

In hippocampal neurons, Ras and Rab interactor 1 (RIN1) hinders the formation of stable synaptic connections by increasing dendritic filopodial motility and regulates long-term depression by enhancing AMPA receptor endocytosis.
Abstract
Ras and Rab interactor 1 (RIN1) is predominantly expressed in the nervous system. RIN1-knockout animals have deficits in latent inhibition and fear extinction in the amygdala, suggesting a critical role for RIN1 in preventing the persistence of unpleasant memories. At the molecular level, RIN1 signals through Rab5 GTPases that control endocytosis of cell-surface receptors and Abl nonreceptor tyrosine kinases that participate in actin cytoskeleton remodeling. Here we report that RIN1 controls the plasticity of cultured mouse hippocampal neurons. Our results show that RIN1 affects the morphology of dendritic protrusions and accelerates dendritic filopodial motility through an Abl kinase–dependent pathway. Lack of RIN1 results in enhanced mEPSC amplitudes, indicating an increase in surface AMPA receptor levels compared with wild-type neurons. We further provide evidence that the Rab5 GEF activity of RIN1 regulates surface GluA1 subunit endocytosis. Consequently loss of RIN1 blocks surface AMPA receptor down-regulation evoked by chemically induced long-term depression. Our findings indicate that RIN1 destabilizes synaptic connections and is a key player in postsynaptic AMPA receptor endocytosis, providing multiple ways of negatively regulating memory stabilization during neuronal plasticity.
INTRODUCTION
Ras and Rab interactor 1 (RIN1), originally identified as a Ras effector protein, is highly expressed in mature neurons, particularly in the cerebral cortex, hippocampus, and amygdala. RIN1 localizes to neuronal cell body and dendrites, with enrichment in postsynaptic densities (Deininger et al., 2008). RIN1-knockout mice are viable and show normal development but possess altered neuronal plasticity, with elevated fear conditioning and conditioned taste aversion. These behaviors are mainly dependent on amygdala functions but are also influenced by other forebrain regions, such as cerebral cortex. RIN1 appears to be a regulator of amygdala-related fear learning and experience-mediated fear extinction (Dhaka et al., 2003; Bliss et al., 2010). Consequently RIN1-knockout mice with enhanced fear acquisition and retention can serve as a useful model for distinct neuropsychiatric conditions such as posttraumatic stress disorder (Bliss et al., 2010). The molecular mechanisms underlying these RIN1 functions, however, have been unclarified.
RIN1 has the ability to signal through two downstream pathways. First, the direct activation of the Abl tyrosine kinases Abl and Arg (Abl-related gene, or Abl2) controls actin cytoskeletal remodeling (Han et al., 1997; Hu et al., 2005). Second, RIN1 can also act as a Rab5-directed guanine nucleotide exchange factor (GEF) protein, thereby regulating Rab5-dependent receptor endocytosis and early endosome formation (Tall et al., 2001). Both of these pathways are involved in neuronal plasticity (Moresco et al., 2003; Brown et al., 2005): Abl kinases are present at hippocampal excitatory synapses, where they control synaptic functions and regulate synaptic plasticity (Moresco et al., 2003; Xiao et al., 2016). The modulation of actin cytoskeletal remodeling, binding to β1 integrin, and synaptic clustering of PSD95 have been all implicated in Abl kinase–mediated activity-dependent synaptic efficacy (for reviews, see Koleske, 2006; Colicelli, 2010; also see Perez de Arce et al., 2010; Warren et al., 2012). On the other hand, the small GTPase Rab5 mediates clathrin-dependent endocytosis in hippocampal neurons (de Hoop et al., 1994) and participates in the activity-dependent removal of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors from hippocampal excitatory synapses during long-term depression (LTD; Brown et al., 2005). Thus RIN1 is an ideal candidate for a negative regulator of synaptic stability and/or potentiation.
Protein interactions and cellular functions of RIN1 are regulated by the phosphorylation of distinct amino acid residues. For example, phosphorylation of serine 351 (S351) enhances the interaction with 14-3-3 adaptor proteins, which sequester active RIN1 in the cytoplasm and prevent its translocation to cellular membranes (Wang et al., 2002). Consequently a RIN1S351A mutant protein is more active with respect to epidermal growth factor receptor interaction (Hu et al., 2008a) and receptor down-regulation than the wild-type protein (Balaji et al., 2012). RIN1 directly interacts with Abl kinases via its proline-rich domain, leading to disengaged autoinhibition of Abl and subsequent phosphorylation of RIN1 at tyrosine 36 (Y36). This process further stabilizes the interaction of the proteins and enhances autocatalytic Abl kinase activity, followed by increased phosphorylation of its substrates (Hu et al., 2005). Of note, mutation of tyrosine 36 together with three secondary tyrosine residues within RIN1 (RIN1QM) blocks the activation of Abl kinases (Balaji et al., 2012; Balaji and Colicelli, 2013). RIN1-mediated activation of Abl kinases is further modulated through protein kinase D–mediated phosphorylation of serine 292 (S292; Ziegler et al., 2011). Finally, a Rab5 GEF–deficient RIN1E574A mutant disrupts RIN1-mediated Rab5 actions (Galvis et al., 2009).
In this study, we investigated the role of RIN1 in cellular mechanisms regulating receptor endocytosis and the morphology and stabilization of dendritic spines. We used embryonic hippocampal neuronal cultures as cellular model systems to investigate RIN1-mediated effects on dendritic spine morphology and motility. Furthermore, we analyzed whether RIN1 regulates plasticity-dependent surface localization of GluA1 AMPA receptor subunits in cultured neurons. Using the aforementioned mutant RIN1 proteins, we sought to distinguish between the contributions of Abl kinase activation and Rab5-dependent signaling pathways to RIN1-mediated effects under normal conditions or during chemically induced long-term depression. Our results indicate that RIN1 weakens synaptic connections by increasing the motility of dendritic protrusions and enhancing AMPA receptor endocytosis through the activation of Abl kinases and its Rab5 GEF activity, respectively.
RESULTS
RIN1 expression increases during the dendritic development of cultured neurons
To visualize endogenous RIN1 in lysates of wild-type brain tissue, we used a previously described murine-specific RIN1 antibody (Deininger et al., 2008). Of note, in lysates obtained from Rin1−/− animals, the protein was not detected, proving the specificity of the RIN1 antibody (Figure 1A). In accordance with the gradual increase in RIN1 levels during embryonic and postnatal forebrain development (Deininger et al., 2008), RIN1 expression in embryonic hippocampal neuronal cultures was low during the first week after plating (Figure 1, B, C, and E). From day in vitro (DIV) 7 on, in parallel with dendritic development and synaptic maturation, RIN1 level strongly increased in cultures prepared from wild-type mice, similar to the observed increase in AMPA receptor subunit GluA1 expression. Because the used Rin1−/− animals have a C57Bl/6 genetic background (Deininger et al., 2008), cultures prepared from C57Bl/6 mice would serve as the most appropriate wild-type controls. However, because this mouse strain is not ideal for the preparation of embryonic hippocampal cultures, we used neuronal cultures from CD1 wild-type embryos instead, showing developmental changes in RIN1, GluA1, and βIII-tubulin expression similar to those of C57Bl/6 cultures (Figure 1, B and C).
FIGURE 1:
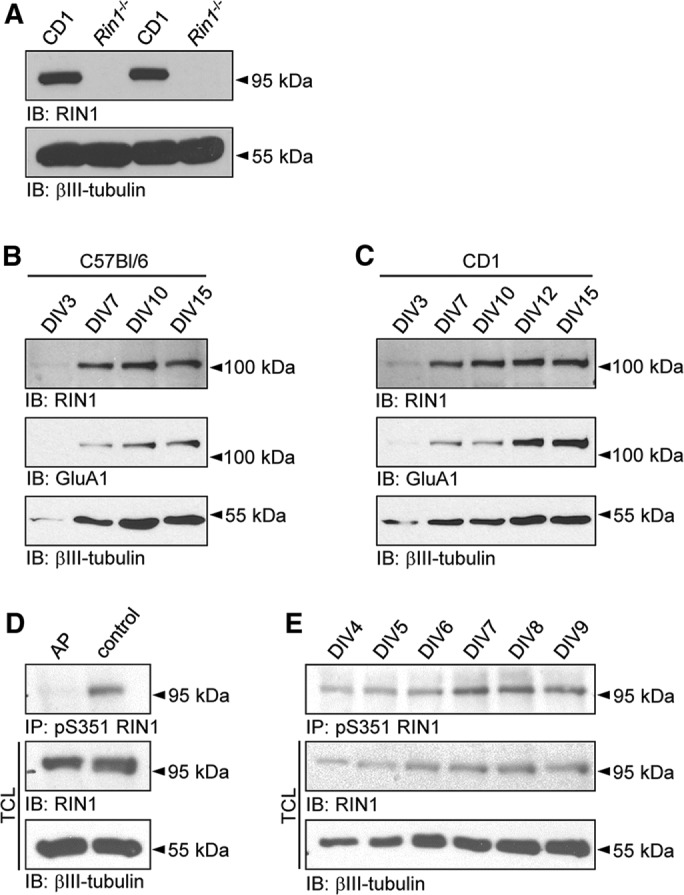
RIN1 levels in developing hippocampal neuronal cultures. (A) The specificity of RIN1 antibody is proved by the lack of RIN1 signal in Rin1−/− samples compared with CD1 lysates. RIN1 and GluA1 expression during development of C57Bl/6 (B) and CD1 (C) wild-type neuronal cultures. (D) Alkaline phosphatase (AP) treatment of cell lysates abolishes anti-pS351 RIN1 precipitation. (E) Changes in the relative phosphorylation of the Ser351 site in RIN1 during in vitro development of CD1 cultures. Neuron-specific βIII-tubulin served as a loading control (A, D) or as an indicator of neurite development (B, C, E). TCL, total cell lysates.
To analyze the phosphorylation level at serine 351 in developing neuronal cultures, we used a phosphospecific antibody (anti-pS351; Ziegler et al., 2011). The specificity of the antibody was proved by alkaline phosphatase treatment of culture lysates before precipitation: whereas the overall level of the endogenous RIN1 protein was the same in total cell lysates (TCLs), the pS351-specific antibody failed to precipitate RIN1 after phosphatase treatment (Figure 1D).
The amount of endogenous RIN1 protein and the level of pS351-RIN1 increased during in vitro development, indicating that phosphorylation of the serine 351 is under continuous regulation in developing neuronal cultures (Figure 1E).
RIN1 activity decreases the ratio of mature spines
To investigate RIN1-mediated effects on dendritic development and spine formation, we cultivated hippocampal neurons from Rin1−/− embryos and transfected them with plasmids encoding enhanced green fluorescent protein (EGFP) or EGFP-tagged wild-type RIN1 or RIN1S351A. Morphology of the transfected neurons was compared 24 h after the transfection, on DIV12–13.
Rin1−/− neurons had similar morphology to wild-type CD1 (Figure 2A) or C57Bl/6 (unpublished data) cells and formed elaborate dendritic trees within the cultures. EGFP-expressing Rin1−/− neurons had all three main types of dendritic protrusions, namely stubby and filamentous protrusions and mature, mushroom-like spines with extended dendritic spine heads (Figure 2B). Protrusion density on secondary dendrites was not affected when either wild type or the S351A mutant RIN1 constructs were reintroduced into the Rin1−/− neurons, indicating that the overall amount of dendritic protrusions is regulated independently of RIN1 expression (Figure 2C). However, restored RIN1 functions significantly reduced the ratio of mushroom-like spines within 24 h after transfection. The relative amount of mushroom-like spines decreased further when the RIN1S351A was present. Strikingly, in this case, the decrease in mushroom-like spines was accompanied by an increase in long, filamentous protrusions (Figure 2, B and D). These data indicate that RIN1 can turn the more stable, mature spines with prominent spine heads into long and thin filamentous protrusions within 1 d of expression and that the phosphorylation at serine 351 decreases this activity.
FIGURE 2:

(A) Representative dendritic outlines of EGFP-expressing CD1 and Rin1−/− hippocampal neurons on DIV13. (B) Dendritic branches of Rin1−/− hippocampal neurons expressing EGFP, RIN1WT-EGFP, or RIN1S351A-EGFP for 24 h. (C) Average protrusion density and (D) ratio of stubby, mushroom-like or filamentous spines within the transfected hippocampal neurons. (E) pCrkL levels in HEK293T cells transfected with EGFP-tagged wild-type RIN1, RIN1S351A, RIN1QM, or RIN1E574A constructs. Anti-GFP signal indicates similar expression levels of RIN1 constructs. α-Tubulin served as a loading control. Data are presented as mean ± SEM. *p < 0.05; **p < 0.01. Asterisk represents significant differences compared with control values. Arrows indicate filamentous protrusions; arrowheads show mushroom-type dendritic spines. Scale bar, 50 μm (A), 1 μm (B).
RIN1 increases filopodial motility depending on the Abl kinase pathway
To investigate the activation of Abl/Arg kinases by RIN1 point mutants, we transfected HEK293T cells with empty EGFP vector, wild-type RIN1, or RIN1 point mutant RIN1S351A, RIN1QM, or RIN1E574A (Figure 2E). RIN1QM blocked the RIN1-dependent activation of Abl/Arg, whereas reduced 14-3-3 binding (RIN1S351A) or lack of Rab5 GEF activity (RIN1E574A) did not influence the phosphorylation of CrkL, a known downstream target of Abl kinases.
The elongated, thin filamentous protrusions in neurons are usually motile filopodia seeking future synaptic partners (Ziv and Smith, 1996; Korkotian and Segal, 2001). The morphology of motile filopodia and that of the more stable, thin spines with already established synaptic connections are quite similar and can be distinguished only by the presence of postsynaptic machinery within the protrusion head in fixed cultures. However, live-cell imaging can provide direct observations on protrusion motility (Tárnok et al., 2015). Therefore we transfected Rin1−/− neurons with fluorescently tagged RIN1 constructs and analyzed the protrusion motility on secondary dendritic branches on DIV 12–13, within 24 h of posttransfection time (Supplemental Movies S1 and S2).
The tip of thin, elongated protrusions was marked manually on every consecutive frame, and average speed of tip movement (Figure 3A) and the covered distance in increasing time steps (cumulative time-dependent displacement functions; Figure 3, B–D) were determined. Overexpression of wild type as well as the RIN1S351A mutant significantly increased the motility of the protrusions. This effect was completely blocked when 5 μM imatinib mesylate (also known as STI-571 or Gleevec), a tyrosine kinase inhibitor used in the treatment of multiple cancers (Savage and Antman, 2002), was applied for 1 h. Because RIN1 directly activates Abl tyrosine kinases, these data already indicate that elevated Abl activity is responsible for the observed increase in filopodial motility upon ectopic expression of RIN1 in Rin1−/− neurons. Phosphorylation of RIN1 at serine 351, however, seems to be dispensable for filopodial motility and/or Abl kinase activation.
FIGURE 3:

Average tip movement within 1 min (A) and cumulative time-dependent displacement functions (B–D) of data obtained on filopodial motility within Rin1−/− hippocampal cultures transfected with the indicated EGFP-tagged constructs. Imatinib 5 μM was applied for 1 h before imaging (A–C). Data are presented as mean ± SEM. $p < 0.05; **,$$p < 0.01; ***,$$$p < 0.001. Asterisk indicates difference from the EGFP values.
To further prove the importance of Abl kinase activation in RIN1-controlled filopodial motility, we introduced RIN1 mutants defective in Abl kinase activation (RIN1QM) or modulation of Abl kinase activity (RIN1S292A) into Rin1−/− neurons. Strikingly, neither of these mutants increased filopodial motility compared with EGFP-transfected, control protrusions. On the contrary, the Rab5 GEF activity of RIN1 did not influence RIN1-evoked increase in filopodial motility, because the RIN1E574A point mutant also enhanced the motility of protrusions (Figure 3, A and D). These data prove that RIN1-mediated Abl kinase activation within dendritic protrusions augments filopodial motility.
The Rab5 GEF activity of RIN1 regulates transferrin endocytosis
The Rab5 GEF activity of RIN1 regulates EphA4 receptor endocytosis in neurons during the first week after plating (Deininger et al., 2008). To prove that RIN1 controls endocytosis in more developed cultured hippocampal neurons as well, we pulse-labeled DIV12–13 neuronal cultures with fluorescently labeled transferrin for 1 min and followed the internalization of the fluorescent transferrin signal by a confocal microscope. As expected, internalization of transferrin in Rin1−/− neurons increased during the 1- to 5-min chasing time (Figure 4). When wild-type RIN1 was reintroduced into Rin1−/− neurons, the rate of transferrin endocytosis was significantly increased after 1 min of chasing, similar to the effect induced by the RIN1S351A mutant (Figure 4B). Of note, endocytosis in RIN1S351A-expressing neurons was more enhanced after 5 min of chasing time compared with the wild-type RIN1. These effects were clearly dependent on the Rab5 GEF activity of RIN1, because the RIN1E574A mutation completely blocked enhanced endocytosis, whereas the RIN1QM mutation did not influence RIN1-mediated elevation in transferrin endocytosis. Thus transferrin receptor endocytosis is regulated exclusively by the Rab5 GEF activity of RIN1, whereas the Abl kinase pathway is not involved in this process in hippocampal neurons. The phosphorylation at serine 351, however, limits Rab5-mediated endocytosis.
FIGURE 4:
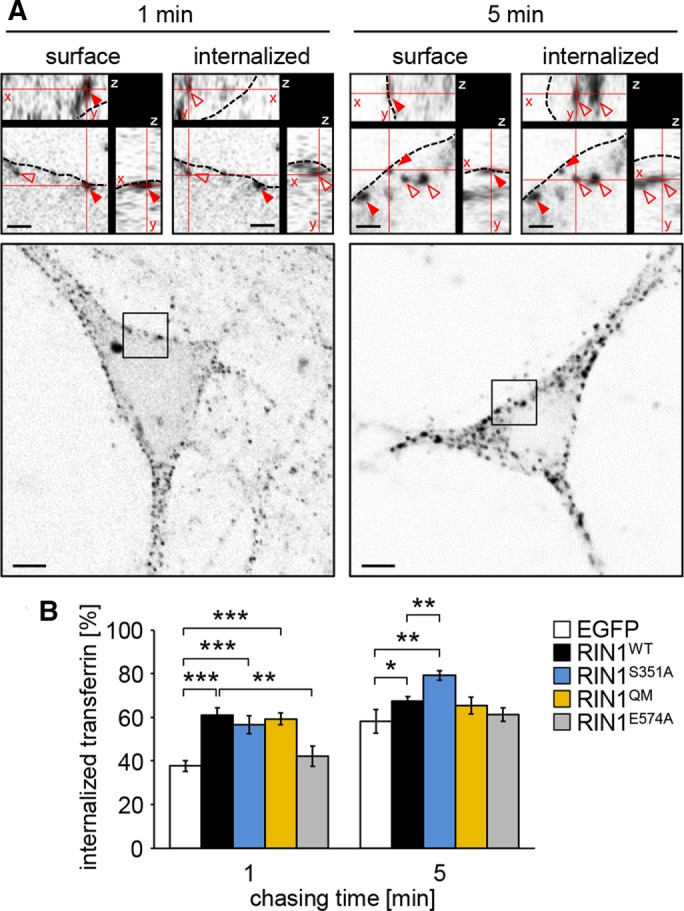
Transferrin uptake in pulse-labeled Rin1−/− hippocampal neurons. (A) Fluorescently labeled transferrin signals associated with the plasma membrane (surface) or internalized after 1 or 5 min of chasing time. Red arrowheads show surface-localized transferrin puncta; empty arrowheads indicate internalized transferrin signals; dashed line indicates plasma membrane. The xyz-planes are represented in cut-view images (top). Scale bars, 1 μm (cut views), 5 μm (overviews). (B) The ratio of internalized transferrin signals compared with the total number of transferrin-positive puncta visible in the equatorial plane of the neuronal soma in Rin1−/− neurons transfected with the indicated fluorescently tagged constructs. Data are presented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Loss of RIN1 increases the amount of AMPA receptors in the plasma membrane
Excitatory AMPA receptors convey fast synaptic transmission and play an important role in synaptic plasticity. The amount of surface AMPA receptors is controlled by the balance between secretory and endocytotic mechanisms and can be specifically regulated within minutes of neuronal plasticity (Derkach et al., 2007). Because RIN1 had a regulatory role in Rab5-dependent transferrin receptor endocytosis in neurons, we tested whether RIN1 plays a role in the regulation of AMPA receptors at the cell surface.
We examined total (TCL) and plasma membrane–localized (surface) GluA1 levels by cell surface biotinylation assay in DIV13–14 CD1 wild-type and Rin1−/− hippocampal neuronal cultures (Figure 5, A–G). Owing to the lack of a suitable reference marker for the precipitation of biotinylated proteins, we could not directly compare the amount of surface GluA1 subunits between Rin1−/− and CD1 cultures. However, by parallel detection and normalization of the GluA1 signal to the neuron-specific βIII-tubulin, we observed that the overall amount of GluA1 subunits was significantly reduced in Rin1−/− compared with CD1 cultures (Figure 5, A and B).
FIGURE 5:

Analysis of AMPA receptor expression and activity at the cell surface. (A–D) Surface biotinylation of GluA1 subunits in CD1 and Rin1−/− hippocampal neuronal cultures. (A) Representative Western blots of TCLs and biotinylated samples precipitated on NeutrAvidin beads (surface) under control conditions or 2 h after cLTD treatment. βIII-tubulin served as a loading control and was absent from the precipitated samples. TCL lysates and βIII-tubulin blots from CD1 and Rin1−/− cultures were developed under the same conditions. In the case of biotinylated GluA1 subunits, exposure time for the CD1 samples was longer than in the case of the Rin1−/− samples. (B) Comparison of the relative GluA1 levels in TCLs from Rin1−/− or CD1 cultures. (C, D) Changes in the relative surface-labeled GluA1 levels after cLTD treatment in CD1 (C) or Rin1−/− (D) cultures. (E) Representative Western blot images of total GluA1 levels in C57Bl/6 neuronal cultures after cLTD induction. (F, G) Comparison of cLTD-evoked decrease in the total GluA1 level between CD1 (F) and C57Bl/6 (G) neuronal cultures. (H) Representative whole-cell patch clamp recordings obtained from CD1 or Rin1−/− neurons. The mEPSCs are indicated by red dots. (I, K) Pooled cumulative probability density distributions of mEPSC interevent intervals (IEIs) and mEPSC amplitudes, respectively. (J, L) Scatter plots showing the medians of IEIs and mEPSC amplitudes, respectively. Median mEPSC amplitude in Rin1−/− cultures is significantly higher than in CD1 cultures. Data are presented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
To compare directly the amount of functional AMPA receptors in the plasma membrane, we performed whole-cell voltage clamp experiments in CD1 and Rin1−/− neurons in the presence of 0.5 μM tetrodotoxin (Figure 5, H–L). When the membrane potential of the recorded cells is held at −60 mV, miniature synaptic events occur due to the spontaneous release of glutamate, and miniature excitatory postsynaptic currents (mEPSCs) are generated by the activation of AMPA receptors (Lisman et al., 2007). The frequency of mEPSCs was slightly but not significantly decreased in Rin1−/− culture (see the cumulative probability functions and the median values of interevent intervals determined in the recorded neurons on Figure 5, I and J). Instead, the cumulative probability distribution of mEPSC amplitudes in Rin1−/− neurons shifted to the right compared with wild-type neurons, indicating a significant increase in mEPSC amplitudes (Figure 5, K and L).
On the basis of these data, we conclude that a greater amount of the available AMPA receptors is localized in the plasma membrane of Rin1−/− neurons than in the control CD1 neurons.
AMPA receptor endocytosis is regulated by the Rab5 GEF activity of RIN1
To analyze GluA1 subunit localization at the cellular level, we applied an antibody-feeding assay to analyze RIN1-dependent changes in the postsynaptic GluA1 levels. DIV13–14 living neurons isolated from CD1 wild-type or Rin1−/− embryos were treated with an antibody recognizing the extracellular N-terminus of GluA1 for 10 min. Cells were rapidly fixed without permeabilization, and the bound anti-GluA1 antibody was visualized by incubation with a fluorescently labeled secondary antibody. Most of the GluA1-specific signal was detected at the plasma membrane (Figure 6A). Postsynaptic areas were visualized after permeabilization with an antibody specific for Shank2, the main scaffold protein within the postsynaptic density (PSD; Naisbitt et al., 1999). GluA1 signal intensities were determined within the Shank2-positive areas located at the plasma membrane within dendritic spines or dendritic shaft synapses (indicated by arrowheads and arrows, respectively, in Figure 6A). Intensity values were compared only between those sister cultures that were labeled and stained at the same time and under identical conditions (Figure 6, B–D).
FIGURE 6:
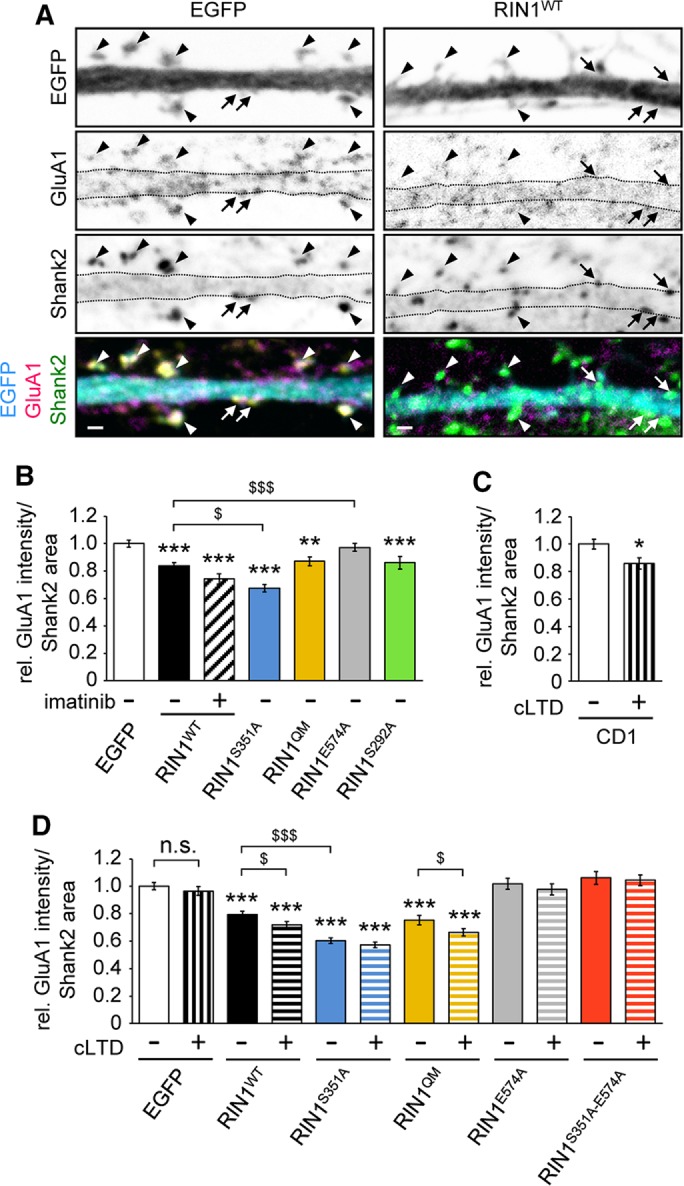
Anti-GluA1 antibody feeding in hippocampal neuronal cultures. Relative anti-GluA1 signal intensity was determined within Shank2-positive postsynaptic densities (PSDs) at the plasma membrane. (A) Representative pictures of Rin1−/− dendrites transfected with EGFP or EGFP-tagged RIN1WT after 10 min of antibody feeding with anti-GluA1. PSD was delineated based on clear Shank2 positivity. Arrows point at shaft synapses; arrowheads indicate spines. Scale bar, 1 μm. (B–D) Quantification of GluA1 signal intensities within Shank2-positive PSD areas in EGFP-transfected neurons of CD1 cultures (C) or in Rin1−/− neurons transfected with the indicated EGFP-tagged constructs (B, D). cLTD was evoked by 3 min of 50 μM NMDA treatment, and neurons were analyzed 2 h later (C, D). Imatinib was applied at 5 μM for 1 h (B). Data are presented as mean ± SEM. *,$p < 0.05; **p < 0.01; ***,$$$p < 0.001. Asterisk indicates difference compared with the EGFP values.
When Rin1−/− neurons were transfected with fluorescently labeled wild-type and mutant RIN1 constructs or EGFP, reintroducing wild-type RIN1 into Rin1−/− neurons led to significantly decreased GluA1 levels within the postsynaptic Shank2-positive areas (Figure 6, A and B). This effect was even more prominent when the RIN1S351A mutant was introduced into the Rin1−/− neurons (Figure 6B). The loss of GluA1 subunits within the postsynaptic areas was dependent on the Rab5 GEF activity of the transfected RIN1 constructs, because the expression of the RIN1E574A point mutant resulted in similar relative GluA1 values as the control, EGFP-expressing neurons. Expression of the Abl-deficient RIN1 point mutant (RIN1QM), lack of phosphorylation at the S292 RIN1 site (RIN1S292A), and treatment of RIN1WT-expressing neurons with the tyrosine kinase inhibitor imatinib, however, led to a similar reduction in surface GluA1 intensities as observed upon wild-type RIN1 expression (Figure 6B). These data suggest that RIN1 regulates the amount of GluA1 subunits within the postsynaptic sites through its Rab5 GEF activity.
Loss of surface AMPA receptors evoked by chemically induced long-term depression depends on the Rab5 GEF activity of RIN1
Brief treatment with N-methyl-d-aspartate (NMDA) strongly activates NMDA receptors, which in turn leads to a rapid and long-lasting loss of surface AMPA receptors, providing a suitable tool to investigate the regulation of LTD formation (Ehlers, 2000; Snyder et al., 2005; Lee et al., 2014). To clarify whether RIN1 is involved in AMPA receptor internalization during LTD, we analyzed the changes in GluA1 surface distribution upon chemically induced LTD (cLTD) in CD1, C57Bl/6, and Rin1−/− cultures (Figures 5, A and C–G, and 6, C and D).
Our Western blot results clearly show that in CD1 and C57Bl/6 cultures, cLTD treatment evoked a significant loss in the overall as well as in the surface GluA1 subunit levels (Figure 5, A, C, and E–G). Strikingly, in the absence of RIN1, neurons were not able to down-regulate surface AMPA receptors or degrade GluA1 subunits after 2 h of cLTD induction (Figure 5, A and D).
To corroborate the role of RIN1 in cLTD-induced elimination of GluA1 receptors from the plasma membrane, we performed an antibody-feeding assay in control CD1 and Rin1−/− neurons left untreated or subjected to cLTD (Figure 6, C and D). In the case of CD1 wild-type neurons, cLTD treatment evoked a 14 ± 0.04% reduction (mean ± SEM) of the relative GluA1 intensity within the Shank2-positive PSD areas compared with nontreated sister cultures (Figure 6C). Conversely, Rin1−/− neurons failed to show any signs of cLTD-induced loss of GluA1 subunits within the PSD areas (see EGFP values in Figure 6D), which is in agreement with the surface biotinylation results (Figure 5, A and D).
Previous reports demonstrated that Rab5 participates in the activity-dependent endocytosis of AMPA receptors during LTD (Brown et al., 2005). Of importance, cLTD-mediated effects on surface GluA1 localization were restored only when Rin1−/− neurons were transfected with RIN1 constructs possessing intact Rab5 GEF activity (see the RIN1WT and RIN1QM values vs. RIN1E574A and RIN1S351A−E574A double-mutant values in Figure 6D). Of note, cLTD treatment could not evoke a further drop in the relative GluA1 levels in Rin1−/− neurons expressing the RIN1S351A mutant (Figure 6D). This indicates that the lack of serine 351 phosphorylation enhances Rab5 GEF activity to the maximal extent, which cannot be further elevated by cLTD.
Taken together, our data clearly indicate that cLTD-evoked loss of surface AMPA receptors depends on the Rab5-GEF activity of RIN1.
DISCUSSION
In this study, we investigated the role of RIN1 in plasticity-related cellular mechanisms in hippocampal neurons. Our results show that RIN1 overexpression enhances dendritic filopodial motility through the regulation of Abl kinase activity. In addition, RIN1 controls receptor turnover through activation of Rab5 and plays a critical role in the endocytosis of GluA1 AMPA receptor subunits. Concomitant lack of RIN1 leads to elevated mEPSC amplitudes by increasing the surface AMPA receptor pools and abolishes the NMDA-dependent down-regulation of these receptors upon chemically induced LTD. Thus our data indicate that RIN1 reduces synaptic strength in cultivated hippocampal neurons through its downstream targets Abl kinase and Rab5, which is in accordance with RIN1’s role in LTD formation or depotentiation (Dhaka et al., 2003; Bliss et al., 2010).
RIN1-dependent increase in dendritic filopodia motility was abolished by imatinib, a widely used inhibitor of Abl kinases, as well as by introducing the QM mutation, which interferes with downstream Abl kinase activation (Hu et al., 2008a). At first sight, these results contradict previous findings obtained from fibroblasts or cancer cells. RIN1 has been shown to negatively regulate growth factor–induced cell migration depending on Abl kinase activation (Hu et al., 2005, 2008a; Ziegler et al., 2011; Balaji and Colicelli, 2013) or on competing with Raf kinase for binding to Ras (Wang et al., 2002; Balaji et al., 2012; Gerarduzzi et al., 2017), indicating an actin-stabilizing role for RIN1. On the other hand, the structure and dynamics of actin filaments in dendritic filopodia differ from those of conventional filopodia in many aspects: branched and straight actin filaments do not form a tight bundle and show mixed polarity (Hotulainen et al., 2009; Korobova and Svitkina, 2010). In addition, local polymerization and depolymerization rates are unbalanced in dendritic filopodia (Tatavarty et al., 2012). Moreover, protrusion motility in dendritic filopodia is not governed primarily by growth factors but depends on an interplay among forces generated by actin retrograde flow, myosin contractility, and substrate adhesion (Tashiro and Yuste, 2004; Tatavarty et al., 2012). Therefore we cannot exclude the possibility that besides interfering with actin dynamics, Abl kinases regulate filopodial motility by other means, including integrin signaling (Warren et al., 2012; Kerrisk et al., 2013).
The regulation of the intracellular localization of RIN1 also differed characteristically between nonneuronal cells and cultivated hippocampal neurons: We could not detect an elevated plasma membrane–associated localization of the RIN1S351A mutant in transfected neurons (Figure 2B; unpublished data) as previously reported in nonneuronal cells (Wang et al., 2002; Balaji et al., 2012). This might suggest that in neurons, additional upstream regulators of RIN1 (e.g., growth factor signaling) participate in enhanced membrane localization (Jozic et al., 2012).
RIN1-mediated effects on the maturation of dendrites or dendritic spines have not been addressed. Abl kinases, on the other hand, have been investigated in relation to neuronal motility. Abl tyrosine kinases positively regulated neurite outgrowth (Zukerberg et al., 2000; Woodring et al., 2002) as well as dendritic motility and sprout formation in developing hippocampal cultures (Jones et al., 2004). These findings are in agreement with our results on RIN1-mediated increase in dendritic filopodia motility. In addition, our findings indicate that RIN1S351A and RIN1WT induced Abl kinase–dependent phosphorylation of CrkL to a similar level, which is in agreement with a similar increase in filopodial motility evoked by these two proteins. Nevertheless, Arg knockdown in cultivated hippocampal neurons evoked spine destabilization (Lin et al., 2013). It is important to note, however, that these investigations focused on the appearance or disappearance of dendritic protrusions within 1 h instead of following their short-term motility changes.
The Koleske group also reported that Abl and Arg stabilize dendrites and dendritic spines in the mouse brain, but only from early adulthood on (Moresco et al., 2005; Sfakianos et al., 2007; Gourley et al., 2012; Warren et al., 2012). Because these studies analyzed fixed brain tissues of knockout animals, we cannot directly compare them with our findings showing that RIN1 overexpression reduced the ratio of mushroom spines in cultivated neurons. In addition, the phenotype of cells with complete loss of Abl kinases is not necessarily expected to be equivalent to the phenotype of cells that lost an Abl kinase upstream activator. Similarly, an acute perturbation of Abl kinase activity by the application of imatinib can lead to different outcomes compared with the genetic ablation of the kinases. Therefore further studies are required to clarify the role of RIN1 in relation to dendritic filopodia motility, spine formation, and stabilization.
Of importance, in vitro, Arg-knockdown neurons exhibited increased mEPSC amplitudes besides having a decreased frequency of these events (Lin et al., 2013), which resembles our electrophysiological results in Rin1−/− neurons. Because Abl and Arg both modulate the efficiency of neurotransmitter release from the presynaptic terminal (Moresco et al., 2003; Xiao et al., 2016), we cannot exclude the possibility that the lack of RIN1 leads to alterations in the presynaptic release in an Abl kinase–dependent manner. Nevertheless, our surface biotinylation and antibody feeding experiments clearly prove that RIN1 is critically involved in the control of the AMPA receptor amount within the postsynaptic plasma membrane. In addition, our data indicate that this effect depends on the Rab5 GEF activity of RIN1.
Dynamic trafficking and dephosphorylation of the GluA1 subunit of the AMPA receptor is a key event during LTD. The major subunit composition of AMPA receptors in hippocampal neurons are GluA1/GluA2 heteromers, which are recruited to the synapse in an activity-dependent manner (Huganir and Nicoll, 2013). Rab5 participates in the activity-dependent endocytosis of AMPA receptors from the synaptic membrane during LTD (Brown et al., 2005). According to our results, NMDA-dependent Rab5 activation within the postsynaptic sites requires intact RIN1 GEF functions but is independent of Abl kinases because cLTD-evoked loss of surface GluA1 receptors was blocked in Rin1−/− neurons and rescued only by the presence of RIN1 constructs with a functional Rab5 GEF domain.
The amygdala plays a central role in the acquisition and extinction of fear memories (Barad et al., 2006). Regulatory endocytosis of AMPA receptors at functional synapses are involved in fear extinction in amygdala neurons (Kim et al., 2007; Lee et al., 2013), which implies that RIN1-dependent endocytosis of AMPA receptors might play a role in this process. Indeed, RIN1-knockout animals have deficits in fear learning and extinction and were proposed as a potential model for posttraumatic stress disorder, characterized by enhanced retention of fear-related memories (Dhaka et al., 2003; Bliss et al., 2010). RIN1 also has been implicated as a regulator of plasticity by inhibiting Ras/MEK/ERK-mediated pathways through a competition with Raf as a Ras-binding partner (Dhaka et al., 2003; Balaji et al., 2012). Ras-dependent signaling is known to be involved in spine morphogenesis and compartmentalization (Arendt et al., 2004; Lee and Yasuda, 2009), and its deficits have been reported in relation to mental retardation (Hu et al., 2008b). The role of RIN1 in psychiatric diseases, therefore, needs further clarification.
In line with the forgoing assumptions and on the basis of our data, we propose the following model on how endogenous RIN1 activity blocks the formation and maintenance of stable synaptic connections. In the immature and transient filopodia, RIN1 promotes dynamic changes through activation of Abl kinases, which regulate actin dynamics and integrin signaling (Figure 7A). In the case of the established dendritic spines, RIN1 activity decreases synaptic strength predominantly through its Rab5 GEF activity and is required for endocytosis of AMPA receptors (Figure 7B). Thus RIN1 has a predominate function in the formation of LTD in neurons.
FIGURE 7:

RIN1 signaling pathways in motile dendritic filopodia (A) and established spines (B). (A) S292 phosphorylation of RIN1 enhances the phosphorylation of the Y36 site by Abl kinases and leads to Abl/Arg activation via disengaged autoinhibition. In turn, Abl kinase activity increases actin remodeling and/or alters integrin signaling. (B) In mature spines, RIN1 is sequestered in the cytoplasm by 14-3-3 proteins through binding to the phosphorylated S351 site, thereby blocking the downstream activation of Rab5. The E574 site of RIN1 is responsible for the Rab5 GEF activity and controls the conversion of Rab5 GDP to Rab5 GTP, leading to enhanced clathrin-mediated endocytosis of AMPA receptors. Both of these pathways counteract the stabilization of synaptic connections and decrease synaptic efficacy. PSD, postsynaptic density.
MATERIALS AND METHODS
Animal handling
CD1, C57Bl/6, or Rin1−/− mice were housed in the animal facility at 22 ± 1°C with 12-h light/dark cycles and ad libitum access to food and water. All experiments complied with local guidelines and regulations for the use of experimental animals (PEI/001/1108-4/2013 and PEI/001/1109-4/2013), in agreement with European Union and Hungarian legislation or approved by the Regierungspräsidium Stuttgart.
Cell cultures and transfection
Primary cultures of embryonic hippocampal cells were prepared from CD1, C57Bl/6, or Rin1−/− mice on embryonic day 17/18 according to Czöndör et al. (2009). Cells were seeded onto poly-l-lysine/laminin (Sigma)–coated glass coverslips in 24-well plates or into six-well plates at 1.3 × 105 or 4.5 × 105 cells/well, respectively. For live-cell imaging experiments, cells were seeded into poly-l-lysine/laminin-coated glass-bottom 35-mm Petri dishes (Greiner) with the same cell density. Cells were transfected using Lipofectamine 2000 (Invitrogen) with the following constructs: pEGFP-N1 (Clontech), pEGFP-C2 hRIN1 wt, pEGFP-C2 hRIN1 S351A, and pEGFP-C2 hRIN1 S292A (described in Ziegler et al., 2011). The plasmids encoding pEGFP-C2-hRIN1 E574A and pEGFP-C2-hRIN1-E574A/S351A were generated by QuikChange site-directed PCR mutagenesis following the manufacturer’s instructions (Stratagene). To generate pEGFP-C2-hRIN1 QM (Y36F, Y121F, Y148F, Y295F), the cDNA encoding hRIN1 QM (generated by PCR using M4-hRIN1 QM as a template) was subcloned into a pEGFP-C2 vector.
HEK293T cells were maintained in RPMI 1640 medium supplemented with 10% fetal calf serum. Cells were transfected using TransIT293 (Mirus Bio, Madison, WI) according to the manufacturer’s instructions with the following constructs: pEGFP-N1, pEGFP-C2 hRIN1 wt, pEGFP-C2 hRIN1 S351A, and pEGFP-C2-hRIN1 E574A.
Live-cell imaging and microscopy in living neurons
Live-cell imaging recordings were carried out on transfected cells. During imaging, cultures were kept in imaging buffer (142 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 1 mM NaH2PO4, 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], 5 mM glucose, 0.8 mM MgCl2; pH 7.4) at 37°C in a chamber. Spine motility was recorded using the Zeiss AxioObserver Z1 system with a Plan Apochromat 63×/1.3 water immersion objective and the Colibri system. Time-lapse series were made with a frame rate of 2 s, using 30% 488-nm LED intensity and 100-ms exposure time. Filopodial motility in the transfected neurons was examined by the Manual Tracking ImageJ plug-in (National Institutes of Health, Bethesda, MD). More than 180 filamentous protrusions were analyzed from eight independent cultures.
Whole-cell patch clamp recordings
Electrophysiological recordings of hippocampal neurons were performed in whole-cell conditions under visual monitoring using a Zeiss A1 Axiovert 200 M microscope. The mEPSCs were recorded at room temperature (21–23°C) under voltage clamp conditions. Signals were amplified using a MultiClamp700B (Molecular Devices) and acquired at 20 kHz using the data acquisition software DASYLab, version 11 (National Instruments). Patch pipettes were pulled from standard wall glass of 1.5 mm outer diameter (Harvard Apparatus) and had input resistances of 7–10 MΩ. The composition of the bath solution (ACSF) was 140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, and 10 mM glucose; pH 7.4. Action potential firing was blocked by 0.5 μM tetrodotoxin (Tocris), also eliminating spike-mediated postsynaptic responses. Patch electrodes were filled with the following solution: 100 mM K-gluconate, 10 mM KCl, 20 mM KOH, 2 mM MgCl2, 2 mM NaCl, 10 mM HEPES, 0.2 mM ethylene glycol tetraacetic acid, and 5 mM glucose; pH 7.3. Membrane potential in voltage clamp experiments was held at −60 mV. Analysis of mEPSC frequency and amplitude was performed using software developed by A. Szu”cs (NeuroExpress). The software detected mEPSC events automatically by evaluating the magnitude and kinetics of local current fluctuations >8 pA in amplitude. Eleven CD1 and 16 Rin1−/− neurons were analyzed on DIV14–15 from three independent cultures.
Chemical treatment, transferrin uptake, and antibody feeding
We carried out cLTD at 37°C. Cells were incubated in ECS buffer (150 mM NaCl, 2 mM CaCl2, 5 mM KCl, 10 mM HEPES, 30 mM glucose; pH 7.4) for 5 min. NMDA (50 μM) and glycine (1 μM) were applied for 3 min in ECS buffer; then the medium was changed back to the original medium, and cells survived for an additional 120 min. The tyrosine kinase inhibitor imatinib was applied 1 h earlier and during the experiments (5 μM imatinib mesylate; Sigma-Aldrich).
Transferrin uptake assays of living neurons were carried out with fluorescently labeled transferrin conjugate (50 μM, transferrin–Alexa 546; Molecular Probes) applied for 1 min at 37°C in Hanks’ balanced salt solution (HBSS; Sigma-Aldrich). Neurons survived for an additional 1 or 5 min before fixation.
To analyze AMPA receptor subunit GluA1 localization and cell surface distribution, N-terminal–specific anti-GluA1 antibody (mouse, 1:100, MAB2263; Merck Millipore) was applied to living neurons for 10 min at 37°C in HBSS. Cultures were fixed with 4% paraformaldehyde for 20 min at room temperature without permeabilization. Anti-mouse secondary antibody labeled with Alexa 546 (goat, 1:500, A11030; Invitrogen) was applied for 1.5 h at room temperature after 1 h of blocking with 2% bovine serum albumin. After washing, bound secondary antibodies were fixed with 4% paraformaldehyde for 10 min.
Immunostaining and quantitative microscopy in fixed cultures
Cultures were immunostained essentially as described by Czöndör et al. (2009). We used 0.1% Triton-X100 to permeabilize the cultures. Shank2-specific primary antibody (guinea pig, 1:2000; 162 204; Synaptic System) was applied overnight at 4°C. Biotinylated secondary antibody (anti-guinea pig, 1:1000; 706-065-148; Jackson) was developed by Cy5-conjugated streptavidin (streptavidin-Cy5, 1:500; 016-170-081; Jackson). Coverslips were mounted with ProLong Diamond Antifade Mountant (Life Technologies). Images were taken with an Olympus Fluoview 500 LSM IX81 microscope with a Plan Apochromat 60×/1.4 immersion objective, using sequential excitation with 488-, 546-, and 633-nm lasers.
Transferrin signals were determined manually at the equatorial section of the neuronal soma by counting only those puncta that could be clearly determined as individual objects in the adjacent z-stacks using the AxioVision Cut View mode. Signals were considered as plasma membrane associated when the transferrin label was in close proximity to the plasma membrane, delineated by EGFP labeling within the cytoplasm. More than 130 cells from four independent cultures were analyzed.
GluA1 signal intensity was evaluated within the PSD areas found along 20-µm-long dendritic branches of transfected neurons. PSD area was outlined manually according to clear Shank2 immunopositivity at the plasma membrane within the dendritic spines or along the dendritic shaft using ImageJ on single z-stacks. After subtraction of the background fluorescence intensity, GluA1 signal intensity values were correlated to the corresponding staining intensity in control neurons, processed in an identical manner and at the same time. In all parallel experiments, images were recorded with the same microscopic settings. We analyzed 476 dendritic branches from 10 independent cultures.
Cell surface biotinylation
To determine surface-localized GluA1 subunits of the AMPA receptor, we carried out cell surface biotinylation in CD1 and Rin1−/− hippocampal neuronal cultures at DIV12–14. Cells were rinsed in PBSCM (0.1 mM CaCl2, 1 mM MgCl2 in phosphate-buffered saline [PBS]; pH 8.0) and incubated with 0.5 mg/ml sulfosuccinimidyl-2-[biotinamido]ethyl-1,3-dithiopropionate (EZ-LinkTM Sulfo-NHS-SS-Biotin; Thermo Scientific) for 30 min at 4°C in PBSCM. Unreacted biotinylation reagent was quenched by PBSCM containing 20 mM glycine (Roth) for 2 × 7 min at 4°C. Cells were lysed in lysis buffer containing 1% NP-40, 0.02% SDS, 50 mM NaF, and protease (Complete Mini Protease Inhibitor Cocktail; Roche) and phosphatase inhibitors (PhosSTOP Phosphatase Inhibitor Cocktail; Roche) in PBS. Each lysate was incubated overnight with 30 µl of NeutrAvidin-coupled agarose beads (NeutrAvidin Agarose Resins; Thermo Scientific). Beads were washed with ice-cold lysis buffer, and then biotinylated proteins were eluted with 2× SDS sample buffer. Cell-surface or total protein was subjected to SDS–PAGE. 4 CD1 and 4 Rin1−/− independent cultures were analyzed.
Protein extraction, immunoprecipitation, and Western blotting
Whole-cell extracts were obtained by solubilizing primary hippocampal neurons in lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, and 1% Triton X-100 containing protease and phosphatase inhibitors). Lysates were clarified by centrifugation at 16,000 × g for 10 min. For immunoprecipitations, equal amounts of protein were incubated with specific antibodies for 1.5 h on ice and then precipitated with protein G–Agarose beads (KPL) and washed three times with lysis buffer. Whole-cell extracts or immunoprecipitated proteins were subjected to SDS–PAGE and blotted onto nitrocellulose or polyvinylidene difluoride (Millipore) membrane. After being blocked with 0.5% blocking reagent (Roche Diagnostics) in Tris buffer containing 0.05% Tween-20 and 0.1% NaN3, membranes were probed with specific antibodies as follows: anti-GluA1 (mouse, 1:1000; ab174785; Abcam), anti–α-tubulin (rabbit, 1:1000; 2125; Cell Signaling), anti–βIII-tubulin (mouse, 1:5000; 11-264-C100; Exbio), anti-RIN1 polyclonal serum 1203 (rabbit, 1:1000; Deininger et al., 2008), anti-pCrkL (rabbit, 1:1000; 3181; Cell Signaling), and anti-GFP (mouse, 1:2000; 11814460001; Roche).
Signals were visualized with horseradish peroxidase (HRP)–coupled secondary antibodies (1:20,000; 115-035-005 and 111-035-003; Jackson) using the Luminata Crescendo Western HRP substrate (Millipore), Supersignal (TM), or West Pico (Thermo Scientific). Average intensity values were calculated using Image Studio Lite 5.0 software. The ratio of surface GluA1/total GluA1/βIII-tubulin signals was normalized to the average of control, non-cLTD values from samples run and developed in parallel. Four CD1 (eight control and nine cLTD-treated samples), three C57Bl/6 (five control and five cLTD-treated samples), and four Rin1−/− (nine control and 10 cLTD-treated samples) independent cultures were analyzed.
Statistical analyses of the data
For statistical evaluation, Student’s t test or nonparametric Mann–Whitney tests were used. SPSS Statistics software was used to calculate statistics. Data are displayed as mean ± SEM.
Supplementary Material
Acknowledgments
We thank John Colicelli (University of California, Los Angeles, CA) for providing us with the Rin1−/− mice and the plasmids M4-RIN1 QM and M4-RIN1 E574A and Rüdiger Klein (Max Planck Institute of Neurobiology, Martinsried, Germany) for the RIN1 antiserum. Research was supported by Grant KTIA_NAP_13-2014-0018 from the National Research, Development and Innovation Office and by Hungarian Scientific Research Foundation Grant K81934 to K.S., by the HA 3557/11-2 Deutsche Forschungsgemeinschaft Project to A.H., and by the German-Hungarian Academic Exchange Service 73539 and PPP Ungarn 57215775 Programs to K.S. and A.H.
Abbreviations used:
- cLTD
chemically induced long-term depression
- LTD
long-term depression
- PSD
postsynaptic density
- RIN1
Ras and Rab interactor 1
Footnotes
The authors declare no conflict of interest.
Conceptualization due to A.H. and K.S. Methodology due to A.H., A.S., and K.S. Formal analysis and investigation by Z.S., H.L., A.I., A.E.R., K.E., C.O.O.M., and G.L. Manuscript by Z.S., A.H., and K.S.
†Present address: Department of Immunology, Institute of Nutritional Medicine, University Hohenheim, D-70599 Stuttgart, Germany.
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-07-0526) on November 16, 2016.
REFERENCES
- Arendt T, Gärtner U, Seeger G, Barmashenko G, Palm K, Mittmann T, Yan L, Hümmeke M, Behrbohm J, Brückner MK, et al. Neuronal activation of Ras regulates synaptic connectivity. Eur J Neurosci. 2004;19:2953–2966. doi: 10.1111/j.0953-816X.2004.03409.x. [DOI] [PubMed] [Google Scholar]
- Balaji K, Colicelli J. RIN1 regulates cell migration through RAB5 GTPases and ABL tyrosine kinases. Commun Integr Biol. 2013;6:e25421. doi: 10.4161/cib.25421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaji K, Mooser C, Janson CM, Bliss JM, Hojjat H, Colicelli J. RIN1 orchestrates the activation of RAB5 GTPases and ABL tyrosine kinases to determine the fate of EGFR. J Cell Sci. 2012;125:5887–5896. doi: 10.1242/jcs.113688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barad M, Gean PW, Lutz B. The role of the amygdala in the extinction of conditioned fear. Biol Psychiatry. 2006;60:322–328. doi: 10.1016/j.biopsych.2006.05.029. [DOI] [PubMed] [Google Scholar]
- Bliss JM, Gray EE, Dhaka A, Dell TJO, Colicelli J. Fear learning and extinction are linked to neuronal plasticity through RIN1 signaling. J Neurosci Res. 2010;88:917–926. doi: 10.1002/jnr.22252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TC, Tran IC, Backos DS, Esteban JA. NMDA receptor-dependent activation of the small GTPase Rab5 drives the removal of synaptic AMPA receptors during hippocampal LTD. Neuron. 2005;45:81–94. doi: 10.1016/j.neuron.2004.12.023. [DOI] [PubMed] [Google Scholar]
- Colicelli J. ABL tyrosine kinases: evolution of function, regulation, and specificity. Sci Signal. 2010;3:1–46. doi: 10.1126/scisignal.3139re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czöndör K, Ellwanger K, Fuchs YF, Lutz S, Gulyás M, Mansuy IM, Hausser A, Pfizenmaier K, Schlett K. Protein kinase D controls the integrity of Golgi apparatus and the maintenance of dendritic arborization in hippocampal neurons. Mol Biol Cell. 2009;20:2108–2120. doi: 10.1091/mbc.E08-09-0957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoop MJ, Huber LA, Stenmark H, Williamson E, Zerial M, Parton RG, Dotti CG. The involvement of the small GTP-binding protein Rab5a in neuronal endocytosis. Neuron. 1994;13:11–22. doi: 10.1016/0896-6273(94)90456-1. [DOI] [PubMed] [Google Scholar]
- Deininger K, Eder M, Kramer ER, Zieglgänsberger W, Dodt HU, Dornmair K, Collicelli J, Klein R. The Rab5 guanylate exchange factor RIN1 regulates endocytosis of the EphA4 receptor in mature excitatory neurons. Proc Natl Acad Sci USA. 2008;105:12539–12544. doi: 10.1073/pnas.0801174105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkach VA, Oh MC, Guire ES, Soderling TR. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci. 2007;8:101–113. doi: 10.1038/nrn2055. [DOI] [PubMed] [Google Scholar]
- Dhaka A, Costa RM, Hu H, Irvin DK, Patel A, Kornblum HI, Silva AJ, Dell TJO, Colicelli J. The RAS effector RIN1 modulates the formation of aversive memories. J Neurosci. 2003;23:748–757. doi: 10.1523/JNEUROSCI.23-03-00748.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Galvis A, Giambini H, Villasana Z, Barbieri MA. Functional determinants of Ras interference 1 mutants required for their inhibitory activity on endocytosis. Exp Cell Res. 2009;315:820–835. doi: 10.1016/j.yexcr.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerarduzzi C, He Q, Zhai B, Antoniou J, Di Battista JA. Prostaglandin E2-dependent phosphorylation of RAS inhibition 1 (RIN1) at ser 291 and 292 inhibits transforming growth factor-β-induced RAS activation pathway in human synovial fibroblasts: role in cell migration. J Cell Physiol. 2017;232:202–2015. doi: 10.1002/jcp.25412. [DOI] [PubMed] [Google Scholar]
- Gourley SL, Olevska A, Warren MS, Taylor JR, Koleske AJ. Arg kinase regulates prefrontal dendritic spine refinement and cocaine-induced plasticity. J Neurosci. 2012;32:2314–2323. doi: 10.1523/JNEUROSCI.2730-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Wong D, Dhaka A, Afar D, White M, Xie W, Herschman H, Witte O, Colicelli J. Protein binding and signaling properties of RIN1 suggest a unique effector function. Proc Natl Acad Sci USA. 1997;94:4954–4959. doi: 10.1073/pnas.94.10.4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotulainen P, Llano O, Smirnov S, K Tanhuanpää, Faix J, Rivera C, Lappalainen P. Defining mechanisms of actin polymerization and depolymerization during dendritic spine morphogenesis. J Cell Biol. 2009;185:323–339. doi: 10.1083/jcb.200809046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Bliss JM, Wang Y, Colicelli J. RIN1 is an ABL tyrosine kinase activator and a regulator of epithelial-cell adhesion and migration. Curr Biol. 2005;15:815–823. doi: 10.1016/j.cub.2005.03.049. [DOI] [PubMed] [Google Scholar]
- Hu H, Milstein M, Bliss JM, Thai M, Malhotra G, Huynh LC, Colicelli J. Integration of transforming growth factor beta and RAS signaling silences a RAB5 guanine nucleotide exchange factor and enhances growth factor-directed cell migration. Mol Cell Biol. 2008a;28:1573–1583. doi: 10.1128/MCB.01087-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Qin Y, Bochorishvili G, Zhu Y, Aelst LV, Zhu JJ. Ras signaling mechanisms underlying impaired GluR1-dependent plasticity associated with Fragile X syndrome. J Neurosci. 2008b;28:7847–7862. doi: 10.1523/JNEUROSCI.1496-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huganir RL, Nicoll RA. AMPARs and synaptic plasticity: the last 25 years. Neuron. 2013;80:704–717. doi: 10.1016/j.neuron.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SB, Lu HY, Lu Q. Abl tyrosine kinase promotes dendrogenesis by inducing actin cytoskeletal rearrangements in cooperation with Rho family small GTPases in hippocampal neurons. J Neurosci. 2004;24:8510–8521. doi: 10.1523/JNEUROSCI.1264-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jozic I, Saliba SC, Barbieri MA. Effect of EGF-receptor tyrosine kinase inhibitor on Rab5 function during endocytosis. Arch Biochem Biophys. 2012;525:16–24. doi: 10.1016/j.abb.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerrisk ME, Greer CA, Koleske AJ. Integrin α3 is required for late postnatal stability of dendrite arbors, dendritic spines and synapses, and mouse behavior. J Neurosci. 2013;33:6742–6752. doi: 10.1523/JNEUROSCI.0528-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lee S, Park K, Hong I, Song B, Son G, Park H, Kim WR, Park E, Choe HK, et al. Amygdala depotentiation and fear extinction. Proc Natl Acad Sci USA. 2007;104:20955–20960. doi: 10.1073/pnas.0710548105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koleske AJ. Regulation of cytoskeletal dynamics and cell morphogenesis by Abl family kinases. In: Koleske AJ, editor. Abl Family Kinases in Development and Disease. New York: Springer-Verlag; 2006. pp. 48–67. [Google Scholar]
- Korkotian E, Segal M. Regulation of dendritic spine motility in cultured hippocampal neurons. J Neurosci. 2001;21:6115–6124. doi: 10.1523/JNEUROSCI.21-16-06115.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korobova F, Svitkina T. Molecular architecture of synaptic actin cytoskeleton in hippocampal neurons reveals a mechanism of dendritic spine morphogenesis. Mol Biol Cell. 2010;21:165–176. doi: 10.1091/mbc.E09-07-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Lee EJ, Song YS, Kim E. Long-term depression-inducing stimuli promote cleavage of the synaptic adhesion molecule NGL-3 through NMDA receptors, matrix metalloproteinases and presenilin/γ-secretase. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130158. doi: 10.1098/rstb.2013.0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Song B, Kim J, Park K, Hong I, An B, Song S, Lee J, Park S, Kim J, et al. GluA1 phosphorylation at serine 831 in the lateral amygdala is required for fear renewal. Nat Neurosci. 2013;16:1436–1444. doi: 10.1038/nn.3491. [DOI] [PubMed] [Google Scholar]
- Lee SJR, Yasuda R. Spatiotemporal regulation of signaling in and out of dendritic spines: CaMKII and Ras. Open Neurosci J. 2009;3:117–127. doi: 10.2174/1874082000903020117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Yeckel MF, Koleske AJ. Abl2/Arg controls dendritic spine and dendrite arbor stability via distinct cytoskeletal control pathways. J Neurosci. 2013;33:1846–1857. doi: 10.1523/JNEUROSCI.4284-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE, Raghavachari S, Tsien RW. The sequence of events that underlie quantal transmission at central glutamatergic synapses. Nat Rev Neurosci. 2007;8:597–609. doi: 10.1038/nrn2191. [DOI] [PubMed] [Google Scholar]
- Moresco EMY, Donaldson S, Williamson A, Koleske AJ. Integrin-mediated dendrite branch maintenance requires Abelson (Abl) family kinases. J Neurosci. 2005;25:6105–6118. doi: 10.1523/JNEUROSCI.1432-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moresco EMY, Scheetz AJ, Bornmann WG, Koleske AJ, Fitzsimonds RM. Abl family nonreceptor tyrosine kinases modulate short-term synaptic plasticity. J Neurophysiol. 2003;89:1678–1687. doi: 10.1152/jn.00892.2002. [DOI] [PubMed] [Google Scholar]
- Naisbitt S, Eunjoon K, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF, Sheng M. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron. 1999;23:569–582. doi: 10.1016/s0896-6273(00)80809-0. [DOI] [PubMed] [Google Scholar]
- Perez de Arce K, Varela-Nallar L, Farias O, Cifuentes A, Bull P, Couch BA, Koleske AJ, Inestrosa NC, Alvarez AR. Synaptic clustering of PSD-95 is regulated by c-Abl through tyrosine phosphorylation. J Neurosci. 2010;30:3728–3738. doi: 10.1523/JNEUROSCI.2024-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage DG, Antman KH. Imatinib mesylate—a new oral targeted therapy. N Engl J Med. 2002;346:683–693. doi: 10.1056/NEJMra013339. [DOI] [PubMed] [Google Scholar]
- Sfakianos MK, Eisman A, Gourley SL, Bradley WD, Scheetz AJ, Settleman J, Taylor JR, Greer CA, Williamson A, Koleske AJ. Inhibition of Rho via Arg and p190RhoGAP in the postnatal mouse hippocampus regulates dendritic spine maturation, synapse and dendrite stability, and behavior. J Neurosci. 2007;27:10982–10992. doi: 10.1523/JNEUROSCI.0793-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Colledge M, Crozier RA, Chen WS, Scott JD, Bear MF. Role for A kinase-anchoring proteins (AKAPS) in glutamate receptor trafficking and long term synaptic depression. J Biol Chem. 2005;280:16962–16968. doi: 10.1074/jbc.M409693200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tall GG, Barbieri MA, Stahl PD, Horazdovsky BF. Ras-activated endocytosis is mediated by the Rab5 guanine nucleotide exchange activity of RIN1. Dev Cell. 2001;1:73–82. doi: 10.1016/s1534-5807(01)00008-9. [DOI] [PubMed] [Google Scholar]
- Tárnok K, Gulyás M, Bencsik N, Ferenc K, Pfizenmaier K, Hausser A, Schlett K. A new tool for the quantitative analysis of dendritic filopodial motility. Cytometry A. 2015;87:89–96. doi: 10.1002/cyto.a.22569. [DOI] [PubMed] [Google Scholar]
- Tashiro A, Yuste R. Regulation of dendritic spine motility and stability by Rac1 and Rho kinase: evidence for two forms of spine motility. Mol Cell Neurosci. 2004;26:429–440. doi: 10.1016/j.mcn.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Tatavarty V, Das S, Yu J. Polarization of actin cytoskeleton is reduced in dendritic protrusions during early spine development in hippocampal neuron. Mol Biol Cell. 2012;23:3167–3177. doi: 10.1091/mbc.E12-02-0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Waldron RT, Dhaka A, Patel A, Riley MM, Rozengurt E, Colicelli J. The RAS effector RIN1 directly competes with RAF and is regulated by 14-3-3 proteins. Mol Cell Biol. 2002;22:916–926. doi: 10.1128/MCB.22.3.916-926.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren MS, Bradley WD, Gourley SL, Lin YC, Simpson MA, Reichardt LF, Greer CA, Taylor JR, Koleske AJ. Integrin β1 signals through Arg to regulate postnatal dendritic arborization, synapse density, and behavior. J Neurosci. 2012;32:2824–2834. doi: 10.1523/JNEUROSCI.3942-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodring PJ, Litwack ED, O’Leary DDM, Lucero GR, Wang JYJ, Hunter T. Modulation of the F-actin cytoskeleton by c-Abl tyrosine kinase in cell spreading and neurite extension. J Cell Biol. 2002;156:879–892. doi: 10.1083/jcb.200110014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Levy AD, Rosenberg BJ, Higley MJ, Koleske AJ. Disruption of coordinated presynaptic and postsynaptic maturation underlies the defects in hippocampal synapse stability and plasticity in Abl2/Arg-deficient mice. J Neurosci. 2016;36:6778–6791. doi: 10.1523/JNEUROSCI.4092-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler S, Eiseler T, Scholz RP, Beck A, Link G, Hausser A. A novel protein kinase D phosphorylation site in the tumor suppressor Rab interactor 1 is critical for coordination of cell migration. Mol Biol Cell. 2011;22:570–580. doi: 10.1091/mbc.E10-05-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv NE, Smith SJ. Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron. 1996;17:91–102. doi: 10.1016/s0896-6273(00)80283-4. [DOI] [PubMed] [Google Scholar]
- Zukerberg LR, Patrick GN, Nikolic M, Humbert S, Wu CL, Lanier LM, Gertler FB, Vidal M, Van Etten RA, Tsai LH. Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine phosphorylation, kinase upregulation, and neurite outgrowth. Neuron. 2000;26:633–646. doi: 10.1016/s0896-6273(00)81200-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.