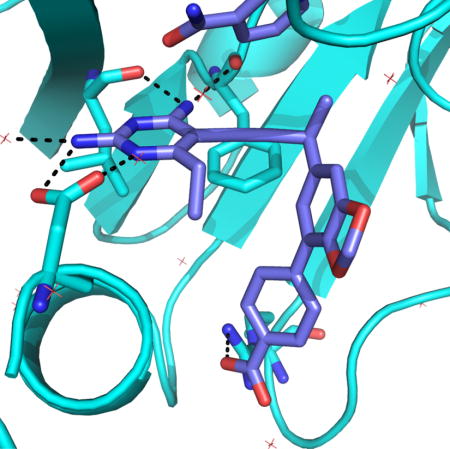
Crystal structure of S. aureus DHFR with UCP1191
A high resolution (1.88 Å) crystal structure of wild-type SaDHFR bound to compound UCP1191 (diffraction data and model statistics in Supplemental Table S5, omit map shown in Supplemental Figure S2 and structural analysis of interactions are shown in Supplemental Table S5) reveals a potential basis for the increased potency of this series of PLA-COOH compounds with both the wild-type and TMP-resistant enzymes. The diaminopyrimidine of the antifolate forms conserved hydrogen bonds with Asp 27 and backbone carbonyl oxygen atoms from Leu 5 and Phe 92 (Figure 3). The propargyl linker and benzodioxalane B-ring form hydrophobic interactions with Phe 92, Thr 46, Leu 28, Val 31 and Ile 50. The phenyl C-ring is positioned well to form hydrophobic interactions with Leu 54, Val 31 and Leu 28. Importantly, the carboxylate moiety forms one direct ionic bond to Arg 57 and one water-mediated hydrogen bond to Arg 57 and Lys 32. In earlier versions of the PLA-COOH compounds that possess a phenyl B-ring (similar to eg. UCP1164) the carboxylate forms an extensive water network with Arg 57 (Reeve et al., 2016) as opposed to this more direct interaction between UCP1191 and Arg 57. Arg 57 is conserved in all of the TMP-resistant enzymes (Figure 3) and forms a similar key contact with dihydrofolate (Scocchera et al., 2016), suggesting that this contact is less likely to mutate to cause resistance to the PLAs.
Figure 3.
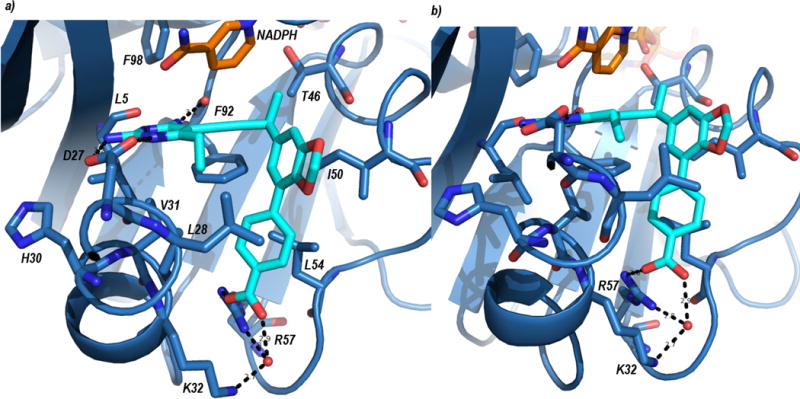
Crystal structure of SaDHFR bound to NADPH (orange) and UCP1191 (cyan). Panel a) shows an overview of the ligand in the active site and panel b) shows a more detailed view of the interaction with Arg 57.
Interestingly, the TMP-resistant enzymes tend to conserve their mechanisms of reducing TMP affinity (Figure 4). Leu 5 is an isoleucine in the TMP-resistant enzymes; this mutation would disturb Phe 92, which is critical both for hydrogen bonding (through the backbone carbonyl to the 4-amino group of the pyrimidine) as well as hydrophobic interactions with the linker. Leu 28 is a tyrosine in DfrG and DfrK and Val 31 is Ile in DfrA; these mutations also perturb Phe 92. The Val 31 Ile mutation was predicted by K* in the OSPREY suite to cause resistance to an earlier PLA, and in fact reduced affinity by 60-fold (Frey et al., 2010b; Reeve et al. 2014). Crystal structures of the double mutant enzyme, F98Y/V31I, show the perturbation of Phe 92. His 30 mutations have been observed clinically in the dfrB gene (Dale et al., 1997). While the mutation His 30 Asn has been shown to disrupt the water network stabilizing the pyrimidine ring (Frey et al., 2010a; Reeve et al., 2016), the TMP-resistant enzymes DfrK and DfrG carry a tyrosine at this position, which may achieve the same goal. Finally, all three TMP-resistant enzymes maintain a tyrosine at position 98 (wt Phe). The tyrosine has been shown to perturb NADPH binding Frey et al., 2009; Keshipeddy et al., 2015) and to decrease synergistic binding between TMP and NADPH (Heaslet et al., 2009).
Figure 4.

Sequence alignment of proteins resulting from chromosomal DHFR (dfrB) and plasmid-acquired DHFR genes, dfrG, dfrK and dfrA.
Previous design efforts focused on achieving inhibitor potency against the mutations observed in the chromosomal copy, dfrB, such as the Phe98Tyr-mutated DHFR enzyme (Keshipeddy et al., 2015; Reeve et al., 2016; Oefner et al., 2009a, 2009b; Heaslet et al., 2009).
These efforts may prove valuable as the TMP-resistant enzymes DfrG, DfrK and DfrA all possess a tyrosine residue at position 98. As shown here, designing inhibitors against the F98Y chromosomal mutant provided a significant advantage in achieving superior potency against these resistant enzymes, as they appear to rely on common mechanisms. Recently, we described potent activity of the COOH-PLA series, specifically UCP1164, UCP1172, UCP1173 and UCP1175 against strains containing these clinically relevant point mutations in dfrB. Including F98Y and F98Y with H30N and H149R. While the COOH-PLAs are more potent against the single F98Y mutant strain than the acquired dfr isoforms, UCP1164 and UCP1172 maintain superior activity against the acquired resistance elements over the double mutants. The remaining compounds display similar inhibitory activity in the acquired and mutant DHFR enzymes (Reeve et al., 2016). As the mutations that confer TMP resistance appear to belong to a conserved and relatively manageable group, future design efforts can capitalize on this group to optimize ligands that inhibit the majority of clinically observed TMP-resistant species.
Discussion
TMP-SMX has been a mainstay for treating S. aureus infections in the community setting for decades. Despite knowledge of the existence of the plasmid-encoded resistance elements dfrG and dfrK since 2005, their importance in clinical strains of MRSA and MSSA in the United States was not thought to be significant and often antifolate development was targeted toward resistant mutants of the chromosomal gene. Herein, we show a surprising preponderance of these genes from a relatively small collection of TMP-resistant MRSA/MSSA isolates from two Connecticut hospitals. Genetic analysis of the strains supports that these elements are potentially easily transferred between bacteria, suggesting that occurrence of these genes may be much wider than reported. The report here of these new elements in the United States is highly concerning as they confer extremely high levels of resistance to TMP, appear to be very mobile and are associated with a wide range of infections.
Building a program to understand the structural mechanisms of TMP resistance has supported the design of propargyl-linked antifolates that potently inhibit both TMP-resistant enzymes and the strains harboring these elements. Here, the identification of the dfrG and dfrK genes in clinical isolates has fostered the refinement of these inhibitors to arrive at highly potent antibacterial agents. Furthermore, the clinical relevancy of the compounds remains acute as timely clinical data drive compound design.
Significance
The continued spread of antibiotic resistance elements between pathogenic bacterial strains is diminishing the lifetime of many first-line antibiotics, leaving limited treatment options for bacterial infections. Only by investigating the contemporary mechanisms of antibacterial resistance is it possible to design new antibiotics that will efficaciously inhibit the resistant bacteria that are currently circulating in hospitals, assisted-care living and the wider community. Here, we report the first identification of two plasmid-borne genes found in clinical Staphylococcus aureus isolates from United States hospitals as well as identify the molecular mechanisms of resistance to several common antibiotics. The clinical impact of these genes, dfrG and dfrK, is significant as the resulting proteins confer high levels of trimethoprim resistance and render this commonly used antibiotic useless. Additionally, the fact that each of the strains presented here possesses plasmid-borne enzymes speaks to the potential widespread existence of these resistant strains. Using a structure-based approach designed to overcome trimethoprim-resistant enzymes, a series of charged propargyl-linked antifolates are presented that directly target the acquired resistance elements and potently inhibit the resistant enzymes and bacteria.
Experimental Procedures
Clonal Analysis
SpaA-typing was performed by Charles River using Accugenix’s AccuGENX-ST service to identify clonality among isolates.
Genomic Sequencing of Clinical isolates
Genomic DNA was isolated using Promega Wizard Genomic DNA Isolation kit. DNA extracts were quantified using the Quant-iT PicoGreen kit (invitrogen, ThermoFisher Scientific). One ng of genomic DNA was fragmented, adapter sequences attached, size selected and cleaned using the Nextera XT Library Preparation kit (Illumina, Inc.) according to the manufacturer’s protocol. Libraries were validated and mean insert length was calculated using a Bioanalyzer High Sensitivity chip (Agilent Technologies). The libraries were sequenced on the MiSeq using v2 2×250 base pair kit (Illumina, Inc). The genome was assembled using CLC Workbench and annotated using the Rast Server (Azuz et al., 2008; Brettin et al., 2015; Overbeek et al., 2014)). The assembled and annotated genomes are available through the corresponding author.
PCR Identification and Sequencing
PCR on gel purified genomic and plasmid DNA was performed to detect the presence of dfrB, dfrA, dfrG and dfrK genes. PCR was performed using rTaq Polymerase (Takara) and reactions were run on 1.2% agarose gels and visualized with ethidium bromide using 2-Log Ladder as a size comparator (Thermo Scientific). PCR product was purified using Promega SV Gel and PCR Clean-Up System and sequenced using the corresponding sense primer to confirm the gene identity.
Minimum Inhibitory Concentrations
Minimum inhibitory concentrations for UCP compounds, trimethoprim, sulfamethoxazole (Dao et al., 2014), levofloxacin, linezolid (in DMSO), erythromycin (in ethanol) and ciprofloxacin (in 0.1N HCl) were determined following CSLI guidelines following been previously described methods (CLSI, 2014).
Clinical Antibiotic Susceptibility
Susceptibilities were determined using Sensititre Gram Positive plates (Remel, ThermoFisher Scientific) using Mueller-Hinton Broth and an inoculum of 1×105 CFU/mL. The plates were incubated for 18 hours at 37°C and MICs were colorimetrically determined using Alamar Blue (ThermoFisher Scientific). Susceptible/Intermediate/Resistant designations were made based on CLSI breakpoint standards (CLSI, 2014).
DfrA, DfrB, DfrG and DfrK Protein Expression and Purification
The expression and purification of dfrB in pET-41a(+) has been previously described (Frey et al., 2009, 2012; Reeve et al., 2016). BL21 (DE3) cells (Invitrogen) were transformed with DfrA and DfrG in pET-41a(+) and DfrK in pET-24-a(+) were transfected into BL21 (DE3) cells (Invitrogen) were transformed with. The cells were grown to mid-log phase at 37°C, induced with 1 mM IPTG and were allowed to grow for an additional 18 hours at 20°C. Cells were pelleted and resuspended to 30 mL using a buffer containing 0.4 M KCl, 25 mM Tris, pH 8.0, 5 mM beta-mercaptoethanol, 5% glycerol, 100 μg/mL lysozyme, 5 mM imidazole and DNase (Fisher Scientific) and lysed via sonication. DfrG was resuspended to 30 mL using a buffer containing 0.5 M KCl, 50 mM Tris, pH 8.0, 5 mM beta-mercaptoethanol, 5% glycerol, 0.8 mg/mL lysozyme, 5 mM imidazole, DNase, RNase and a protease inhibitor tablet (Life Technologies) and lysed via French Press.
Protein was purified using Ni-NTA agarose using a wash buffer containing 25 mM Tris, pH 8.0, 0.4 M KCl, 5 mM Imidazole, 5 mM beta-mercaptoethanol and 5% glycerol and protein was eluted using a buffer containing 25 mM Tris pH 8.0, 0.3 M KCl, 20% glycerol, 0.1 mM EDTA, 250 mM imidazole and 5 mM beta-mercaptoethanol. Clean protein was pooled and desalted into a buffer containing 25 mM Tris pH 8.0, 0.1 mM KCl, 15% glycerol (20% for DfrG), 0.1 mM EDTA and 2 mM DTT. Protein was flash frozen and stored at −80°C.
IC50 Determination
IC50s were determined following a standard method-that has been previously described (Reeve et al., 2014, 2016).
DfrB:NADPH:UCP1191 Crystallography
Purified DfrB at 13 mg/mL protein was co-crystallized with 2 mM NADPH and 1 mM UCP1191 in DMSO via the hanging drop method. The mixture of protein and cofactor was incubated on ice for 3 hours. Equal volumes of protein solution were added to an optimized buffer solution containing 0.1 M MES, pH 5.0, 0.3 M sodium acetate, 17% PEG 10,000 and 12.5% gamma-butyrolactone. When stored at 4°C, crystals typically formed within 7 days. Crystals were harvested and frozen in cryo-protectant buffer containing 25% glycerol. Data were collected remotely on beamline 14-1 at Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory. Data were indexed and scaled using HKL2000. Phaser was used to identify molecular replacement solutions using PDB ID: 3F0Q (Frey et al, 2009) as a probe. Coot (Emsley et al., 2004) and Phenix (Adams et al., 2010) were used for structure refinement until acceptable RWork and RFree were achieved.
Doubling Time Determination
A volume of 1 mL of overnight culture was used to inoculate 50 mL of LB media. Culture was grown at 37°C at 225 RPM. Growth was monitored at A600 every 30 minutes. The doubling time was determined from the linear portion of the growth curve using the following equation:
Synthetic Methods
The 1H and 13C NMR spectra were recorded on Bruker instruments at 400 MHz. Chemical shifts are reported in ppm and are referenced to residual DMSO solvent; 2.50 and 39.51 ppm for 1H and 13C respectively. The high-resolution mass spectrometry was provided by University of Connecticut Mass Spectrometry Laboratory using AccuTOF mass spectrometer with a DART source. Optical rotation was measured on a Jasco P-2000 polarimeter at 589nm. TLC analyses were performed on Sorbent Technologies silica gel HL TLC plates. All glassware was oven-dried and allowed to cool under an argon atmosphere. Anhydrous dichloromethane, ether, and tetrahydrofuran were used directly from Baker Cycle-Tainers. Anhydrous dimethylformamide was purchased from Acros and degassed by purging with argon. All reagents were used directly from commercial sources unless otherwise stated. A premixed heterogeneous mixture of CuI (10%/w) in Pd(PPh3)2Cl2–(Pd/Cu) was used for the Sonogashira coupling.
Procedure for the synthesis of (S)-4-(6-(4-(2,4-diamino-6-ethylpyrimidin-5-yl)but-3-yn-2-yl)benzo[d][1,3]dioxol-4-yl)benzoic acid
To a 20 mL screw cap vial with stirbar was added (0.57mmol, 0.15g, 1eq) ethyl-iododiaminopyrimidine, (0.05mmol, 0.03g, 0.08eq) Pd/Cu and (5.7mmol, 0.55g, 10eq) KOAc. Argon purged anhydrous DMF (0.05M, 11.3mL) was added followed by alkyne (0.73mmol, 0.25g, 1.3 eq). The reaction mixture was stirred under argon for 15 min and degassed once using freeze/pump/thaw method. The vial was sealed under argon, heated at 60 °C and reaction monitored by TLC. At the end of the reaction, the dark reddish brown solution was concentrated and product purified by flash column chromatography (for preabsorption of crude mixture −SiO2 in 10%/w of cysteine −1.5g, NH2 capped SiO2–1.5g), 13g SiO2 for column, 2% MeOH/CH2Cl2) to afford the coupled pyrimidine as pale brown solid. (0.2 g, 72% yield). TLC Rf = 0.4 (5% MeOH/CH2Cl2). The pyrimidine coupled t-butyl ester product (0.0411mmol, 0.02g, 1eq) in (0.02M, 2mL) d-CHCl3 cooled to 0 °C was deprotected using trifluoroacetic acid (TFA) (8.22mmol, 200 eq, 0.63mL). After dropwise addition, the reaction mixture was brought to room temperature. At the end of the reaction, monitored by NMR, the reaction mixture was rotoevaporated at 20 °C, kept under vacuum for 15 mins to remove excess TFA. To the product mixture containing a small amount of TFA was added anhydrous CH2Cl2 for preabsorption onto silica gel (1g). Flash column chromatography was performed (5g silica gel) initially with 100% EtOAc followed by 0.01% TFA in EtOAc; TLC Rf = 0.3 (10% MeOH/CH2Cl2 with 0.01%TFA). The clean fractions were rotoevaporated at 20 °C ensuring complete removal of solvent. The oily TFA salt was neutralized with phosphate buffer at pH 7. The resulting white precipitate along with buffer solution was transferred to an Eppendorf tube and centrifuged to separate the water from the precipitate. After decanting the water layer, the white precipitate was rinsed with diethyl ether and methanol to remove the water. The dried white solids with a tinge of pink color (0.01g, 57% yield) were subjected to characterization and biological evaluation. NMR spectra are shown in Fig. S3. 1H NMR (400 MHz, DMSO-d) δ 8.03 (d, J = 8.4 Hz, 2H), 7.86 (d, J = 8.4 Hz, 2H), 7.28 (s, 1H), 7.09 (s, 1H),6.28 (broad, 2H), 6.18 (s, 2H), 6.12 (s, 2H), 4.12 (q, J = 7 Hz, 1H), 2.55 (q, J = 7.6 Hz, 2H), 1.54 (d, J = 7.1 Hz, 3H), 1.11 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, DMSO-d) δ171.5, 167.1, 164.2, 161.1, 148.1, 143.4, 139.5, 138.4, 129.8, 129.7, 127.5, 120.2, 118.8, 107.3, 101.2, 100.5, 87.8, 76.0, 32.0, 28.8, 24.6, 12.4; HRMS (DART, M+ + H) m/z 431.1708 (calculated for C24H23N4O4, 431.1719); [α]24 +3.3° (c, 0.146, DMSO)
(R)-4-(6-(4-(2,4-diamino-6-ethylpyrimidin-5-yl)but-3-yn-2-yl)benzo[d][1,3]dioxol-4-yl)benzoic acid
To a 20 mL screw cap vial with stirbar was added (0.45mmol, 0.12g, 1eq) ethyl-iododiaminopyrimidine, (0.04mmol, 0.025g, 0.08eq) Pd/Cu and (4.47mmol, 0.44g, 10eq) KOAc. Argon purged anhydrous DMF (0.05M, 8.9mL) was added followed by alkyne (0.58mmol, 0.20g, 1.3 eq). Following the same workup as the (S) enantiomer, (R) enantiomer was obtained as a pale brown solid (0.164g, 75% yield). TLC Rf = 0.4 (5% MeOH/CH2Cl2); The pyrimidine coupled t-butyl ester product (0.062mmol, 0.03g, 1eq) in (0.02M, 3mL) d-CHCl3 cooled to 0 °C was deprotected using trifluoroacetic acid (TFA) (18.50mmol, 300 eq, 1.42mL). Repeating the same deprotection workup as above, (R) carboxylic acid was obtained as a white solid with a tinge of pink color (0.015g, 56% yield). TLC Rf = 0.3 (10% MeOH/CH2Cl2 with 0.01%TFA). NMR spectra are shown in Fig. S4. 1H NMR (400 MHz, DMSO-d) δ 8.03 (d, J = 7.6 Hz, 2H), 7.87 (d, J = 7.2 Hz, 2H), 7.27 (s, 1H), 7.09 (s, 1H),6.26 (broad, 2H), 6.15 (s, 2H), 6.12 (s, 2H), 4.11 (q, J = 7 Hz, 1H), 2.56 (m, 2H), 1.53 (d, J = 7.0 Hz, 3H), 1.11 (t, J = 7.5 Hz, 3H); 13C NMR (100 MHz, DMSO-d) δ171.6, 167.0, 164.2, 161.1, 148.1, 143.4, 139.5, 138.4, 129.8, 129.7, 127.5, 120.2, 118.8, 107.3, 101.2, 100.5, 87.8, 76.0, 32.0, 28.8, 24.6, 12.4; HRMS (DART, M+ + H) m/z 431.1708 (calculated for C24H23N4O4, 431.1719); [α]24 −5.2°(c, 0.143, DMSO).
Supplementary Material
Figure S1: Related to Table 1. Composite PCR Gel for Gene Identification
Figure S2. Related to Figure 3. OMIT Map of crystal structure of DfrB bound to NADPH and UCP1191
Figure S3. Related to Experimental. 1H and 13C NMR of UCP1205
Figure S4. Related to Experimental. 1H and 13C NMR of UCP1206
Highlights.
Three trimethoprim resistance genes have been identified in US MRSA isolates
dfrG and dfrK are identified for the first time in United States hospitals
Propargyl-linked antifolates inhibit resistant enzymes and bacteria
Structural studies indicate conserved arginine is responsible for potent activity
Acknowledgments
We thank Lidia Beka and Dr. Joerg Graf for assistance in assembling the genomes and the UCONN MARS facilities, specifically Kendra Maas, for assistance with sequencing. We also acknowledge NIH grants AI104841 and AI 111957.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
S.M.R. evaluated biological activity, characterized the strains, analyzed the genomes, determined the crystal structure; E.W.S. synthesized compound UCP1175; S.K. synthesized compounds UCP1164, UCP1172 and UCP1173; N.D. synthesized compounds UCP1039, UCP1191, UCP1205 and UCP1206; J.K expressed and purified DfrG, B.H expressed and purified DfrA, J.F. assisted in enzymatic assays, M.N. provided strains from Hartford Hospital and consulted on clinical impact; J.A. provided strains from UConn Health and consulted on clinical impact, strain typing and susceptibility analysis; D.L.W. directed all synthetic studies; A.C.A. directed all biological and structural studies. S.M.R, D.L.W. and A.C.A. wrote and edited the manuscript.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aeschlimann JR, Dresser LD, Kaatz GW, Rybak MJ. Effects of NorA inhibitors on in vitro antibacterial Activities and postantibiotic effects of levofloxacin,cirpofloxacin and norfloxacin in genetically related strains of Staphylococcus aureus. Antimicrob Agents Chemother. 1999;43:225–340. doi: 10.1128/aac.43.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann R, Van Der Linden M, Chhatwal GS, Nitsche-Schmitz DP. Factors that cause trimethoprim resistance in Streptococcus pyogenes. Antimicrob Agents Chemother. 2014;58:2281–2288. doi: 10.1128/AAC.02282-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettin T, Davis JJ, Disz T, Edwards RA, Gerdes S, Olsen GJ, Olson R, Overbeek R, Parrello B, Pusch GD, et al. RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci Rep. 2015;5:1–6. doi: 10.1038/srep08365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush K, Leal J, Fathima S, Li V, Vickers D, Chui L, Louie M, Taylor G, Henderson E. The molecular epidemiology of incident methicillin-resistant Staphylococcus aureus cases among hospitalized patients in Alberta, Canada: a retrospective cohort study. Antimicrob Resist Infect Control. 2015;4:35. doi: 10.1186/s13756-015-0076-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Center for Disease Control and Prevention. Centers for Disease Control and Prevention Outpatient antibiotic prescriptions — United States. 2013. [Google Scholar]
- Clin. Lab. Stand. Inst. CLSI performance standard of antimicrobial susceptibility testing: twenty-fourth international supplement. CLSI Doc. 2014 M100-S24. [Google Scholar]
- Daigle DM, Hughes DW, Wright AC. Prodigious substrate specificity of AAC(6′)-APH(2″), an aminoglycoside antibiotic resistance determinant in enterococci and staphylococci. Chem Biol. 1999;6:99–110. doi: 10.1016/S1074-5521(99)80006-4. [DOI] [PubMed] [Google Scholar]
- Dale GE, Broger C, Hartman PG, Langen H, Page MGP, Then RL, Stuber D. Characterization of the gene for the chromosomal dihydrofolate reductase (DHFR) of Staphylococcus epidermidis ATCC 14990: The origin of the trimethoprim-resistant S1 DHFR from Staphylococcus aureus. J Bacteriol. 1995;177:2965–2970. doi: 10.1128/jb.177.11.2965-2970.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale GE, Broger C, D’Arcy A, Hartman PG, DeHoogt R, Jolidon S, Kompis I, Labhardt AM, Langen H, Locher H, et al. A single amino acid substitution in Staphylococcus aureus dihydrofolate reductase determines trimethoprim resistance. J Mol Biol. 1997;266:23–30. doi: 10.1006/jmbi.1996.0770. [DOI] [PubMed] [Google Scholar]
- Dao BD, Barreto JN, Wolf RC, Dierkhising RA, Plevak MF, Tosh PK. Serum peak sulfamethoxazole concentrations demonstrate difficulty in achieving a target range: A retrospective cohort study. Curr Ther Res Clin Exp. 2014;76:104–109. doi: 10.1016/j.curtheres.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Gopegui ER, Juan C, Zamorano L, Pérez JL, Oliver A. Transferable multidrug resistance plasmid carrying cfr associated with tet (L), ant (4)-Ia, and dfrK genes from a clinical methicillin-resistant Staphylococcus aureus ST125 strain. Antimicrob Agents Chemother. 2012;56:2139–2142. doi: 10.1128/AAC.06042-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr Sect D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Frei CR, Miller ML, Lewis JS, Lawson Ka, Hunter JM, Oramasionwu CU, Talbert RL. Trimethoprim-sulfamethoxazole or clindamycin for community-associated MRSA (CA-MRSA) skin infections. J Am Board Fam Med. 2010;23:714–719. doi: 10.3122/jabfm.2010.06.090270. [DOI] [PubMed] [Google Scholar]
- Frey KM, Liu J, Lombardo MN, Bolstad DB, Wright DL, Anderson AC. Crystal structures of wild-type and mutant methicillin-resistant Staphylococcus aureus dihydrofolate reductase reveal an alternate conformation of NADPH that may be linked to trimethoprim resistance. J Mol Biol. 2009;387:1298–1308. doi: 10.1016/j.jmb.2009.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey KM, Lombardo MN, Wright DL, Anderson AC. Towards the understanding of resistance mechanisms in clinically isolated trimethoprim-resistant, methicillin-resistant Staphylococcus aureus dihydrofolate reductase. J Struct Biol. 2010;170:93–97. doi: 10.1016/j.jsb.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey KM, Georgiev I, Donald BR, Anderson AC. Predicting resistance mutations using protein design algorithms. Proc Natl Acad Sci USA. 2010;107:13707–13712. doi: 10.1073/pnas.1002162107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey KM, Viswanathan K, Wright DL, Anderson AC. Prospective screening of novel antibacterial inhibitors of dihydrofolate reductase for mutational resistance. Antimicrob Agents Chemother. 2012;56:3556–3562. doi: 10.1128/AAC.06263-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorwitz RJ, Jernigan DB, Powers JH, Jernigan JA. Participants in the CDC-convened experts’ meeting on management of MRSA in the community: Summary of an experts meeting convened by the Centers for Disease Control and Prevention. 2006. [Google Scholar]
- Hampele IC, D’Arcy A, Dale GE, Kostrewa D, Nielsen J, Oefner C, Page MG, Schönfeld HJ, Stüber D, Then RL. Structure and function of the dihydropteroate synthase from Staphylococcus aureus. J Mol Biol. 1997;268:21–30. doi: 10.1006/jmbi.1997.0944. [DOI] [PubMed] [Google Scholar]
- Heaslet H, Harris M, Fahnoe K, Sarver R, Putz H, Chang J, Subramanyam C, Barreiro G, Miller JR. Structural comparison of chromosomal and exogenous dihydrofolate reductase from Staphylococcus aureus in complex with the potent inhibitor trimethoprim. Proteins Struct Funct Bioinforma. 2009;76:706–717. doi: 10.1002/prot.22383. [DOI] [PubMed] [Google Scholar]
- Huovinen P, Sundström L, Swedberg G, Sköld O. Trimethoprim and sulfonamide resistance. Antimicrob Agents Chemother. 1995;39:279. doi: 10.1128/aac.39.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juda M, Chudzik-Rzad B, Malm A. The prevalence of genotypes that determine resistance to macrolides, lincosamides, and streptogramins B compared with spiramyin susceptibility among erythromycin-resistant Staphylococcus eidermidis. Mem Inst Oswaldo Cruz. 2016;111:155–160. doi: 10.1590/0074-02760150356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaatz GW, Seo SM. Inducible NorA-mediated multidrug resistance in Staphylococcus aureus. Antimicrob Agents Chemother. 1995;39:2650–2655. doi: 10.1128/aac.39.12.2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadlec K, Schwarz S. Identification of a plasmid-borne resistance gene cluster comprising the resistance genes erm(T), dfrK, and tet(L) in a porcine methicillin-resistant Staphylococcus aureus ST398 strain. Antimicrob Agents Chemother. 2010;54:915–918. doi: 10.1128/AAC.01091-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshipeddy S, Reeve SM, Anderson AC, Wright DL. Non-racemic antifolates stereo-selectively recruit alternate cofactors and overcome resistance in S. aureus. J Am Chem Soc. 2015;137:8983–8990. doi: 10.1021/jacs.5b01442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SA, Nawasz MS, Khan AA, Cerniglia CE. Simultaneous detection of erythrmycin-resistant methylase genes ermA and ermC from Staphylococcus spp. by multiplex-PCR. Mol and Cell Probes. 1999;13:381–387. doi: 10.1006/mcpr.1999.0265. [DOI] [PubMed] [Google Scholar]
- Lamb KM, Lombardo MN, Alverson J, Priestley ND, Wright DL, Anderson AC. Crystal structures of Klebsiella pneumoniae dihydrofolate reductase bound to propargyl-linked antifolates reveal features for potency and selectivity. Antimicrob Agents Chemother. 2014;58:7484–7491. doi: 10.1128/AAC.03555-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Feng W, Zhang Z, Xue H, Zhao X. Macolide-lincosamide-streptogramin resistance phenotypes and genotypes of coagulase-positive Staphylococcus aureus and coagulase-negative staphylococcal isolates from bovine mastitis. BMC Vet Res. 2015;11:1–8. doi: 10.1186/s12917-015-0492-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lina G, Piemont Y, Godail-Gomot F, Bes M, Peter M, Gauduchon V, Vandenesch F, Etienne J. Panton-Valenitine Leukocidin in staphylococcal infections. Clin Infect Dis. 1999;29:1128–1132. doi: 10.1086/313461. [DOI] [PubMed] [Google Scholar]
- Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, Kaplan SL, Karchmer AW, Levine DP, Murray BE, et al. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011;52:18–55. doi: 10.1093/cid/ciq146. [DOI] [PubMed] [Google Scholar]
- Lodise TP, Jr, McKinnon PS. Burden of methicillin-resistant Staphylococcus aureus: focus on clinical and economic outcomes. Pharmacotherapy. 2007;27:1001–1012. doi: 10.1592/phco.27.7.1001. [DOI] [PubMed] [Google Scholar]
- Lombardo MN, G-Dayanandan N, Wright DL, Anderson AC. Crystal structures of trimethoprim-resistant DfrA1 rationalize potent Inhibition by propargyl-linked antifolates. ACS Infect Dis. 2016;2:149–156. doi: 10.1021/acsinfecdis.5b00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka M, Inoue M, Endo Y, Nakajima Y. Characterization and expression of three genes, msr(A), mph(C) and erm(Y), that confer resistance to macrolide antibiotics in Staphylococcus aureus. FEMS Micro Lett. 2003;220:287–293. doi: 10.1016/S0378-1097(03)00134-4. [DOI] [PubMed] [Google Scholar]
- Nathwani D, Morgan M, Masterton RG, Dryden M, Cookson BD, French G, Lewis D. Guidelines for UK practice for the diagnosis and management of methicillin-resistant Staphylococcus aureus (MRSA) infections presenting in the community. J Antimicrob Chemother. 2008;61:976–994. doi: 10.1093/jac/dkn096. [DOI] [PubMed] [Google Scholar]
- Nurjadi D, Olalekan AO, Layer F, Shittu AO, Alabi A, Ghebremedhin B, Schaumburg F, Hofmann-Eifler J, Van Genderen PJJ, Caumes E, et al. Emergence of trimethoprim resistance gene dfrG in Staphylococcus aureus causing human infection and colonization in sub-Saharan Africa and its import to Europe. J Antimicrob Chemother. 2014;69:2361–8. doi: 10.1093/jac/dku174. [DOI] [PubMed] [Google Scholar]
- Nurjadi D, Friedrich-Jänicke B, Schäfer J, Van Genderen PJJ, Goorhuis A, Perignon A, Neumayr A, Mueller A, Kantele A, Schunk M, et al. Skin and soft tissue infections in intercontinental travellers and the import of multi-resistant Staphylococcus aureus to Europe. Clin Microbiol Infect. 2015;21:567. doi: 10.1016/j.cmi.2015.01.016. [DOI] [PubMed] [Google Scholar]
- Oefner C, Parisi S, Schulz H, Lociuro S, Dale GE. Inhibitory properties and X-ray crystallographic study of the binding of AR-101, AR-102 and iclaprim in ternary complexes with NADPH and dihydrofolate reductase from Staphylococcus aureus. Acta Crystallogr Sect D Biol Crystallogr. 2009;65:751–757. doi: 10.1107/S0907444909013936. [DOI] [PubMed] [Google Scholar]
- Oefner C, Bandera M, Haldimann A, Laue H, Schulz H, Mukhija S, Parisi S, Weiss L, Lociuro S, Dale GE. Increased hydrophobic interactions of iclaprim with Staphylococcus aureus dihydrofolate reductase are responsible for the increase in affinity and antibacterial activity. J Antimicrob Chemother. 2009;63:687–698. doi: 10.1093/jac/dkp024. [DOI] [PubMed] [Google Scholar]
- Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M, et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST) Nucleic Acids Res. 2014;42:D206–D214. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Hamlyn PJ, Talens-Visconti R, Alovero FL, Manzo RH, Fisher LM. Small-colony mutants of Staphylococcus aureus allow selection of gyrase-mediated resistance to dualt-target fluoroquinolones. Antimicrob Agents Chemother. 2002;46:2498–2506. doi: 10.1128/AAC.46.8.2498-2506.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pate AJ, Terribilini RG, Ghobadi F, Azhir A, Barber A, Pearson JM, Kalantari H, Hassen GW. Antibiotics for methicillin-resistant Staphylococcus aureus skin and soft tissue infections: the challenge of outpatient therapy. Am J Emerg Med. 2014;32:135–138. doi: 10.1016/j.ajem.2013.10.026. [DOI] [PubMed] [Google Scholar]
- Petersson AC, Olsson-Liljequist B, Miörner H, Haeggman S. Evaluating the usefulness of spa typing, in comparison with pulsed-field gel electrophoresis, for epidemiological typing of methicillin-resistant Staphylococcus aureus in a low-prevalence region in Sweden 2000–2004. Clin Microbiol Infect. 2010;16:456–462. doi: 10.1111/j.1469-0691.2009.02881.x. [DOI] [PubMed] [Google Scholar]
- Reeve SM, Gainza P, Frey KM, Georgiev I, Donald BR, Anderson AC. Protein design algorithms predict viable resistance to an experimental antifolate. Proc Natl Acad Sci USA. 2014;112:749–754. doi: 10.1073/pnas.1411548112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve SM, Scocchera EW, Ferriera JF, Dayanadan NG, Keshipeddy S, Wrght DL, Anderson AC. Charged propargyn-linked antifolates reveal mechanisms of antifolate resistance and inhibit trimethoprim-resistant MRSA strains possessing clinically relevant mutations. J Med Chem. 2016;59:6493–6500. doi: 10.1021/acs.jmedchem.6b00688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sader HS, Farrell DJ, Flamm RK, Jones RN. Activity of ceftaroline and comparator agents tested against Staphylococcus aureus from patients with bloodstream infections in US medical centres (2009–13) J Antimicrob Chemother. 2015;70:2053–2056. doi: 10.1093/jac/dkv076. [DOI] [PubMed] [Google Scholar]
- Schmitz F, Jones ME, Hofmann B, Hansen B, Scheuring S, Luckefahr M, Fluit A, Verhoef J, Hadding U, Heinz H, Kohrer K. Characterization of grlA, grlB, gyrA and gyrB mutations in 116 unrelated isolates of Staphylococcus aureus and effects of mutations on ciprofloxacin. Antimicrob Agents Chemother. 1998;42:1249–1252. doi: 10.1128/aac.42.5.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scocchera E, Reeve SM, Keshipeddy S, Lombardo MN, Hajian B, Sochia AE, Alverson JB, Priestley ND, Anderson AC, Wright DL. Charged nonclassical antifolates with activity against gram-positive and gram-negative pathogens. ACS Med Chem Lett. 2016;7:692–696. doi: 10.1021/acsmedchemlett.6b00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi J, Tharavichitkul P, Miyoshi-Akiyama T, Chupia V, Fujino T, Araake M, Irie A, Morita K, Kuratsuji T, Kirikae T. Cloning and characterization of a novel trimethoprim-resistant dihydrofolate reductase from a nosocomial isolate of Staphylococcus aureus CM. S2 (IMCJ1454) Antimicrob Agents Chemother. 2005;49:3948–3951. doi: 10.1128/AAC.49.9.3948-3951.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzcinski K, Cooper BS, Hryniewicz W, Dowson CG. Expression of resistance to tetracyclines in strains of methicillin-resistant Staphylococcus aureus. J Antimicrob Chemother. 2000;45:763–770. doi: 10.1093/jac/45.6.763. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Related to Table 1. Composite PCR Gel for Gene Identification
Figure S2. Related to Figure 3. OMIT Map of crystal structure of DfrB bound to NADPH and UCP1191
Figure S3. Related to Experimental. 1H and 13C NMR of UCP1205
Figure S4. Related to Experimental. 1H and 13C NMR of UCP1206