

Abstract
Purpose
The purpose of this study was to determine the effects of metformin on dysfunctional retinas in obesity-induced type 2 diabetic mice.
Methods
A high-fat diet (HFD)-induced diabetic mouse model (C57BL/6J) was used in this study. After 2 months of the HFD regimen, HFD mice were given daily metformin through oral gavage. Body weights, glucose tolerance, and retinal light responses were monitored regularly. Fluorescein angiography (FA) was used to assess changes in retinal vasculature. Ocular tissues (retina, vitreous, and lens) were harvested and analyzed for molecular changes as determined by immunofluorescent staining, Western blot analysis, and cytokine profiling.
Results
Starting 1 month after the diet regimen, mice fed the HFD had mildly compromised retinal light responses as measured by electroretinography (ERG), which worsened over time compared to that in the control. In HFD mice treated with metformin, systemic glucose levels reverted back to normal, and their weight gain slowed. Metformin reversed HFD-induced changes in phosphorylated protein kinase B (pAKT), extracellular signal-regulated kinase (pERK), and 5′AMP-activated protein kinase (pAMPK) in the retina. However, metformin treatments for 3 months did not restore the retinal light responses nor lessen the HFD-induced retinal neovascularization, even though it did reduce intraocular inflammation.
Conclusions
Although metformin was able to reverse systemic changes induced by HFD, it was not able to restore HFD-caused retinal light responses or deter neovascularization.
Keywords: diabetic retinopathy, high-fat diet, inflammation, metformin, obesity
Diabetic retinopathy (DR) is one of the major secondary complications of diabetes and a leading cause of blindness worldwide.1 Glycemic control is a critical strategy for diabetic patients to prevent the development of secondary complications.2 The Diabetes Control and Complications Trial showed that long-term extensive control of serum glucose reduces the incidence and progression of diabetic complications such as retinopathy, nephropathy, and neuropathy.3 One drug that effectively controls systemic glycemia is metformin.4,5 Metformin has been used as an antihyperglycemic agent in diabetic patients,6 and it is recommended in combination therapies to control the level of glycated hemoglobin (HbA1c) in patients receiving ineffective monotherapy.7 The effectiveness of metformin as an antihyperglycemic agent is based on its ability to suppress gluconeogenesis in the liver.5,8 In addition, metformin has been identified as an activator of the AMP-activated protein kinase (AMPK) pathway in numerous cell types including hepatocytes,9 endothelial cells,10 cardiomyocytes,11 cancer cells,12 and adipose tissue.13 In addition to its use as an antihyperglycemic agent, one beneficial effect from metformin is mild weight loss.14,15 The action of metformin on insulin signaling has made metformin viable for treating nonalcoholic fatty liver disease16 and polycystic ovary syndrome.17 Furthermore, the effectiveness of metformin to treat other diabetic complications, such as nephropathy18 and neuropathy has been investigated19; however, whether metformin is able to prevent or reverse DR is not known.
The high-fat diet (HFD) mouse model has been used to study type 2 diabetes due to the development of obesity, glucose intolerance, and insulin resistance.20 Unlike other mouse models of metabolic syndromes and type 2 diabetes that use genetic mutations to induce insulin resistance and obesity,21 the HFD model is a diet-induced obesity model that resembles human obesity-associated type 2 diabetes. Mice develop hyperglycemia, hyperinsulinemia, hyperlipidemia, and chronic inflammation after several months of HFD regimen and are suitable for study of long-term diabetic complications.22,23 These HFD mice show similar phenotypical deficits found in other DR animal models, such as lesions in the retinal vasculature and thickening of Bruch's membrane.24 Furthermore, mice fed a HFD containing 42% fat calories for 12 months have significantly greater numbers of atrophic capillaries and pericyte ghosts than mice fed a normal diet.25 High-fat diet mice emulate the systematic dysfunction that occurs in type 2 diabetes and further show retinal symptoms found in DR, making HFD mice a suitable animal model to study type 2 diabetes and diabetic retinopathy.
Previously, we reported that mice fed a HFD (59% fat calories) develop obesity, hyperglycemia, insulin resistance, glucose intolerance, and decreased retinal light sensitives 26. These mice have retinal neovascularization after 7 months of HFD regimen 27. Because metformin is able to maintain normal systemic glycemia in diabetic animals and patients,4,5 in this study, we investigated whether metformin was able to reverse or minimize HFD-induced retinal dysfunction. We combined electroretinogram (ERG) recordings, immunofluorescent staining, fluorescein angiography (FA), and cytokine profiling to determine the effects of metformin in HFD-induced diabetic retina.
Materials and Methods
Animals
Four-week-old male C57BL/6J mice were purchased from Harlan Laboratory (Houston, TX, USA). All animal experiments were approved by the Institutional Animal Care and Use Committee of Texas A&M University and were performed in compliance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Mice were housed under temperature- and humidity-controlled, 12L:12D cycle conditions. All mice were given food and water ad libitum. At 5 weeks of age (body weight, 20 g), mice were fed a standard laboratory chow (control diet: 10% fat calories, 20% protein calories, and 70% carbohydrate calories; Research Diets, Inc., New Brunswick, NJ, USA) or an HFD (59.4% fat calories, 18.1% protein calories, and 22.5% carbohydrate calories; TestDiet, St. Louis, MO, USA). After 2 months of HFD regimen, half of the HFD mice were given daily metformin treatments at a dose of 150 mg/kg through oral gavage. Body weight and food intake were measured weekly. Nonfasting blood glucose levels and glucose tolerance were measured monthly by taking blood from the tail vein. Glucose levels were measured using Clarity Plus blood glucose monitoring system (Diagnostic Test Group, Boca Raton, FL, USA).
Glucose Tolerance Test
Mice were fasted for 8 hours and given a single intraperitoneal (IP) injection of D-glucose (Sigma-Aldrich Corp., St. Louis, MO, USA) at a dose of 2 g per kg of body weight. Blood samples drawn from the tail vein were used to measure glucose levels, using Clarity Plus blood glucose monitoring system (Diagnostic Test Group) at 0, 30, 60, 90, and 120 minutes following the glucose injection.
In Vivo Electroretinogram
In vivo ERG retinal light responses were recorded as described previously.26 Mice were dark-adapted for a minimum of 3 hours and anesthetized with an IP injection of Avertin (2% 2,2,2-tribromoethanol, 1.25% tert-amyl alcohol; Fisher Scientific, Pittsburgh, PA, USA) solution (12.5 mg/ml) at a dose of 500 μl per 25 g of body weight. Pupils were dilated using a single drop of 1% tropicamide/2.5% phenylephrine mixture for 5 minutes. Mice were placed on a heating pad to maintain body temperature at 37°C. The ground electrode was placed on the tail, and the reference electrode was placed under the skin in the cheek below the eye. A thin drop of Goniovisc (Hub Pharmaceuticals, Rancho Cucamonga, CA, USA) was applied to the surface of the cornea to keep it moist, and a threaded recording electrode conjugated to a minicontact lens (Ocuscience, Henderson, NV, USA) was placed on top of the cornea. All preparatory procedures were done under dim red light, and the light was turned off during the recording. A portable ERG device (OcuScience) was used to measure scotopic ERG recordings at light intensities of 0.1, 0.3, 1, 3, 10, and 25 candela.second/meter2 (cd.s/m2). Responses to 4 light flashes were averaged at the lower light intensities (0.1, 0.3, 1.0, and 3.0 cd.s/m2), whereas only 1 light flash was applied for the higher light intensities (10 and 25 cd.s/m2). A 1-minute recovery period was programmed between different light intensities. The amplitudes and implicit times of the a-wave, b-wave, and oscillatory potentials (OPs) were recorded and analyzed using ERGView 4.4 software (OcuScience). Both eyes were included in the analyses.
Immunofluorescent Staining
Mouse eyes were excised and prepared as previously described.26 Briefly, eyes were fixed with Zamboni fixative and processed for paraffin sectioning at 4 μm. Each glass slide contained single paraffin sections from the control, HFD, and HFD plus metformin (HFD+Met) groups. After deparaffinization and antigen retrieval, sections were washed in phosphate-buffered saline (PBS), blocked with 10% goat serum for 2 hours at room temperature and then incubated overnight with primary antibodies at 4°C. The next day, sections were washed with PBS several times and incubated with fluorescent-conjugated secondary antibodies for 2 hours at room temperature and mounted with ProLong Gold antifade reagent containing 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen/Life Technologies, Grand Island, NY, USA). The primary antibodies used were anti-phospho-protein kinase B (pAKTThr308; 1:100 dilution; Cell Signaling Technology, Danvers, MA, USA), anti-AKT (total AKT; 1:100 dilution; Cell Signaling Technology), anti-di-phospho-extracellular signal-regulated kinase (pERK; 1:100 dilution; Sigma-Aldrich Corp.), anti-ERK (total ERK; 1:100 dilution; Santa Cruz Biotechnology, Dallas, TX, USA), anti-phospho-AMPK (pAMPKThr172; 1:100 dilution; Cell Signaling Technology), and anti-AMPK (total AMPK; 1:100 dilution; Cell Signaling Technology). The secondary antibodies used were Alexa Fluor 488 goat anti-rabbit immunoglobulin G (IgG; 1:150 dilution; Molecular Probes/Life Technologies, Grand Island, NY, USA) and Cy5 goat anti-mouse IgG (1:150 dilution; Abcam, Cambridge, MA, USA). Images were obtained using a Stallion microscope (Carl Zeiss AG, Oberkochen, Germany). Each fluorescent image from control, HFD, and HFD+Met groups was taken under identical settings, including the same exposure time and magnification. Image analysis consisted of selecting and analyzing three fluorescent images from each retinal tissue section, which included all retinal layers (from the photoreceptor outer segment to the ganglion cell layer). The averaged fluorescence intensity per pixel for each image was quantified without any modification, using the luminosity channel of the histogram function in the Photoshop 6.0 software (Adobe Systems, San Jose, CA, USA), and the green or red fluorescence intensities were measured on a scale of 0 to 255 brightness levels. A total of 3 to 5 retinal sections from each animal were processed for immunostaining and image analyses.
Fluorescein Angiography (FA)
Mice were anesthetized using an IP injection of Avertin (12.5 mg/ml) at a dose of 500 μl per 25 g of body weight. Pupils were dilated using a single drop of 1% tropicamide/2.5% phenylephrine mixture for 5 minutes. Immediately following pupil dilation, 10% sodium fluorescein (Akorn, Lake Forest, IL, USA) was injected IP at a dose of 50 μl per 25 g of body weight. Images were taken using iVivo Funduscope for small animals (Ocuscience). The vascular parameters were further analyzed using Photoshop version 6.0 (Adobe Systems) and AngioTool software, a free software developed by the US National Institutes of Health/National Cancer Institute (Bethesda, MD, USA).28 Areas of 289 × 289 pixel2 in the central retina (400 pixels from the optic nerve), as well as in the peripheral retinal region (800 pixels from the optic nerve), were cropped using Photoshop. For each FA-cropped image, at least 2 areas from the central and peripheral retinal regions were obtained to analyze the microvascular density (the percentage of vascular area compared to the retinal area), vessel area, vessel branch points, and average nonvascular area (average lacunarity), using AngioTool. The primary retinal arteries and veins were not included in the analyses.
Western Immunoblot Analysis
Retina samples were collected as previously described.26,27 Briefly, 2 retinas from a single mouse were pooled and counted as 1 sample. Intact retinas were homogenized in a Tris lysis buffer (50 mM Tris, 1 mM EGTA, 150 mM NaCl, 1% Triton X-100, 1% β-mercapto-ethanol, 50 mM NaF, 1 mM Na3VO4; pH, 7.5). Samples were separated using 10%-sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. The primary antibodies used were anti-phosphorylated nuclear factor κ-light-chain enhancer of activated B cells complex (NFκB) P65 at Ser536 (pP65; Cell Signaling Technology) and NFκB P65 (Total P65; Cell Signaling Technology). Blots were visualized using appropriate secondary antibodies conjugated to horseradish peroxidase (Cell Signaling Technology) and an enhanced chemiluminescence detection system (Pierce, Rockford, IL, USA).
Bio-plex Assay for Cytokine Profiling
Because of the small size of mouse eyes, the vitreous and lens from both eyes were harvested and pooled (as n = 1) for Bio-plex analyses of intraocular cytokines. The assay was performed according to instructions from the manufacturer (EMD Millipore, Darmstadt, Germany). The concentrations of chosen cytokines were determined using the Milliplex Map kit (EMD Millipore) and detected using the Bio-Plex 200 system (Bio-Rad, Hercules, CA, USA). Data were analyzed using Bio-Plex Manager software (Bio-Rad). The cytokine profile included interleukin-6 (IL-6), IL-2, monocyte chemoattractant protein-1 (MCP-1), granulocyte colony-stimulating factor (G-CSF), and vascular endothelial growth factor (VEGF).
Statistical Analyses
All data are mean ± standard error of the mean (SEM). Statistical analyses were carried out using Origin 8.6 software (OriginLab, Northampton, MA, USA). The Student's t-test was used for statistical comparisons between the control and HFD groups. 1-way analysis of variance (ANOVA) followed by Tukey's post hoc test was used for statistical analyses among the control, HFD, and HFD+Met groups. Throughout, both eyes from the same animal were used in the analyses, and the sample size n was the number of animals per group. P values <0.05 were regarded as significant.
Results
Metformin Decelerated Body Weight Gain and Reversed Hyperglycemia in HFD Diabetic Animals
Mice fed a HFD for 2 weeks (Fig. 1, open circle) already had a significant weight gain compared to the controls (Fig. 1A, open square). Treatment with metformin in HFD-induced obese mice (Fig. 1, gray triangles) slowed down their weight gain compared to HFD mice without metformin intervention (Fig. 1A). Mice under the HFD regimen for 2 months developed hyperglycemia (Fig. 1B), but after metformin for only 1 month, the nonfasting blood glucose levels of these HFD mice returned to normal levels (Fig. 1B). The glucose tolerance test results after 1 month of metformin treatment further verified the effectiveness of metformin in antihyperglycemia and reversing the glucose intolerance in HFD obese mice back to the control level (Fig. 1C). Hence, metformin was effective in controlling systemic glycemia and weight gain.
Figure 1.
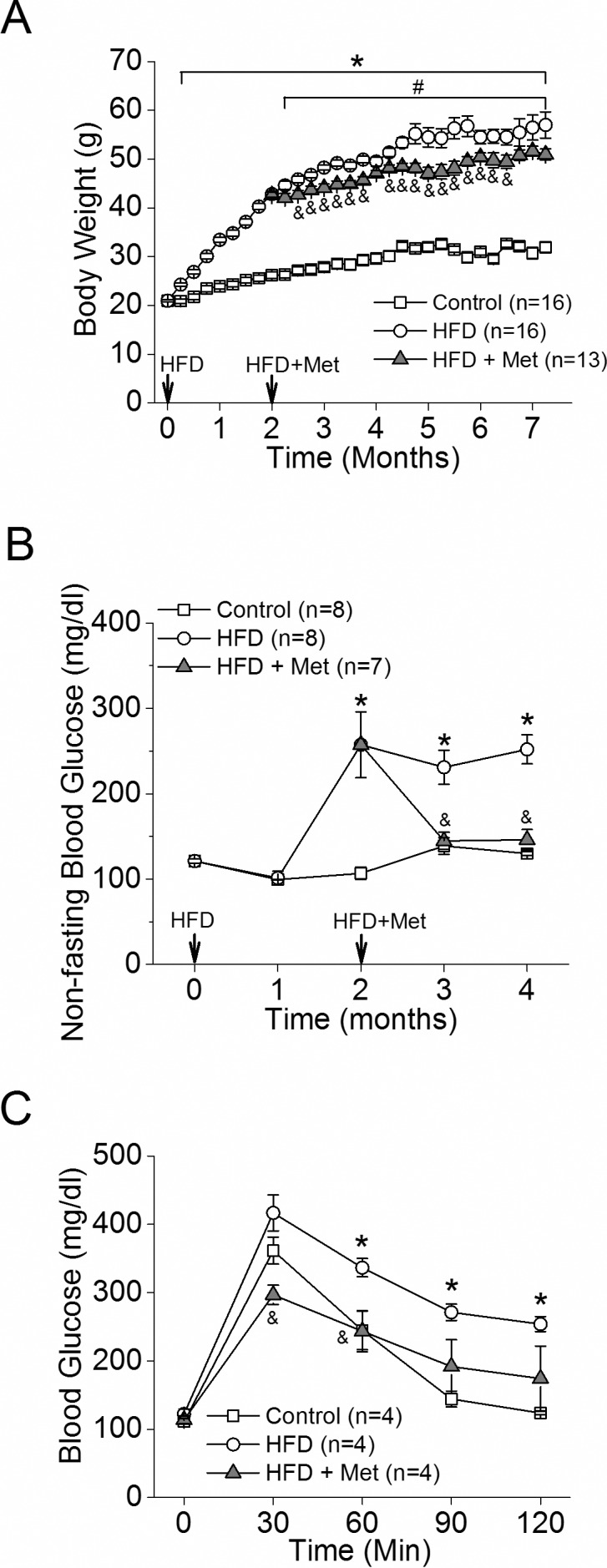
Metformin slows the rate of weight gain and controls diet-induced hyperglycemia in HFD mice. Mice were fed a normal chow diet (Control [open square]) or HFD (open circle). Two months after the diet regimen, half of the HFD mice were given daily oral metformin treatments (gray triangle). (A) Mice fed HFD had a significant weight gain starting 1 week after HFD compared to the control (*). The HFD-fed mice given daily metformin (HFD+Met) showed decelerated weight gain compared to the HFD mice without metformin intervention (HFD). # indicates statistical significance between the control and HFD+Met groups. & indicates statistical significance between the HFD and HFD+Met groups. (B) HFD-fed mice had an increase of nonfasting blood glucose after 2 months of the diet regimen. Following 1 month of metformin treatment, the resting blood glucose levels of HFD+Met mice were back to control levels. (C) The ability of metformin to control HFD-induced hyperglycemia is shown by the glucose tolerance test. *,#,&P < 0.05.
HFD-Induced Retinal Dysfunction Began With Dampened Oscillatory Potential Responses
We previously showed that retinal light responses decreased after mice were fed a HFD for 3 months.26 To further determine when HFD-induced obesity caused retinal dysfunction, we measured the retinal light sensitivities with scotopic ERG recordings after the mice were fed the HFD for only 1 month. We found that the amplitudes of ERG a- and b-waves in HFD mice (Fig. 1, gray circles) were similar to those of the controls (Figs. 2A, 2B, open squares), but the HFD mice had longer a- and b-wave implicit times (Fig. 2B). These HFD mice also had significantly decreased OP amplitudes and delayed OP implicit times compared with the control mice (Figs. 2A, 2C), which indicates a possible early sign of obesity-induced retinal dysfunction, as a delayed OP latency is the first sign of an early diabetic retina in both rodents and humans.29–31
Figure 2.

Scotopic ERG implicit times and oscillatory potential responses are decreased in mice given HFD for 1 month. (A) Representative scotopic ERG and oscillatory potential waveforms recorded from control and HFD-fed mice at light intensity of 25 cd.s/m2 are shown. (B) The average scotopic ERG a- and b-wave implicit times were delayed in HFD-fed mice after 1 month of HFD compared to that in controls, but there were no differences in ERG amplitudes. (C) The oscillatory potential amplitudes (OP1–OP4) of HFD mice were significantly decreased, and the implicit times were delayed compared to those of the controls. Student's t-test was used for statistical analysis. *P < 0.05.
Metformin Treatment Did Not Improve the HFD-Induced Decreases in Retinal Light Responses
After 2 months of HFD, the retinal light responses in HFD mice (Fig. 3, gray circles) were further deteriorated (Fig. 3). Compared to the control (Fig. 3, open squares), these HFD mice had decreased a- and b-wave amplitudes (Figs. 3A, 3B) in addition to the functional deficits previously observed after 1 month of HFD (Figs. 3A, 3C). After 5 months of HFD, the retinal light responses in these HFD mice (Fig. 3, open circles) worsened and were significantly lower than those of the control mice (Fig. 4, open squares). However, the retinal light responses in HFD+Met mice (Fig. 4, gray triangles) did not improve, as measured by ERG a- and b-waves (Figs. 4A, 4B), and their OPs were further aggravated (Figs. 4A, 4C), even though these HFD mice had been treated with metformin for the first 3 months (Fig. 4D).
Figure 3.

Scotopic ERG amplitudes, implicit times, and oscillatory potential responses were further decreased in mice given HFD for 2 months. (A) Representative scotopic ERG and oscillatory potential waveforms recorded from control and HFD-fed mice at light intensity of 25 cd.s/m2 are shown. (B) The average scotopic ERG a- and b-wave amplitudes were decreased, and implicit times were delayed in mice under 2 months of HFD compared to controls. (C) The oscillatory potential amplitudes of HFD mice are also significantly decreased, and the implicit times are delayed compared to those of the controls. Student's t-test was used for statistical analysis. *P < 0.05.
Figure 4.

Metformin treatment for 3 months does not decrease HFD-induced retinal deficiencies. Control mice were fed standard chow for 5 months. Test mice were fed HFD for 5 months. The HFD+Met mice were given HFD for 5 months and metformin for the last 3 months. (A) Representative scotopic ERG and oscillatory potential waveforms recorded from controls, HFD-fed mice, and HFD mice treated with metformin at light intensity of 25 cd.s/m2 are shown. (B) HFD-fed mice have decreased a- and b-wave amplitudes and delayed b-wave implicit times compared to those of the controls (*). The HFD+Met mice have decreased a-wave and b-wave amplitudes and delayed b-wave implicit times compared to those of controls (#). The HFD+Met mice had delayed b-wave implicit times compared to those of HFD mice (&), but there were no statistical differences between the in a-wave amplitudes, b-wave amplitudes, and a-wave implicit times in the HFD group and those in the HFD+Met group. (C) The oscillatory potential amplitudes were decreased, and implicit times were delayed in HFD-fed mice compared to those in the controls (*). The oscillatory potential amplitudes were decreased and implicit times were delayed in HFD+Met mice compared to those in the controls (#). Furthermore, HFD+Met mice had decreased OP amplitudes and delayed implicit times compared to those in the HFD-fed mice (&). (D) Maximal scotopic a- and b-wave amplitudes are unchanged in control mice over time. However, maximal scotopic a- and b-wave amplitudes of HFD-fed mice are decreased after 2 months and further decreased after 5 months of the diet regimen. Treatments with metformin for 3 months (HFD+Met) did not reverse the a- and b-wave amplitudes back to control levels. *,#,&P < 0.05.
Metformin Restored Cell-Signaling Proteins in the Retina That Were Affected by HFD
Although metformin did not recover HFD-induced retinal dysfunction, we next examined whether oral administration of metformin impacted the retina of HFD mice at the molecular level. We determined the activation/phosphorylation of AKT,32,33 ERK,34 and AMPK35 signaling, as these kinases are critical in cell metabolism, growth, and survival. Mice receiving a HFD for 6 months had a decrease in the phosphorylation of AKT (pAKT) and AMPK (pAMPK) but an increase in activated ERK (pERK) in the retina, and there were no apparent changes in the total amounts of AKT, ERK, and AMPK in the HFD retina compared with control retinas (Fig. 5, Con). The retinas from HFD+Met mice treated for 3 months with HFD and metformin for 4 months had a recovery in these signaling molecules: their pAKT and pAMPK expression levels were no longer dampened and were similar to those of the controls, whereas the level of pERK was decreased and comparable to that of the control level (Fig. 5). Interestingly, the retinas from these HFD+Met mice had an apparent up-regulation of total AMPK and pAMPK compared to the controls, which indicated that metformin might have a direct effect on AMPK in the neural retina, as metformin is known to up-regulate the expression and activation of AMPK in the kidney,36 adipose tissue,13,37 and heart.38
Figure 5.

Metformin reverses HFD-induced effects on the immunofluorescent intensities of several proteins in mice retina. Mouse retinal sections (4 μm) were processed for immunofluorescent staining. Control mice (Con) were given standard chow for 7 months. The HFD-fed mice (HFD) were given HFD for 7 months. The HFD+Met (HFD+Met) mice were given HFD for 7 months and treated with metformin for the last 5 months. (A–C) The fluorescent images of pAKT (A) and total AKT (B) and the statistical analyses of the fluorescent intensities in the control, HFD, and HFD+Met retinas (C) are shown. The pAKT fluorescent intensity of HFD retinas is significantly lower (*) than those in the other two groups. (D–F) The fluorescent images of pERK (D) and total ERK (E) and the statistical analyses of the fluorescent intensities in the control, HFD, and HFD+Met retinas (F) are shown. The fluorescent intensity of pERK in the HFD-fed mouse retina is significantly higher (*) than those in the control and HFD+Met retinas. (G–I) The fluorescent images of pAMPK (G) and total AMPK (H) and the statistical analyses of the fluorescent intensities in the control, HFD, and HFD+Met retinas (I) are shown. The fluorescent intensity of pAMPK in the HFD retinas is significantly lower (*) than those in the control and HFD+Met retinas (*), whereas pAMPK is significantly higher in the HFD+Met mouse retina (#) than those of the control and HFD retinas. The fluorescent intensity of total AMPK is significantly higher in the HFD+Met retina (#) than those of the other two groups. Scale bar: 50 μm. INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outer nuclear layer; OPL, outer plexiform layer. (C, F, I) Box plots represent the distribution of fluorescent intensities within a specific group. The black line represents the median, and the gray line represents the mean of the specific group. N is the number of animals in the group. *,#P < 0.05.
Metformin Treatment Did Not Rescue HFD-Induced Neovascularization in the Retina
We recently demonstrated that after 6 months of HFD, the HFD mouse retinas developed “neovascularization” with an apparent increase of retinal vasculature density and microaneurysm-like structures,27 so we next examined whether metformin treatments could stop the process of neovascularization in the HFD mouse retina. We used FA28 and AngioTool to compare changes in the central and peripheral retinal vasculature of the control, HFD, and HFD+Met mice (Fig. 6A). In the central retina, there were no differences in any vascular parameters among the three experimental groups (Fig. 6B). However, in the peripheral retina, mice receiving HFD for 6 months had a significant increase in vascular density, vessel area, and the number of branch points compared to those of the control mice (Fig. 6C), whereas the average retinal area without detectable vasculature (average lacunarity) was decreased in HFD mice and HFD+Met (Fig. 6C). Thus, mice eating HFD for 6 months had retinal neovascularization, but treatment with metformin for 4 months did not improve HFD-caused retinal neovascularization (Fig. 6C).
Figure 6.

Metformin does not reverse HFD-induced neovascularization. Fluorescein angiography was used to determine the retinal vasculature in mice fed normal chow (control), fed HFD for 6 months (HFD), or HFD-fed mice treated with metformin (HFD+Met). (A) Commercial software (AngioTool) was used to determine the vascular parameters including vascular density, vessel area, number of vessel branch points, and average nonvascular space (average lacunarity). (B) There were no statistical differences between vascular density, vessel area, the number of branch points, and average lacunarity of the HFD retinas and those of HFD+Met mice in the central region of retinas. (C) In the peripheral regions of retinas, there are statistical differences in HFD mice vascular density, vessel area, number of branch points, and average lacunarity between the control and HFD-fed mice (*), as well between the control and HFD+Met mice (*). Box plots represent the distribution of fluorescent intensities within a specific group. The black line represents the median, and the gray line represents the mean of the specific group. N is the number of animals in the group. *P < 0.05.
Metformin Treatment Decreased Inflammation
Because metformin has anti-inflammatory properties,39 we next determined whether metformin could reverse HFD-induced intraocular inflammation. Western blot analysis showed a significantly higher expression of pP65, a subunit of NFκB transcription complex and a biomarker for inflammation,40 in HFD mouse retina than that in controls, but the retinas of HFD mice treated with metformin had less pP65 than the HFD mouse retinas (Fig. 7A). We further analyzed the intraocular inflammatory cytokines from mouse vitreous and lenses by using the Bio-plex assays. Generally, all cytokines analyzed in this study (including IL-6, IL-12, MCP-1, G-CSF, and VEGF) were increased in the HFD mice with significant increases in the levels of IL-6, G-CSF, and VEGF compared to those in the controls (Figs. 7B, 7E, 7F). Metformin treatments were able to reduce the levels of all cytokines, with IL-6 and VEGF significantly decreased, back to control levels (Figs. 7B, 7F). These data provided evidence that metformin indeed reduced intraocular inflammation in HFD mice.
Figure 7.

Metformin reduces HFD-induced intraocular inflammation. The retinas, vitreous, and lenses were collected from mice fed normal chow, or HFD for 6 months, or HFD mice treated with metformin for the last 4 months. (A) Retinas were harvested and subjected to Western blot analysis of phosphorylated P65 (pP65) and P65 (Total P65; loading control). The HFD retina has a significantly higher pP65 (*) than those of the other two groups. (B–F) Inflammatory cytokine profiles were analyzed from the vitreous and lenses obtained from the control, HFD, and HFD+Met groups: (B) interleukin-6 (IL-6), (C) IL-12, (D) MCP-1, (E) G-CSF, and (F) VEGF. The HFD-fed mice have a significantly higher IL-6 (B) and VEGF (F) than those of the other two groups (*). The HFD-fed mice also have a significantly higher G-CSF (E) than that of the control (*). Box plots represent the distribution of data within a specific group. The black line represents the median, and the gray line represents the mean of the specific group. N is the number of animals in the group. *P < 0.05.
Discussion
In this report, we examined the effects of metformin, an antihyperglycemic agent, on retinal function and physiology in mice with HFD-induced diabetes. We hypothesized that controlling systemic glycemia with metformin could recover HFD-induced retinal dysfunction. As seen in human patients, where ERG OPs are more sensitive to diabetic stress,41,42 we observed that 1 month after HFD regimen, the HFD mice had decreased OPs and delayed OP implicit times, even though these HFD mice had not yet developed systemic hyperglycemia. These data indicate that retinal function may be compromised under prediabetic conditions, preceding systemic hyperglycemia. With the development of diabetes, both the ERG a- and the b-waves were dampened after 2 months of HFD, which were concurrent with the development of hyperglycemia. Treatment with metformin significantly decelerated weight gain and controlled systemic blood glucose levels in HFD mice, but it was not able to restore the retinal function. The ERG OP amplitudes were further decreased and implicit time was delayed more in HFD+Met mice than in HFD mice without metformin intervention. It is possible that, if metformin was given at a higher dose or at an earlier stage, it might be able to reverse HFD-induced retinal dysfunction. However, the dose we chose was previously used to successfully reverse obesity-induced hyperglycemia and liver damage.43 We started the metformin treatments in these HFD mice after hyperglycemia developed to simulate human diabetic patients where treatments often start after systemic hyperglycemia is detected. However, metformin successfully acted as a protective drug in various disease models when used to pretreat animals prior to the induction of diseases, including acute kidney injury44 and cerebral forebrain ischemia.45,46 Thus, it is possible that metformin is more beneficial if it is given at an earlier, prediabetic stage as a “preventive” strategy rather than as a “treatment” for diabetic retinas.
In a recent report,25 mice fed a HFD containing 42% fat calories developed diabetes by 6 months of the diet regimen, and by 12 months decreased OPs with delayed OP implicit times, as well as vascular complications including atrophic capillaries and pericyte ghosts were apparent. Interestingly, there were no significant changes in the ERG a- and b-waves recorded from those HFD mice.25 We consistently observed that mice fed a HFD containing 59.4% fat calories developed glucose intolerance and insulin resistance at the end of 3 months of the HFD regimen,26,27 which is consistent with other reports using a HFD with the same fat calories.43,47 These HFD mice had significant weight gain only 2 weeks after the diet regimen compared to the controls. As we previously reported,26,27 the ERG a- and b-waves in these HFD mice have decreased amplitudes and delayed implicit times. In the current study, we further demonstrated that OPs were affected in the HFD mice even before systemic hyperglycemia. These results indicate that a higher fat content in the diet might exaggerate the progression of DR by adversely impacting the neural retina prior to the “symptoms” of diabetes, such as hyperglycemia. Thus, we postulate that obesity due to consumption of higher fat-content food might negatively affect vision even before the diagnosis of diabetes. Further clinical research to correlate human dietary habits and vision impairment will be needed to determine this possibility.
Despite metformin's inability to recover retinal function, it had significant effects on several cell-signaling proteins in the retina that were altered by HFD. In context of the pathogenesis of DR, the phosphoinositide 3-kinase-AKT (PI3K-AKT) pathway has been shown to regulate angiogenesis.48 Previously, we showed that HFD-induced type 2 diabetic retinas have reduced pAKT, which is also seen in STZ-induced type 1 diabetic mice.49 In this report, we found that metformin treatments restored pAKT and increased AMPK and pAMPK in HFD mouse retinas. AMPK is a cellular energy sensor, and activated AMPK further stimulates catabolic processes for increasing ATP production.35 In muscles, AMPK activates the PI3K-AKT pathway that leads to increased glucose uptake into muscle cells.50 We previously showed that activation of AMPK leads to activation of AKT and its downstream signaling in the avian retina.51 Metformin is known to up-regulate the expression and activation of AMPK in the kidney,36 adipose tissue,13,37 and heart.38 Hence, the effects of metformin on pAKT could be a downstream effect from its activation of AMPK-dependent signaling.
In addition, increased pERK is correlated with the presence of proinflammatory cytokines.52 Activation of ERK is involved in the up-regulation of VEGF, an angiogenic protein that causes microvascular complications and neovascularization in DR.53 We showed that there was an increase of pERK in the retinas of HFD mice and that metformin reduced pERK in HFD-mouse retina. Furthermore, HFD mouse retinas had increased expression of pP65 compared to controls, which was reduced in retinas of HFD+Met mice. Obesity is known to induce systemic inflammation,54,55 but induction of systemic inflammation does not correlate with retinal inflammation,56 which is why vitreous biopsy samples are often used from patients to determine intraocular inflammation clinically. However, due to the small size and limited vitreous of mouse eyes, we had to analyze the status of intraocular inflammation from both vitreous and lenses to mimic human clinical settings, which was technically challenging. The expression of proinflammatory cytokines in HFD mice was increased, and metformin was able to reverse HFD-induced intraocular inflammation. Unfortunately, metformin treatment was unsuccessful in recovering retinal light sensitivities and hindering neovascularization.
In summary, metformin treatment successfully reversed hyperglycemic conditions and decreased intraocular inflammation in HFD mice but was unable to restore the retinal light responses. Development of therapeutic treatments for DR might require a multi-pronged approach in addition to antihyperglycemia and anti-inflammation.
Acknowledgments
The authors thank Chaodong Wu, MD, PhD, Nutrition, Texas A&M University, for comments and suggestions. We also thank Kathryn Klotz for technical assistance.
Supported by US National Institutes of Health/National Eye Institute Grant NIHR21EY023339 (GY-PK) and a College of Veterinary Medicine and Biomedical Sciences at Texas A&M University graduate research grant (JY-AC).
Disclosure: A.J. Kim, None; J.Y.-A. Chang, None; L. Shi, None; R.C.-A. Chang, None; M.L. Ko, None; G.Y.-P. Ko, None
References
- 1. Wild S,, Roglic G,, Green A,, Sicree R,, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004; 27: 1047–1053. [DOI] [PubMed] [Google Scholar]
- 2. American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care. 2013; 36 (suppl 1): S11–S66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993; 329: 977–986. [DOI] [PubMed] [Google Scholar]
- 4. UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet. 1998; 352: 854–865. [PubMed] [Google Scholar]
- 5. Song S,, Andrikopoulos S,, Filippis C,, Thorburn AW,, Khan D,, Proietto J. Mechanism of fat-induced hepatic gluconeogenesis: effect of metformin. Am J Physiol Endocrinol Metab. 2001; 281: E275–282. [DOI] [PubMed] [Google Scholar]
- 6. Inzucchi SE,, Bergenstal RM,, Buse JB,, et al. Management of hyperglycaemia in type 2 diabetes: a patient-centered approach. Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia. 2012; 55: 1577–1596. [DOI] [PubMed] [Google Scholar]
- 7. Yu S,, Schwab P,, Bian B,, Radican L,, Tunceli K. Use of Add-on treatment to metformin monotherapy for patients with type 2 diabetes and suboptimal glycemic control: a U.S. database study. J Manag Care Spec Pharm. 2016; 22: 272–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stumvoll M,, Nurjhan N,, Perriello G,, Dailey G,, Gerich JE. Metabolic effects of metformin in non-insulin-dependent diabetes mellitus. N Engl J Med. 1995; 333: 550–554. [DOI] [PubMed] [Google Scholar]
- 9. Cao J,, Meng S,, Chang E,, et al. Low concentrations of metformin suppress glucose production in hepatocytes through AMP-activated protein kinase (AMPK). J Biol Chem. 2014; 289: 20435–20446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yu JW,, Deng YP,, Han X,, Ren GF,, Cai J,, Jiang GJ. Metformin improves the angiogenic functions of endothelial progenitor cells via activating AMPK/eNOS pathway in diabetic mice. Cardiovasc Diabetol. 2016; 15: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hu M,, Ye P,, Liao H,, Chen M,, Yang F. Metformin protects H9C2 cardiomyocytes from high-glucose and hypoxia/reoxygenation injury via inhibition of reactive oxygen species generation and inflammatory responses: role of AMPK and JNK. J Diabetes Res. 2016; 2016: 2961954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sun Y,, Tao C,, Huang X,, et al. Metformin induces apoptosis of human hepatocellular carcinoma HepG2 cells by activating an AMPK/p53/miR-23a/FOXA1 pathway. Onco Targets Ther. 2016; 9: 2845–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Luo T,, Nocon A,, Fry J,, et al. AMPK activation by metformin suppresses abnormal adipose tissue extracellular matrix remodeling and ameliorates insulin resistance in obesity. Diabetes. 2016; 65: 2295–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Diabetes Prevention Program Research Group. Long-term safety, tolerability, and weight loss associated with metformin in the Diabetes Prevention Program Outcomes study. Diabetes Care. 2012; 35: 731–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Malin SK,, Kashyap SR. Effects of metformin on weight loss: potential mechanisms. Curr Opin Endocrinol Diabetes Obes. 2014; 21: 323–329. [DOI] [PubMed] [Google Scholar]
- 16. Guigas B,, Bertrand L,, Taleux N,, et al. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside and metformin inhibit hepatic glucose phosphorylation by an AMP-activated protein kinase-independent effect on glucokinase translocation. Diabetes. 2006; 55: 865–874. [DOI] [PubMed] [Google Scholar]
- 17. Valsamakis G,, Lois K,, Kumar S,, Mastorakos G. Metabolic and other effects of pioglitazone as an add-on therapy to metformin in the treatment of polycystic ovary syndrome (PCOS). Hormones (Athens). 2013; 12: 363–378. [DOI] [PubMed] [Google Scholar]
- 18. Alhaider AA,, Korashy HM,, Sayed-Ahmed MM,, Mobark M,, Kfoury H,, Mansour MA. Metformin attenuates streptozotocin-induced diabetic nephropathy in rats through modulation of oxidative stress genes expression. Chem Biol Interact. 2011; 192: 233–242. [DOI] [PubMed] [Google Scholar]
- 19. Wile DJ,, Toth C. Association of metformin, elevated homocysteine, and methylmalonic acid levels and clinically worsened diabetic peripheral neuropathy. Diabetes Care. 2010; 33: 156–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Winzell MS,, Ahren B. The high-fat diet-fed mouse: a model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes. 2004; 53 (suppl 3): S215–219. [DOI] [PubMed] [Google Scholar]
- 21. Fellmann L,, Nascimento AR,, Tibirica E,, Bousquet P. Murine models for pharmacological studies of the metabolic syndrome. Pharmacol Ther. 2013; 137: 331–340. [DOI] [PubMed] [Google Scholar]
- 22. Ahren B,, Simonsson E,, Scheurink AJ,, Mulder H,, Myrsen U,, Sundler F. Dissociated insulinotropic sensitivity to glucose and carbachol in high-fat diet-induced insulin resistance in C57BL/6J mice. Metabolism. 1997; 46: 97–106. [DOI] [PubMed] [Google Scholar]
- 23. Weisberg SP,, McCann D,, Desai M,, Rosenbaum M,, Leibel RL,, Ferrante AW., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003; 112: 1796–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vinores SA,, Campochiaro PA,, May EE,, Blaydes SH. Progressive ultrastructural damage and thickening of the basement membrane of the retinal pigment epithelium in spontaneously diabetic BB rats. Exp Eye Res. 1988; 46: 545–558. [DOI] [PubMed] [Google Scholar]
- 25. Rajagopal R,, Bligard GW,, Zhang S,, Yin L,, Lukasiewicz P,, Semenkovich CF. Functional deficits precede structural lesions in mice with high-fat diet-induced diabetic retinopathy. Diabetes. 2016; 65: 1072–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chang RC,, Shi L,, Huang CC,, et al. High-fat diet-induced retinal dysfunction. Invest Ophthalmol Vis Sci. 2015; 56: 2367–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shi L,, Kim AJ,, Chang RC,, et al. Deletion of miR-150 exacerbates retinal vascular overgrowth in high-fat-diet induced diabetic mice. PLoS One. 2016; 11: e0157543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zudaire E,, Gambardella L,, Kurcz C,, Vermeren S. A computational tool for quantitative analysis of vascular networks. PLoS One. 2011; 6: e27385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pardue MT,, Barnes CS,, Kim MK,, et al. Rodent hyperglycemia-induced inner retinal deficits are mirrored in human diabetes. Trans Vis Sci Tech. 3 (3): 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shirao Y,, Kawasaki K. Electrical responses from diabetic retina. Prog Retin Eye Res. 1998; 17: 59–76. [DOI] [PubMed] [Google Scholar]
- 31. Yonemura D,, Aoki T,, Tsuzuki K. Electroretinogram in diabetic retinopathy. Arch Ophthalmol. 1962; 68: 19–24. [DOI] [PubMed] [Google Scholar]
- 32. Dilly AK,, Rajala RV. Insulin growth factor 1 receptor/PI3K/AKT survival pathway in outer segment membranes of rod photoreceptors. Invest Ophthalmol Vis Sci. 2008; 49: 4765–4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li G,, Rajala A,, Wiechmann AF,, Anderson RE,, Rajala RV. Activation and membrane binding of retinal protein kinase Balpha/Akt1 is regulated through light-dependent generation of phosphoinositides. J Neurochem. 2008; 107: 1382–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cobb MH. MAP kinase pathways. Prog Biophys Mol Biol. 1999; 71: 479–500. [DOI] [PubMed] [Google Scholar]
- 35. Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007; 8: 774–785. [DOI] [PubMed] [Google Scholar]
- 36. Kim D,, Lee JE,, Jung YJ,, et al. Metformin decreases high-fat diet-induced renal injury by regulating the expression of adipokines and the renal AMP-activated protein kinase/acetyl-CoA carboxylase pathway in mice. Int J Mol Med. 2013; 32: 1293–1302. [DOI] [PubMed] [Google Scholar]
- 37. Li A,, Zhang S,, Li J,, Liu K,, Huang F,, Liu B. Metformin and resveratrol inhibit Drp1-mediated mitochondrial fission and prevent ER stress-associated NLRP3 inflammasome activation in the adipose tissue of diabetic mice. Mol Cell Endocrinol. 2016. [DOI] [PubMed]
- 38. Lai YC,, Tabima DM,, Dube JJ,, et al. SIRT3-AMP-Activated protein kinase activation by nitrite and metformin improves hyperglycemia and normalizes pulmonary hypertension associated with heart failure with preserved ejection fraction. Circulation. 2016; 133: 717–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Saisho Y. Metformin and inflammation: its potential beyond glucose-lowering effect. Endocr Metab Immune Disord Drug Targets. 2015; 15: 196–205. [DOI] [PubMed] [Google Scholar]
- 40. Karin M,, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005; 5: 749–759. [DOI] [PubMed] [Google Scholar]
- 41. Bresnick GH,, Palta M. Predicting progression to severe proliferative diabetic retinopathy. Arch Ophthalmol. 1987; 105: 810–814. [DOI] [PubMed] [Google Scholar]
- 42. Bresnick GH,, Palta M. Oscillatory potential amplitudes. Relation to severity of diabetic retinopathy. Arch Ophthalmol. 1987; 105: 929–933. [DOI] [PubMed] [Google Scholar]
- 43. Woo SL,, Xu H,, Li H,, et al. Metformin ameliorates hepatic steatosis and inflammation without altering adipose phenotype in diet-induced obesity. PLoS One. 2014; 9: e91111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li J,, Gui Y,, Ren J,, et al. Metformin protects against cisplatin-induced tubular cell apoptosis and acute kidney injury via AMPKalpha-regulated autophagy induction. Sci Rep. 2016; 6: 23975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ashabi G,, Khalaj L,, Khodagholi F,, Goudarzvand M,, Sarkaki A. Pre-treatment with metformin activates Nrf2 antioxidant pathways and inhibits inflammatory responses through induction of AMPK after transient global cerebral ischemia. Metab Brain Dis. 2015; 30: 747–754. [DOI] [PubMed] [Google Scholar]
- 46. Ghadernezhad N,, Khalaj L,, Pazoki-Toroudi H,, Mirmasoumi M,, Ashabi G. Metformin pretreatment enhanced learning and memory in cerebral forebrain ischaemia: the role of the AMPK/BDNF/P70SK signalling pathway. Pharm Biol. 2016; 1–9. [DOI] [PubMed]
- 47. Zhuang G,, Meng C,, Guo X,, et al. A novel regulator of macrophage activation: miR-223 in obesity-associated adipose tissue inflammation. Circulation. 2012; 125: 2892–2903. [DOI] [PubMed] [Google Scholar]
- 48. Ackah E,, Yu J,, Zoellner S,, et al. Akt1/protein kinase Balpha is critical for ischemic and VEGF-mediated angiogenesis. J Clin Invest. 2005; 115: 2119–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jiang T,, Chang Q,, Cai J,, Fan J,, Zhang X,, Xu G. Protective effects of melatonin on retinal inflammation and oxidative stress in experimental diabetic retinopathy. Oxid Med Cell Longev. 2016; 2016: 3528274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sajan MP,, Bandyopadhyay G,, Miura A,, et al. AICAR and metformin, but not exercise, increase muscle glucose transport through AMPK-, ERK-, and PDK1-dependent activation of atypical PKC. Am J Physiol Endocrinol Metab. 2010; 298: E179–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Huang CC,, Shi L,, Lin CH,, Kim AJ,, Ko ML,, Ko GY. A new role for AMP-activated protein kinase in the circadian regulation of L-type voltage-gated calcium channels in late-stage embryonic retinal photoreceptors. J Neurochem. 2015; 135: 727–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Busik JV,, Mohr S,, Grant MB. Hyperglycemia-induced reactive oxygen species toxicity to endothelial cells is dependent on paracrine mediators. Diabetes. 2008; 57: 1952–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jin J,, Yuan F,, Shen MQ,, Feng YF,, He QL. Vascular endothelial growth factor regulates primate choroid-retinal endothelial cell proliferation and tube formation through PI3K/Akt and MEK/ERK dependent signaling. Mol Cell Biochem. 2013; 381: 267–272. [DOI] [PubMed] [Google Scholar]
- 54. Kim MS,, Yamamoto Y,, Kim K,, et al. Regulation of diet-induced adipose tissue and systemic inflammation by salicylates and pioglitazone. PLoS One. 2013; 8: e82847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ying W,, Kanameni S,, Chang CA,, et al. Interferon tau alleviates obesity-induced adipose tissue inflammation and insulin resistance by regulating macrophage polarization. PLoS One. 2014; 9: e98835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tamura H,, Kiryu J,, Miyamoto K,, et al. In vivo evaluation of ocular inflammatory responses in experimental diabetes. Br J Ophthalmol. 2005; 89: 1052–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]