

The polarity regulator Par3 directs the reorganization of the apical membrane in polarized cells through the action of phosphatidylinositol 3-kinase and the Rac1 exchange factor Tiam1. This mechanism may be exploited by pathogens to bypass the epithelial barrier.
Abstract
Pathogens can alter epithelial polarity by recruiting polarity proteins to the apical membrane, but how a change in protein localization is linked to polarity disruption is not clear. In this study, we used chemically induced dimerization to rapidly relocalize proteins from the cytosol to the apical surface. We demonstrate that forced apical localization of Par3, which is normally restricted to tight junctions, is sufficient to alter apical membrane identity through its interactions with phosphatidylinositol 3-kinase (PI3K) and the Rac1 guanine nucleotide exchange factor Tiam1. We further show that PI3K activity is required upstream of Rac1, and that simultaneously targeting PI3K and Tiam1 to the apical membrane has a synergistic effect on membrane remodeling. Thus, Par3 coordinates the action of PI3K and Tiam1 to define membrane identity, revealing a signaling mechanism that can be exploited by human mucosal pathogens.
INTRODUCTION
The mucosal barrier, composed of polarized epithelial cells with distinct apical and basolateral membranes, represents the first line of defense against most microbes (Weitnauer et al., 2016). Understanding how successful pathogens overcome this barrier may provide unique insights into how cell polarity is established and maintained, as well as shed light on other disease processes such as cancer metastasis and tissue repair (Ruch and Engel, 2017). When the important human pathogen Pseudomonas aeruginosa binds to the apical surface of epithelial cells, it forms large bacterial aggregates and induces a dramatic remodeling of the apical membrane. The patch of membrane in contact with the bacterial aggregate loses apical proteins, acquires basolateral membrane characteristics, including accumulation of basolateral proteins and lipids, and forms an actin-rich “protrusion” (Kierbel et al., 2007). This change in membrane structure is dependent upon the activity and recruitment of the polarity protein Par3, phosphatidylinositol 3-kinase (PI3K), and the small GTPase Rac1. Remarkably, protrusion formation is associated with localized activation of the innate immune response, suggesting that the innate immune system may detect changes in cell polarity (Tran et al., 2014). Similarly, Neisseria meningitidis, an important cause of fatal meningitis, recruits polarity proteins, including Par3, to the apical membrane of endothelial cells leading to the formation of an ectopic junction underneath N. meningitidis. The end result of this process is disruption of adherens junction function and bacterial dissemination across the endothelial barrier (Coureuil et al., 2009). Thus, relocalization of Par3 to the apical membrane is associated with dramatic changes in apical membrane identity in the context of host–pathogen interactions, but the mechanism underlying this change is incompletely understood.
Par3 plays a critical role in organizing signaling events during establishment and maintenance of apical–basal polarization. In mammalian cells, Par3 localizes to the apical membrane initiation site (AMIS) and helps to recruit aPKC and Par6. As the AMIS matures, Par3 moves away from the nascent apical membrane and localizes to the tight junction (Bryant et al., 2010). Thus, while Par3 plays a role at the apical membrane early in polarization, it is excluded from the apical domain in a stable epithelium. The effect of targeting Par3 to the apical membrane in well-polarized cells is completely unknown, but based on the data from pathogens, it may drive changes to lipid or protein components of the membrane.
PI3K and Rac1 signaling are crucial for both cell polarity and P. aeruginosa–induced protrusion formation (Rodriguez-Boulan and Macara, 2014; Tran et al., 2014). PI3K is a key regulator of apical and basolateral membrane identity by virtue of its ability to convert phosphatidylinositol 4,5-phosphate (PIP2), which is enriched at the apical membrane, to phosphatidylinositol 3,4,5-phosphate (PIP3), which is restricted to the basolateral membrane (Shewan et al., 2011). The Rho GTPase Rac1 contributes to cell polarity by regulating actomyosin contractility and cell–cell adhesion and establishing and maintaining junctional structures (Iden and Collard, 2008; Mack and Georgiou, 2014). Par3 modulates PIP identity by activating the lipid kinase PI3K and spatially restricting the activity of the cognate lipid phosphatase, phosphatase and tensin homologue (PTEN; Wu et al., 2007; Feng et al., 2008; Itoh et al., 2010; Krahn et al., 2010). This property, combined with the observation that Par3 can bind directly to both PIP2 and PIP3, has led to speculation that Par3 may act as a PIP gate at the tight junction (Wu et al., 2007; Krahn et al., 2010). Par3 controls Rac1 activity by directly interacting with the Rac1 guanine nucleotide exchange factor Tiam1 (Chen and Macara, 2005; Mertens et al., 2005). This interaction localizes Rac1 activity to the tight junction (Mack et al., 2012). Par3 also directly interacts with the Rac1 GTPase-activating protein Bcr1, which restricts the activity of Rac1 and aPKC (Narayanan et al., 2013). By binding to Tiam1 and Bcr1, Par3 can finely tune and spatially localize Rac1 activity within polarized epithelial cells and in turn modulate actin dynamics. Thus, Par3 cross-talks extensively with both PI3K and Rac1 signaling.
To illuminate how Par3 hijacking by pathogens affects cell polarity, we use chemically induced dimerization (CID) in polarized Madin–Darby canine kidney (MDCK) cells. We show that forced apical localization of Par3 is sufficient to remodel apical membrane polarity by coordinating the action of PI3K and Tiam1, which function synergistically to change apical membrane identity. These results define a mechanism for dynamic membrane remodeling in polarized epithelial cells that can be targeted by pathogens, and provides a rationale as to why Par3 is restricted from the apical membrane in polarized cells.
RESULTS AND DISCUSSION
Forced apical localization of Par3 is sufficient to transform the apical membrane and recapitulate aspects of P. aeruginosa–induced protrusions
To understand how the localization of Par3 affects membrane identity, we used CID to rapidly target Par3 to the apical surface. CID uses two proteins (FRB domain of mTor and FKBP12) that interact only in the presence of a small-molecule dimerizer called Rapalog (Putyrski and Schultz, 2012). To generate an apical membrane–targeting construct, we fused the FRB domain of mTOR to the apically targeted integral membrane protein Crb3a lacking the C-terminal PDZ-interacting motif (AP-FRB; Zheng et al., 2010). Par3 was fused to monomeric red fluorescent protein (mRFP)–FKBP12 (FK-Par3), or, as a control, a triple-hemagglutinin (3xHA) tag was fused to mRFP-FKBP12 (FK-HA; Supplemental Figure S1A). The constructs were simultaneously introduced via transient transfection into MDCK cells, resulting in production of full-length chimeric proteins (Supplemental Figure S2B). These MDCK cells also stably express PH-Akt-GFP, which binds to the basolateral lipid PIP3 (Watton and Downward, 1999). We specifically chose transient transfection because of its low efficiency in MDCK cells, resulting in “mosaic” monolayers in which a few transfected cells were surrounded by untransfected cells. This approach allowed us to closely mimic bacterial infections in which P. aeruginosa forms bacterial aggregates on the apical surface of only a small fraction of cells (Tran et al., 2014).
In the absence of the dimerizing agent Rapalog, AP-FRB was tightly localized to the apical membrane, whereas FK-Par3 and FK-HA were primarily cytosolic, likely due to overexpression (Figure 1A). Within 60 min of Rapalog addition, FK-HA or FK-Par3 was concentrated at the apical membrane, confirming that this system allows for the rapid relocalization of target proteins to the apical membrane (Figure 1A). As an additional control, we verified that transfection of each construct individually did not alter apical PIP3 or actin morphology (Supplemental Figure S1B). Forced apical localization of FK-Par3 but not FK-HA was accompanied by accumulation of PH-Akt-GFP on the apical surface and a striking “bulging” or “pinching” of apical actin (Figure 1A and Supplemental Figure S1C). These qualitative observations were confirmed by quantitation in which Rapalog-induced targeting of Par3 to the apical membrane caused a robust and statistically significant increase in apical PIP3 and change in apical actin morphology, which were not observed in the absence of Rapalog (Figure 1B; see Materials and Methods for details).
FIGURE 1:
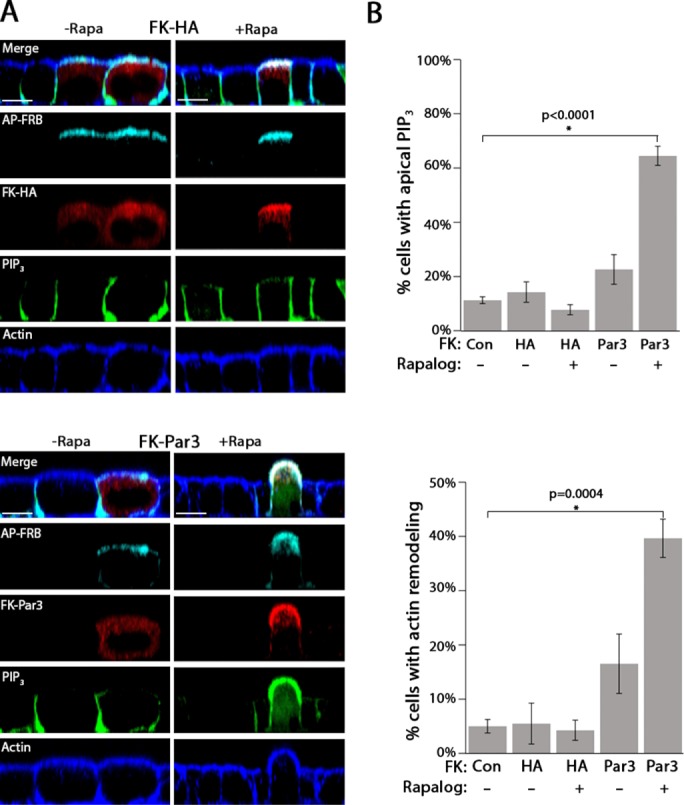
Par3 drives apical membrane remodeling. (A) Single XZ-slice from a confocal micrograph of MDCK cells expressing PH-Akt-GFP (PIP3; green) transfected with AP-FRB (cyan; FLAG) and either FK-HA or FK-Par3 (red; HA) treated with vehicle (–Rapa) or 200 nM Rapalog (+Rapa) for 60 min. Actin was visualized with phalloidin staining (blue). (B) Quantitation of apical PIP3 and actin rearrangement in control cells (not expressing a construct) or cells expressing FK-HA or FK-Par3 in the presence or absence of Rapalog (see Materials and Methods for details). Error bars, SEM. The p value was determined by one-way ANOVA followed by post hoc Tukey’s test. n = 4. Scale bar, 10 µm.
The aforementioned morphological alterations resemble the changes observed upon apical addition of P. aeruginosa to polarized epithelial cells (Tran et al., 2014). In that context, apical membrane polarity is inverted at the site of bacterial aggregate binding, accompanied by Par3 recruitment and formation of an actin-rich protrusion (Kierbel et al., 2007; Tran et al., 2014). To explore further how forced apical Par3 localization alters cell polarity, we quantified the surface intensity of the apical membrane protein podocalyxin (PodxI), the apical cortex protein ezrin, and the basolateral membrane proteins p58 and E-cadherin, after targeting FK-HA or FK-Par3 to the apical membrane for 60 min (see Materials and Methods for details of the quantitation). Apical localization of FK-Par3 resulted in a significant decrease in apical intensity of PodxI and an increase in the apical intensity of p58, E-cadherin, and ezrin (Figure 2). These results show that some degree of polarity inversion occurs after apical targeting of Par3. The fact that ezrin shows increased intensity at the apical membrane may reflect the development of generalized rather than apical-specific cortical ezrin targeting upon apical Podxl loss (Bryant et al., 2014). Of importance, these changes were not associated with apoptosis, as assessed by staining for activated caspase-3, or with disruption of the tight junctions, as judged by ZO-1 distribution (Supplemental Figure S3, A and B), ruling out the possibility that forced apical localization of Par3 leads to cell death or cell extrusion. We conclude that apical localization of FK-Par3 is sufficient to alter the lipid identity, protein identity, and actin morphology of the apical membrane without affecting tight junction integrity. We refer to this change as “apical membrane remodeling.”
FIGURE 2:

Forced apical targeting of Par3 changes the distribution of proteins at the apical membrane. Confocal images (single XZ-slice and maximum intensity XY-projection) showing the apical surface intensity of PodxI (A), ezrin (B), p58 (C), or E-cadherin (D; cyan) in MDCK cells transfected with AP-FRB and either FK-HA (red, HA) or FK-Par3 (red, HA) and treated with 200 nM Rapalog for 60 min. Actin was visualized with phalloidin staining (blue). The bar graphs show quantitation (see Materials and Methods for details). Cells with >25% drop (PodxI) or >25% increase (Ezrin, E-Cadherin, p58) in the indicated marker were scored as positive. Error bars, SEM. The p value was determined by one-way ANOVA followed by post hoc Tukey’s test. n ≥ 3. Scale bar, 10 µm.
We previously showed that P. aeruginosa aggregate binding and protrusion formation is associated with Par3-dependent NFκB activation (Tran et al., 2014). We tested whether apical membrane remodeling was sufficient for NFκB activation by staining for NFκB in cells undergoing CID. Nuclear localization of NFκB in cells was not observed upon forced apical localization of Par3. As a positive control, addition of purified flagellin was sufficient to induce NFκB nuclear translocation (Supplemental Figure S3C). We conclude that additional signals beyond apical membrane remodeling, such as bacterial molecules, are required for innate immune activation after P. aeruginosa aggregate binding. We hypothesize that the change in membrane identity may allow pathogen recognition receptors that are normally restricted to the basolateral surface to access the apical space and detect pathogens (Abreu, 2010). Alternatively, activation of the host innate immune response may require detection of two or more signals, such as apical actin remodeling combined with the presence of a pathogen-associated molecular pattern (Keestra et al., 2013; Rajamuthiah and Mylonakis, 2014). Indeed, such a finely tuned mechanism might allow sensitive and accurate discrimination of pathogens from commensals or nonpathogenic microbes (Keestra and Baumler, 2014).
The CC1 region of Par3 is necessary but not sufficient for apical membrane remodeling
We tested the hypothesis that the first coiled-coil region in the C-terminus of Par 3 (Supplemental Figure S2A), which has been shown to interact with PI3K and Tiam1 and thus affect both PI3K and Rac1 signaling, is required for apical membrane remodeling (Chen and Macara, 2005; Itoh et al., 2010). We generated a mutant version of FK-Par3 in which the CC1 domain was deleted (FK-ΔCC1). Full-length FK-ΔCC1 was detected by Western blot, showed similar localization to FK-Par3 in the absence of Rapalog, and was efficiently recruited to the apical membrane upon Rapalog addition (Figure 3A and Supplemental Figure S2B). However, apical translocation of FK-∆CC1 did not significantly change the level of apical PIP3 or significantly alter apical actin morphology (Figure 3B).
FIGURE 3:

The CC1 region of Par3 is necessary but not sufficient to drive apical membrane remodeling. (A) Confocal XZ-slices of MDCK cells expressing PH-Akt-GFP (PIP3; green) transfected with AP-FRB and FK-∆CC1 (red; HA) and treated with vehicle (–Rapa) or 200 nM Rapalog (+Rapa) for 60 min. Actin was visualized with phalloidin staining (blue). (B) Quantitation of the appearance of apical PIP3 and actin rearrangement in cells transfected with the indicated constructs in the presence or absence of Rapalog. (C) Confocal XZ-slices of cells expressing FK-CC1. (D) Quantitation of the appearance of apical PIP3 and actin rearrangement in cells transfected with the indicated constructs in the presence or absence of Rapalog. Error bars, SEM. The p value was determined by one-way ANOVA followed by post hoc Tukey’s test. n = 3. Scale bar, 10 µm.
To determine whether the CC1 region was sufficient to drive apical membrane remodeling, we cloned the CC1 region of Par3 behind mRFP-FKBP to generate FK-CC1. Following transfection, FK-CC1 was expressed as a full-length protein, showed cytosolic/membrane localization, and was robustly recruited to the apical membrane upon Rapalog addition (Figure 3C and Supplemental Figure S2B). Quantitation revealed that Rapalog-induced recruitment of FK-CC1 to the apical membrane did not significantly increase apical PIP3 or apical actin rearrangement compared with control cells (Figure 3D). Taken together, these results show that forced apical localization of the CC1 region of Par3 is necessary but not sufficient to drive apical membrane remodeling.
Rac1 and PI3K are necessary for Par3-induced apical membrane remodeling
We tested whether PI3K or Rac1/Tiam1 activity was required for Par3-mediated apical membrane remodeling, as they both interact with Par3 via the CC1 domain, and their activity is essential for mediating apical membrane changes underneath P. aeruginosa aggregates (Chen and Macara, 2005; Kierbel et al., 2007; Itoh et al., 2010; Tran et al., 2014). We performed CID in the presence of chemical inhibitors of PI3K (LY294002) or Rac1 (Rac1 inhibitor II). LY294002 blocked both accumulation of apical PIP3 and apical actin rearrangement. The Rac1 inhibitor blocked apical actin rearrangement, but accumulation of apical PIP3 was not affected (Figure 4, A and B). We conclude that PI3K activity is required for apical PIP3 generation, which occurs before Rac1-mediated apical actin rearrangement.
FIGURE 4:

PI3K and Rac1 activity are required for Par3-mediated apical membrane remodeling. (A) Confocal XZ-slices of MDCK cells expressing PH-Akt-GFP (PIP3; green) transfected with AP-FRB and FK-Par3 (red; HA), treated with 200 nM Rapalog (+Rapa) for 60 min, and, where indicated, treated with Rac1 inhibitor (200 µM) or LY294002 (50 µM). Actin was visualized with phalloidin staining (blue). (B) Quantitation of the appearance of apical PIP3 and actin rearrangement in cells transfected with the indicated constructs in the presence of the indicated drugs. Error bars, SEM. The p value was determined by one-way ANOVA followed by post hoc Tukey’s test. n ≥ 3. Scale bar, 10 µm.
We next determined whether apical targeting of PI3K or Tiam1 was sufficient to drive apical membrane remodeling. For PI3K, the iSH2 domain of the p85 subunit was cloned behind mRFP-FKBP to generate FK-iSH2 (Suh et al., 2006). Tiam1 was cloned behind mRFP-FKBP to generate FK-Tiam1. Both constructs were expressed as full-length proteins (Supplemental Figure S2C). Upon Rapalog addition, FK-iSH2 or FK-Tiam1 was recruited from the cytosol to the apical membrane (Figure 5, A and D). Quantitation revealed that forced apical localization of FK-iSH2 significantly increased apical PIP3 without altering apical actin morphology (Figure 5D). The opposite results were observed for Rapalog-induced forced apical localization of FK-Tiam1: there was a significant increase in apical actin rearrangement but no significant change in apical PIP3 (Figure 5, B and D). Next we coexpressed both FK-iSH2 and FK-Tiam1 in cells. To ensure equal expression of both constructs, a dual-expression plasmid was generated using a P2A sequence from porcine teschovirus-1 (Kim et al., 2011). In this system, a single mRNA is generated that encodes both proteins, and after translation, it self-cleaves to release the two proteins. Cells transfected with this dual-expression plasmid produced both FK-iSH2 and FK-Tiam1 as full-length proteins (Supplemental Figure S2D). In cells expressing both FK-iSH2 and FK-Tiam1, addition of Rapalog resulted in a significant increase in both apical PIP3 and apical actin rearrangement (Figure 5, C and D). Remarkably, the increase in apical actin rearrangement was significantly greater than that observed for FK-Tiam1 alone (Figure 5D), suggesting that PI3K and Tiam1 act synergistically to drive apical membrane remodeling. We conclude that coordinated activity of PI3K and Tiam1 is required for apical membrane remodeling.
FIGURE 5:

PI3K and Tiam1 are sufficient to drive apical membrane remodeling. (A–C) Confocal XZ-slices of MDCK cells expressing PH-Akt-GFP (PIP3; green), stained for actin (blue; phalloidin), and transfected with AP-FRB and (A) FK-iSH2 (red; HA), (B) FK-Tiam1 (red; HA), or (C) both constructs (red, FK-iSH2; cyan, FI-Tiam1) and treated with vehicle control (–rapa) or 200 nM Rapalog (+rapa) for 60 min. (D) Quantitation of immunofluorescence of apical PIP3 or apical actin rearrangement for the indicated constructs and treatments. Error bars, SEM. The p value was determined by one-way ANOVA followed by post hoc Tukey’s test. n ≥ 3. Scale bar, 10 µm.
By applying CID to direct apical membrane protein targeting, we interrogated how protein localization affects polarity with high spatiotemporal resolution. To the best of our knowledge, our experiments represent the first example of using CID to target proteins to the apical membrane in polarized cells. Our work demonstrates that that forced apical localization of Par3 reprograms apical membrane identity by integrating PI3K and Tiam1 signaling. We further show that the requirement of PI3K activity is upstream of Tiam1, the action of both proteins is required for apical membrane remodeling, and PI3K and Tiam1 act synergistically to reinforce membrane changes. Par3 has been implicated in both the activation and suppression of Rac1 activity (Chen and Macara, 2005; Zhang and Macara, 2006; Pegtel et al., 2007; Mack et al., 2012; Matsuzawa et al., 2016); our work suggests that Par3 tethers Tiam1 to specific cellular locations and then relies on the local protein environment to dictate whether Rac1 is activated or repressed. It is also remarkable that targeting Tiam1 and PI3K to the apical membrane could drive membrane remodeling, and expression of both proteins had a synergistic effect on apical membrane remodeling. Thus it seems likely that a positive feedback loop exists between PI3K and Rac1 in this context, as previously suggested (Balla, 2013). Note that targeting the CC1 region of Par3, which binds to both proteins, is not sufficient to drive apical membrane remodeling. Because Par3 is believed to function as a dimer, it may be that the dimeric form of the CC1 region is necessary to bind Tiam1 and PI3K in vivo (Mizuno et al., 2003).
Par3 is at the apical membrane early during polarity establishment but is later localized to the tight junction (Bryant et al., 2010; Morais-deSa et al., 2010). Why Par3 is restricted from the apical domain after polarity establishment has been an important unanswered question. Our results demonstrate that the exclusion of Par3 is necessary for apical membrane stability. We speculate that Par3 may direct changes in protein trafficking at the apical membrane, likely through endocytosis of apical proteins and transcytosis of basolateral proteins, processes known to be regulated by Par3 and by P. aeruginosa (Kierbel et al., 2007; Nishimura and Kaibuchi, 2007; Horikoshi et al., 2009; Bryant et al., 2010; Laketa et al., 2014; Matsui et al., 2015; Sun et al., 2016).
Par3 is increasingly recognized as a pathogen target. In addition to P. aeruginosa, both N. meningitidis and human papillomavirus-18 target Par3 to disrupt polarity and junction formation (Coureuil et al., 2009; Facciuto et al., 2014). Thus, understanding how Par3 is linked to changes in membrane identity and innate immunity is an important question. Our approach allowed us to compare P. aeruginosa–induced apical membrane remodeling with Par3-induced apical membrane remodeling. Although there were similarities in the biochemical and morphological changes to the apical membrane, apical targeting of Par3 was not sufficient to induce localized NFκB activation. This finding suggests that additional or alternate bacterial or host signals are required. Of note, others have reported that Par3 inhibits NFκB activation in polarized epithelial cells by restricting aPKC activation. Thus Par3 may intersect with NFκB signaling in multiple ways (Mashukova et al., 2011; Forteza et al., 2013; Guyer and Macara, 2015). It may be important to examine the interplay between aPKC and Par3 in future studies. In summary, using CID, we defined a molecular signaling cascade that can drive dynamic changes in membrane identity in polarized epithelial cells that can be usurped by pathogens.
MATERIALS AND METHODS
Antibodies and reagents
Antibodies were obtained from the following sources. Rat anti-HA monoclonal antibody (mAb; 3F10) was from Roche Life Science (Indianapolis, IN); mouse anti-HA mAb was from Covance (Princeton, NJ); mouse anti-FLAG mAb was from ThermoFisher (Waltham, MA); rat anti-ZO1 antibody and mouse monoclonal anti-p65 antibody were from Santa Cruz Biotechnology (Santa Cruz, CA); monoclonal rabbit anti-Caspase-3 antibody was from Cell Signaling Technology (Danvers, MA); monoclonal mouse anti-ezrin antibody was from BD Biosciences (San Jose, CA); monoclonal mouse anti–E-cadherin antibody was a gift from W. Gallin (University of Alberta, Edmonton, Canada); mouse anti-PodxI was from G. Ojakian, The State University of New York; and mouse anti-p58 was obtained from the Mostov laboratory. Phalloidin-405 was from Biotium (Hayward, CA). Secondary antibodies used for immunofluorescence were from Molecular Probes (Eugene, OR). Secondary antibodies used for Western blotting were from ThermoFisher.
LY294002 and Rac1 Inhibitor II were from Calbiochem (San Diego, CA). Rapalog (AP21967, A/C heterodimerizer) was from Clontech (Mountain View, CA). Etoposide was from Tokyo Chemical Industry (Tokyo, Japan). Flagellin was purified from P. aeruginosa as previously described (Bucior et al., 2012).
Cell culture and transient transfection
MDCK cells expressing PH-Akt-GFP were grown in MEM supplemented with 10% fetal bovine serum (FBS) and maintained as previously described (Tran et al., 2014). For transient transfection, 6 × 105 cells were plated on 12-mm polycarbonate filters (0.4-µm pore size; Costar). At 2–6 h postplating, cells were transfected via the apical chamber with Lipofectamine 2000 (ThermoFisher) per manufacturer’s instructions. Medium was replaced 20 h posttransfection, and cells were grown for an additional 2 d for a total of 3 d of growth on filters. No mycoplasma was detected in the cells used in this study. Cells are routinely checked for contamination every 12 mo.
Immunofluorescence
Cells were fixed on filters in phosphate-buffered saline (PBS)/4% paraformaldehyde for 10 min and blocked in PBS/0.7% fish-skin gelatin for 10 min. Then filters were excised using a scalpel. Samples were permeabilized in PBS/0.2% Triton X-100 for 3 min and then laid out onto Parafilm cell side facing up. Antibodies were diluted in PBS/0.7% fish-skin gelatin, and 80 µl of antibody solution was pipetted onto each sample. Samples were stained with primary antibodies for 1–24 h, depending on antibodies used. After washing, samples were incubated with secondary antibodies and phalloidin-405 for 2 h. Slides were imaged using either a Nikon Ti-E microscope with a Yokagawa CSU22 spinning-disk confocal unit using a 60× Plan Apo VC oil lens or a Nikon Ti with a wide-field, high-speed Yokogawa CSUW1 spinning-disk unit and a 40× Plan Apo oil lens. Microscopes were controlled via MicroManager 1.4 or Nikon Elements (Edelstein et al., 2014).
Chemically induced dimerization
For chemically induced dimerization, MDCK cells transiently expressing CID constructs were washed with PBS and then incubated with a solution containing 200 nM Rapalog diluted into MEM or an equal volume of ethanol as a vehicle control. Cells were incubated at 37°C/5% CO2 for 60 min and then prepared for immunofluorescence as described.
For inhibitor experiments, MDCK cells were pretreated for 60 min with either Rac1 inhibitor (200 µM) or LY294002 (50 µM). Next Rapalog was added to the inhibitor-containing medium for an additional 60 min, and the cells were then prepared for immunofluorescence as described. As a control for NFκB activation, MDCK cells were incubated with 200 ng/ml flagellin purified from P. aeruginosa to induce NFκB activation (Bucior et al., 2012). As a control for caspase-3 experiments, MDCK cells were incubated with 500 µM etoposide for 3 h before staining to induce caspase-3 activation.
Cloning
All Par3 constructs were generated using human Par3 isoform 2 (GeneID: 56288). The version used has three silent mutations: C1666T, G1668A, and G3870A.
To generate AP-FRB, the FRB region of human mTOR1 (GeneID: 2475; amino acids 2021–2113) was PCR amplified from PM-FRB-CFP (a gift from Tamas Balla, National Institutes of Health; Varnai et al., 2006) with two mutations that allowed the FRB domain to bind to the rapamycin analogue AP21967 (Rapalog) introduced via QuikChange (A6292C and C6293T, leading to the T2098L amino acid change). Next Crb3 lacking the C-terminal PDZ binding motif was PCR amplified from pTRE2-Crb3-Venus (a kind gift of Quansheng Du, Georgia Health Sciences University; Zheng et al., 2010) with homology to pcDNA4 on the 5′ end. pcDNA was cut with HindIII and NotI. The PCR products and cut vector were assembled via InFusion (Clontech) according to the manufacturer’s protocol.
All FK constructs contain the human FKBP12 gene (GeneID: 2280). To generate FK-Par3, mRFP-FKBP12 was PCR amplified from mRFP-FKBP (a gift from Tamas Balla; Varnai et al., 2006) with 5′ homology to pcDNA4 and 3′ homology to Par3. Par3-3xHA was PCR amplified from pQCXIH-Par3-3xHA with 3′ homology to pcDNA4. pcDNA4 was digested with HindIII and NotI. The vector and PCR products were assembled using an InFusion reaction according to the manufacturer’s protocol. To generate FK-HA, mRFP-FKBP12 was PCR amplified from mRFP-FKBP with 5′ homology to pcDNA4. A 3xHA tag was PCR amplified from Par3-3xHA with 5′ homology to FKBP and 3′ homology to pcDNA4. pcDNA4 was digested with NotI and HindIII. The PCR products and cut vector were assembled via InFusion according to the manufacturer’s protocol.
FK-Par3 ΔCC1 was generated as described above starting with Par3 ΔCC1 (a kind gift from Ira Mellman, Yale University; Sfakianos et al., 2007). FK-CC1 was generated by PCR amplifying mRFP-FKBP with 5′ homology to pcDNA4, PCR amplifying the CC1 region of Par3 (aa936-1039) with 5′ homology to mRFP-FKBP, and PCR amplifying 3xHA with 3′ homology to CC1 and 5′ homology to pcDNA4. The PCR products were mixed with cut pcDNA4 (NotI, HindIII) and assembled via InFusion reaction.
The FK-iSH2 construct is based on human p85α (GeneID: 5295). The construct was generated using overlap PCR. The iSH2 domain of p85 (GeneID: 5295; amino acids 441–601) was PCR amplified from pSV-hp85α-HA (a gift from Ronald Kahn, Harvard Medical School; Ueki et al., 2003; Addgene plasmid #11499). The PCR added a 3xHA tag and a NotI site on the 3′ end. mRFP-FKBP was amplified by PCR using a 5′ primer containing a 5′ HindIII site and 3′ homology to iSH2. The two PCRs were mixed, and 15 cycles of PCR were carried out to generate a full-length construct. The full-length construct was amplified in another PCR, purified, cut, and ligated into pcDNA4 using HindIII and NotI restriction sites.
For FK-Tiam1, a codon-optimized version of mouse Tiam1 C1199 was used (lacking the N-terminal PEST sequences). The FK-Tiam1 construct was generated by GenScript.
For coexpression of FK-Tiam1 and FK-iSH2, a dual-expression plasmid was generated. First, a 3xMyc tag was cloned in place of the 3xHA tag on FK-iSH2 using InFusion cloning. Then a construct was generated that had FK-Tiam1-3xHA-P2A-FK-iSH2-3xMyc, using InFusion cloning. The incorporation of the P2A sequence allows the generation of single mRNA that produces a self-cleaving peptide and ensures equal expression of both constructs (Kim et al., 2011).
Western blotting and trichloroacetic acid precipitation
MDCK cells were incubated with 100 µl of detergent solution (DS; 1% Triton X-100, 0.4% deoxycholic acid, 50 mM Tris, pH 8) on ice for 5 min. Samples were centrifuged at 12,000 rpm for 5 min to pellet cell debris. For FK-HA–, AP-FRB–, and FK-iSH2–transfected cells, supernatants were loaded onto 4–12% Bis-Tris protein gels (Novex, Waltham, MA) and run with 3-(N-morpholino)propanesulfonic acid buffer (Novex). Gels were transferred to polyvinylidene difluoride membranes using an iBlot (Invitrogen, Waltham, MA) according to manufacturer’s protocol. Membranes were blocked in 3% milk in Tris-buffered saline (TBS) with tween (TBST; 20 mM Tris, pH 7.6, 138 mM NaCl, 0.1% Tween-20), incubated with primary antibodies diluted into TBST 3% bovine serum albumin (BSA), washed with TBST, incubated with horseradish peroxidase–conjugated secondary antibodies diluted into TBST 3% BSA, and developed with HyGlo Quick Spray (Denville).
For the FK-Par3 constructs and FK-Tiam1, lysates were precipitated with trichloroacetic acid (TCA) before blotting. Briefly, cold TCA was added to a final concentration of 5%. Samples were incubated on ice for 10 min and then centrifuged at 12,000 rpm for 10 min at 4°C. The supernatant was discarded, and the pellet was washed twice with −20°C acetone, resuspended in 50 µl DS, and then run on a gel as described.
Acquisition and quantification of immunofluorescence
Images were acquired by first adjusting laser power and exposure time on the control sample. The areas to image were selected by viewing the HA channel out of focus to avoid acquisition bias. Cells expressing high levels of the constructs were selected for imaging. After addition of Rapalog, only cells that showed apical localization of constructs were imaged and used for analysis. The laser power and exposure time were held constant for all images taken on the same day so that approximately equal expression levels of the various constructs were observed.
Quantitation of immunofluorescence images was done using Nikon Elements software. Apical PIP3 was scored as qualitatively “positive” if the apical signal intensity of PH-Akt-GFP was greater than or equal to the basolateral intensity signal. To quantify apical actin rearrangement, we examined the morphology of the apical cortical actin. If the signal for actin protruded above the nearest neighbors or showed a pinch near the apical membrane, the cell was scored as positive. For all immunofluorescence quantification, any cells that were in the process of being extruded from the monolayer (i.e., had lost contact with the basal surface) were excluded from analysis. Cells not expressing any transgene but from the same field were used as control cells. As a control for acquisition bias, control experiments were performed using an automated image acquisition method that acquired images blindly. As a control for scoring bias, scoring was performed on blinded samples. Both controls showed the same results as the data presented here. The number, n, of biological repeats for each experiment not explicitly stated in the figure legends is as follows: Figure 4, apical PIP3: –Rapa, 6; +Rapa, 7; RacI Inhib, 6; LY, 6. Figure 4, actin remodeling: –Rapa, 3; +Rapa, 4; RacI Inhib, 3; LY, 3. Figure 5, FK-iSH2, 5; FK-Tiam1, 5’; FK-P2A, 3.
To determine the apical intensity of PodxI (n = 4), ezrin (n = 3), p58 (n = 4), and E-cadherin (n = 3), we calculated the apical fluorescence intensity of each protein under the various experimental conditions and divided that value by the fluorescence intensity in untransfected cells from the same field. For each field, a cropped image containing the Z-sections of the apical membrane was generated. Using the region of interest (ROI) tool in Nikon Elements, transfected cells were outlined (ROIexp). Next untransfected cell(s) were outlined (ROICON). For each ROIEXP and ROICON, the average fluorescence intensity of the marker protein was calculated. To calculate the change in fluorescence intensity, the signal for each marker (PodxI, Ezrin, E-cadherin, or p58) in an experimental cell was divided by the intensity of untransfected cell(s) in the same field (ROIEXP/ROICON). Cells were scored as significantly changed if there was a 25% drop in PodxI signal or a 25% gain in p58, ezrin, or E-cadherin signal. We performed this analysis using other cutoffs and found similar results. Control cells that were not expressing a transgene were selected from the same fields and compared with the same background as experimental cells and represent the “control” sample in each graph.
Statistics
Statistical analysis was performed using Prism6 (GraphPad). A one-way analysis of variance (ANOVA) was used to determine whether the means were significantly different. If the ANOVA showed a statistically significant difference (p < 0.05), a post hoc Tukey’s multiple comparison test was performed. p < 0.05 was considered significant. A multiplicity-adjusted p value was calculated for each comparison, which is the “exact” p value reported here.
Supplementary Material
Acknowledgments
We thank the members of the Engel and Mostov labs for thoughtful critiques and ideas that were important for our results. Imaging was performed at the University of California, San Francisco, Nikon Imaging Center, which is supported by National Institutes of Health Grant 1S10OD017993. This project was funded by grants from the National Institutes of Health to J.E. (AI065902), D.M.B. (K99CA163535), and K.E.M. (DK074398 and DK091530). T.R. was supported by the National Institutes of Health (T32) and the Cystic Fibrosis Research Elizabeth Nash Memorial Fellowship.
Abbreviations used:
- CID
chemically induced dimerization
- PI3K
phosphatidylinositol 3-kinase
- PIP2
phosphatidylinositol 4,5-phosphate
- PIP3
phosphatidylinositol 3,4,5-phosphate
- PTEN
phosphatase and tensin homologue
Footnotes
The authors declare no competing interests.
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-07-0541) on November 23, 2016.
REFERENCES
- Abreu MT. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol. 2010;10:131–144. doi: 10.1038/nri2707. [DOI] [PubMed] [Google Scholar]
- Balla T. Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev. 2013;93:1019–1137. doi: 10.1152/physrev.00028.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant DM, Datta A, Rodriguez-Fraticelli AE, Peranen J, Martin-Belmonte F, Mostov KE. A molecular network for de novo generation of the apical surface and lumen. Nat Cell Biol. 2010;12:1035–1045. doi: 10.1038/ncb2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant DM, Roignot J, Datta A, Overeem AW, Kim M, Yu W, Peng X, Eastburn DJ, Ewald AJ, Werb Z, Mostov KE. A molecular switch for the orientation of epithelial cell polarization. Dev Cell. 2014;31:171–187. doi: 10.1016/j.devcel.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucior I, Pielage JF, Engel JN. Pseudomonas aeruginosa pili and flagella mediate distinct binding and signaling events at the apical and basolateral surface of airway epithelium. PLoS Pathog. 2012;8:e1002616. doi: 10.1371/journal.ppat.1002616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Macara IG. Par-3 controls tight junction assembly through the Rac exchange factor Tiam1. Nat Cell Biol. 2005;7:262–269. doi: 10.1038/ncb1226. [DOI] [PubMed] [Google Scholar]
- Coureuil M, Mikaty G, Miller F, Lecuyer H, Bernard C, Bourdoulous S, Dumenil G, Mege RM, Weksler BB, Romero IA, et al. Meningococcal type IV pili recruit the polarity complex to cross the brain endothelium. Science. 2009;325:83–87. doi: 10.1126/science.1173196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, Stuurman N. Advanced methods of microscope control using muManager software. J Biol Methods. 2014;1:e10. doi: 10.14440/jbm.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facciuto F, Bugnon Valdano M, Marziali F, Massimi P, Banks L, Cavatorta AL, Gardiol D. Human papillomavirus (HPV)-18 E6 oncoprotein interferes with the epithelial cell polarity Par3 protein. Mol Oncol. 2014;8:533–543. doi: 10.1016/j.molonc.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W, Wu H, Chan LN, Zhang M. Par-3-mediated junctional localization of the lipid phosphatase PTEN is required for cell polarity establishment. J Biol Chem. 2008;283:23440–23449. doi: 10.1074/jbc.M802482200. [DOI] [PubMed] [Google Scholar]
- Forteza R, Wald FA, Mashukova A, Kozhekbaeva Z, Salas PJ. Par-complex aPKC and Par3 cross-talk with innate immunity NF-kappaB pathway in epithelial cells. Biol Open. 2013;2:1264–1269.. doi: 10.1242/bio.20135918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyer RA, Macara IG. Loss of the polarity protein PAR3 activates STAT3 signaling via an atypical protein kinase C (aPKC)/NF-kappaB/interleukin-6 (IL-6) axis in mouse mammary cells. J Biol Chem. 2015;290:8457–8468. doi: 10.1074/jbc.M114.621011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikoshi Y, Suzuki A, Yamanaka T, Sasaki K, Mizuno K, Sawada H, Yonemura S, Ohno S. Interaction between PAR-3 and the aPKC-PAR-6 complex is indispensable for apical domain development of epithelial cells. J Cell Sci. 2009;122:1595–1606. doi: 10.1242/jcs.043174. [DOI] [PubMed] [Google Scholar]
- Iden S, Collard JG. Crosstalk between small GTPases and polarity proteins in cell polarization. Nat Rev Mol Cell Biol. 2008;9:846–859. doi: 10.1038/nrm2521. [DOI] [PubMed] [Google Scholar]
- Itoh N, Nakayama M, Nishimura T, Fujisue S, Nishioka T, Watanabe T, Kaibuchi K. Identification of focal adhesion kinase (FAK) and phosphatidylinositol 3-kinase (PI3-kinase) as Par3 partners by proteomic analysis. Cytoskeleton. 2010;67:297–308. doi: 10.1002/cm.20444. [DOI] [PubMed] [Google Scholar]
- Keestra AM, Baumler AJ. Detection of enteric pathogens by the nodosome. Trends Immunol. 2014;35:123–130. doi: 10.1016/j.it.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keestra AM, Winter MG, Auburger JJ, Frassle SP, Xavier MN, Winter SE, Kim A, Poon V, Ravesloot MM, Waldenmaier JF, et al. Manipulation of small Rho GTPases is a pathogen-induced process detected by NOD1. Nature. 2013;496:233–237. doi: 10.1038/nature12025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierbel A, Gassama-Diagne A, Rocha C, Radoshevich L, Olson J, Mostov K, Engel J. Pseudomonas aeruginosa exploits a PIP3-dependent pathway to transform apical into basolateral membrane. J Cell Biol. 2007;177:21–27. doi: 10.1083/jcb.200605142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Lee SR, Li LH, Park HJ, Park JH, Lee KY, Kim MK, Shin BA, Choi SY. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS One. 2011;6:e18556. doi: 10.1371/journal.pone.0018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krahn MP, Klopfenstein DR, Fischer N, Wodarz A. Membrane targeting of Bazooka/PAR-3 is mediated by direct binding to phosphoinositide lipids. Curr Biol. 2010;20:636–642. doi: 10.1016/j.cub.2010.01.065. [DOI] [PubMed] [Google Scholar]
- Laketa V, Zarbakhsh S, Traynor-Kaplan A, Macnamara A, Subramanian D, Putyrski M, Mueller R, Nadler A, Mentel M, Saez-Rodriguez J, Pepperkok R, et al. PIP(3) induces the recycling of receptor tyrosine kinases. Sci Signal. 2014;7:ra5. doi: 10.1126/scisignal.2004532. [DOI] [PubMed] [Google Scholar]
- Mack NA, Georgiou M. The interdependence of the Rho GTPases and apicobasal cell polarity. Small GTPases. 2014;5:10. doi: 10.4161/21541248.2014.973768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack NA, Porter AP, Whalley HJ, Schwarz JP, Jones RC, Khaja AS, Bjartell A, Anderson KI, Malliri A. beta2-syntrophin and Par-3 promote an apicobasal Rac activity gradient at cell-cell junctions by differentially regulating Tiam1 activity. Nat Cell Biol. 2012;14:1169–1180. doi: 10.1038/ncb2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashukova A, Wald FA, Salas PJ. Tumor necrosis factor alpha and inflammation disrupt the polarity complex in intestinal epithelial cells by a posttranslational mechanism. Mol Cell Biol. 2011;31:756–765. doi: 10.1128/MCB.00811-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Watanabe T, Matsuzawa K, Kakeno M, Okumura N, Sugiyama I, Itoh N, Kaibuchi K. PAR3 and aPKC regulate Golgi organization through CLASP2 phosphorylation to generate cell polarity. Mol Biol Cell. 2015;26:751–761. doi: 10.1091/mbc.E14-09-1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzawa K, Akita H, Watanabe T, Kakeno M, Matsui T, Wang S, Kaibuchi K. PAR3-aPKC regulates Tiam1 by modulating suppressive internal interactions. Mol Biol Cell. 2016;27:1511–1523. doi: 10.1091/mbc.E15-09-0670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens AE, Rygiel TP, Olivo C, van der Kammen R, Collard JG. The Rac activator Tiam1 controls tight junction biogenesis in keratinocytes through binding to and activation of the Par polarity complex. J Cell Biol. 2005;170:1029–1037. doi: 10.1083/jcb.200502129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno K, Suzuki A, Hirose T, Kitamura K, Kutsuzawa K, Futaki M, Amano Y, Ohno S. Self-association of PAR-3-mediated by the conserved N-terminal domain contributes to the development of epithelial tight junctions. J Biol Chem. 2003;278:31240–31250. doi: 10.1074/jbc.M303593200. [DOI] [PubMed] [Google Scholar]
- Morais-deSa E, Mirouse V, St Johnston D. aPKC phosphorylation of Bazooka defines the apical/lateral border in Drosophila epithelial cells. Cell. 2010;141:509–523. doi: 10.1016/j.cell.2010.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan AS, Reyes SB, Um K, McCarty JH, Tolias KF. The Rac-GAP Bcr is a novel regulator of the Par complex that controls cell polarity. Mol Biol Cell. 2013;24:3857–3868. doi: 10.1091/mbc.E13-06-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T, Kaibuchi K. Numb controls integrin endocytosis for directional cell migration with aPKC and PAR-3. Dev Cell. 2007;13:15–28. doi: 10.1016/j.devcel.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Pegtel DM, Ellenbroek SI, Mertens AE, van der Kammen RA, de Rooij J, Collard JG. The Par-Tiam1 complex controls persistent migration by stabilizing microtubule-dependent front-rear polarity. Curr Biol. 2007;17:1623–1634. doi: 10.1016/j.cub.2007.08.035. [DOI] [PubMed] [Google Scholar]
- Putyrski M, Schultz C. Protein translocation as a tool: the current rapamycin story. FEBS Lett. 2012;586:2097–2105. doi: 10.1016/j.febslet.2012.04.061. [DOI] [PubMed] [Google Scholar]
- Rajamuthiah R, Mylonakis E. Effector triggered immunity. Virulence. 2014;5:697–702. doi: 10.4161/viru.29091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Boulan E, Macara IG. Organization and execution of the epithelial polarity programme. Nat Rev. Mol Cell Biol. 2014;15:225–242. doi: 10.1038/nrm3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruch TR, Engel JN. Targeting the mucosal barrier: how pathogens modulate the cellular polarity network. Cold Spring Harb Perspect Biol. 2017 doi: 10.1101/cshperspect.a027953. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfakianos J, Togawa A, Maday S, Hull M, Pypaert M, Cantley L, Toomre D, Mellman I. Par3 functions in the biogenesis of the primary cilium in polarized epithelial cells. J Cell Biol. 2007;179:1133–1140. doi: 10.1083/jcb.200709111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shewan A, Eastburn DJ, Mostov K. Phosphoinositides in cell architecture. Cold Spring Harb Perspect Biol. 2011;3:a004796. doi: 10.1101/cshperspect.a004796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC, Inoue T, Meyer T, Hille B. Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science. 2006;314:1454–1457. doi: 10.1126/science.1131163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Asghar SZ, Zhang H. The polarity protein Par3 regulates APP trafficking and processing through the endocytic adaptor protein. Numb. Neurobiol Dis. 2016;93:1–11. doi: 10.1016/j.nbd.2016.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran CS, Eran Y, Ruch TR, Bryant DM, Datta A, Brakeman P, Kierbel A, Wittmann T, Metzger RJ, Mostov KE, Engel JN. Host cell polarity proteins participate in innate immunity to Pseudomonas aeruginosa infection. Cell Host Microbe. 2014;15:636–643. doi: 10.1016/j.chom.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueki K, Fruman DA, Yballe CM, Fasshauer M, Klein J, Asano T, Cantley LC, Kahn CR. Positive and negative roles of p85 alpha and p85 beta regulatory subunits of phosphoinositide 3-kinase in insulin signaling. J Biol Chem. 2003;278:48453–48466. doi: 10.1074/jbc.M305602200. [DOI] [PubMed] [Google Scholar]
- Varnai P, Thyagarajan B, Rohacs T, Balla T. Rapidly inducible changes in phosphatidylinositol 4,5-bisphosphate levels influence multiple regulatory functions of the lipid in intact living cells. J Cell Biol. 2006;175:377–382. doi: 10.1083/jcb.200607116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watton SJ, Downward J. Akt/PKB localisation and 3′ phosphoinositide generation at sites of epithelial cell-matrix and cell-cell interaction. Curr Biol. 1999;9:433–436. doi: 10.1016/s0960-9822(99)80192-4. [DOI] [PubMed] [Google Scholar]
- Weitnauer M, Mijosek V, Dalpke AH. Control of local immunity by airway epithelial cells. Mucosal Immunol. 2016;9:287–298. doi: 10.1038/mi.2015.126. [DOI] [PubMed] [Google Scholar]
- Wu H, Feng W, Chen J, Chan LN, Huang S, Zhang M. PDZ domains of Par-3 as potential phosphoinositide signaling integrators. Mol Cell. 2007;28:886–898. doi: 10.1016/j.molcel.2007.10.028. [DOI] [PubMed] [Google Scholar]
- Zhang H, Macara IG. The polarity protein PAR-3 and TIAM1 cooperate in dendritic spine morphogenesis. Nat Cell Biol. 2006;8:227–237. doi: 10.1038/ncb1368. [DOI] [PubMed] [Google Scholar]
- Zheng Z, Zhu H, Wan Q, Liu J, Xiao Z, Siderovski DP, Du Q. LGN regulates mitotic spindle orientation during epithelial morphogenesis. J Cell Biol. 2010;189:275–288. doi: 10.1083/jcb.200910021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.