

Abstract
Studies were undertaken to determine the genetic needs for the survival of Pseudomonas fluorescens Pf0-1, a gram-negative soil bacterium potentially important for biocontrol and bioremediation, in soil. In vivo expression technology (IVET) identified 22 genes with elevated expression in soil relative to laboratory media. Soil-induced sequences included genes with probable functions of nutrient acquisition and use, and of gene regulation. Ten sequences, lacking similarity to known genes, overlapped divergent known genes, revealing a novel genetic organization at those soil-induced loci. Mutations in three soil-induced genes led to impaired early growth in soil but had no impact on growth in laboratory media. Thus, IVET studies have identified sequences important for soil growth and have revealed a gene organization that was undetected by traditional laboratory approaches.
Pseudomonas spp. are common inhabitants of soil and water. In terrestrial environments, they can be found in close association with the plant root zone (rhizosphere) and living free in soil. Further, the fluorescent pseudomonads can also be found in association with plants and animals as commensals and pathogens. The observation that some species, such as Pseudomonas fluorescens and Pseudomonas aeruginosa, can be found in a range of environments points to the evolution of an array of strategies to cope with environmental perturbations. This idea is supported by the description of a large number of known and predicted environmentally responsive regulators in the complete genome sequence of P. aeruginosa (43).
Pseudomonas spp. are being evaluated for applications in biocontrol, plant growth promotion, and bioremediation. Coupled with the phenotype of interest, the presumed adaptability of these isolates makes them ideal choices. However, relatively little is known about the factors contributing to their success in soil and in the rhizosphere. Soil is a complex and challenging environment with a variety of fluctuating conditions, such as nutrient level, temperature, water content, and pH. To survive and persist in soil, microbes must be able to respond and adapt rapidly to the environmental changes.
Our laboratory has been investigating the genetic basis of environmental persistence by using P. fluorescens Pf0-1. Selection for adherence defects led to the discovery of AdnA, a regulator affecting flagellum production, motility, attachment to sand and seeds (8, 9), and biofilm formation (5). A mutation in adnA reduced the ability of Pf0-1 to spread and persist in soil (21).
To further understand the underlying basis for Pf0-1 survival in soil, we sought to identify genes whose expression is induced in soil. The promoter-trapping strategy, termed in vivo expression technology (IVET), was chosen for the study. IVET was described in 1993 by Mahan et al. (19) and is similar to a method reported earlier by Osbourn et al. (27). Such promoter traps allow the positive selection of niche-induced promoters by using genetic complementation of a conditionally lethal mutation, mediated by the fusion of an in vivo-expressed promoter to the coding sequence that complements the mutation. The advantage of the IVET approach over other genetic methods is that it allows the selection of environmentally induced sequences independent of whether the loss of those sequences would be lethal. Thus, IVET helps recover both essential and nonessential genes that contribute to ecological success.
Rainey and Preston (34) suggested that the requirement for diaminopimelate and lysine by dapB mutant bacteria would be a useful phenotype around which to base an IVET selection. Diaminopimelate is essential but is not present in soils, and diaminopimelate auxotrophy is lethal to growing cells, while nongrowing cells can remain viable for long periods. The selection is therefore not too stringent to prevent the recovery of genes that are not induced immediately upon introduction to the soil. Here, we describe the development of a dapB-based IVET system and its application to unraveling the genetic basis of ecological success of P. fluorescens Pf0-1 in soil.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. P. fluorescens and Escherichia coli were grown at 30 and 37°C, respectively. E. coli was routinely cultured in Luria-Bertani medium (39), while P. fluorescens strains were grown in either LB medium or Pseudomonas minimal medium (PMM) (15). When required, the media were solidified by the addition of 1.5% agar. Antibiotics and supplements were added as required, at the following concentrations (unless otherwise stated): ampicillin, 50 μg/ml; kanamycin, 25 μg/ml; streptomycin, 25 μg/ml; tetracycline, 15 μg/ml for plasmids in E. coli and 5 μg/ml for IVET plasmids integrated into the Pf0-1 genome; and X-Gal (5-bromo-4-chloro-3-indoyl-β-d-galactopyranoside), 50 μg/ml. Diaminopimelic acid (DAP) was added at 1 mg/ml, and lysine was used at 100 μg/ml. E. coli strains were transformed by electroporation with a Bio-Rad Micropulser following the recommendations of the manufacturer, while DNA was introduced to P. fluorescens strains by conjugation from E. coli S17-1.
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Genotype or description | Reference or source |
|---|---|---|
| E. coli | ||
| DH5α λpir | φ80dlacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 deoR λ-pir | J. J. Mekalanos |
| GM2163 | F−ara-14 leuB6 fhuA31 lacY1 tsx78 glnV44 galK2 galT22 mcrA dcm-6 hisG4 rfbD1 rpsL136 dam-13::Tn9 xylA5 mtl-1 thi-1 mcrB1 hsdR2 | New England Biolabs |
| S17.1 | recA pro hsdR RP4-2-Tc::Mu-Km::Tn7 λ-pir | 42 |
| P. fluorescens | ||
| Pf0-1 | Wild type, Ampr | 6 |
| Pf0-2x | Pf0-1 adnA::Sm/Sp | 38 |
| Pf0-1Δdap | Pf0-1 ΔdapB | This study |
| Pf0-1::iiv2Sm | Pf0-1 with partial deletion of iiv2 | This study |
| Pf0-1::iiv3Sm | Pf0-1 with partial deletion of iiv3 | This study |
| Pf0-1::iiv7Sm | Pf0-1 with partial deletion of iiv7 | This study |
| Plasmids | ||
| pGEM-T Easy | Apr; cloning vector for PCR products | Promega |
| pHRP315 | pMB1 origin; Apr; ΩSm/Sp cassette | 30 |
| pHRP317 | pMB1 origin; Apr; ΩSm/Sp cassette | 30 |
| pSR47s | Kmr; sacB-containing suicide vector (π-requiring R6K replication origin) | 23 |
| pSRΔdap | pSR47s derivative containing ΔdapB | This study |
| pSRiiv2Sm | pSR47s derivative containing iiv2::Sm | This study |
| pSRiiv3Sm | pSR47s derivative containing iiv3::Sm | This study |
| pSRiiv7Sm | pSR47s derivative containing iiv7::Sm | This study |
| pUIC3 | Universal IVET construct; lacZY′, bla, Tcr, R6K origin | 32 |
| pIVETdap | dapB′ cloned in SpeI site of pUIC3 | This study |
DNA manipulation and sequencing.
Recombinant DNA techniques were carried out as described previously (39). Restriction and DNA-modifying enzymes were purchased from Invitrogen, Inc. (Carlsbad, Calif.) and Promega (Madison, Wis.). Recombinant plasmids were routinely harvested from E. coli DH5α λpir. Plasmid DNA was prepared by using a QIAprep Spin Miniprep Kit (QIAGEN, Valencia, Calif.), and total genomic DNA was prepared by using a Wizard genomic DNA purification kit (Promega). When required, DNA was recovered from agarose gel slices by using a QIAEX II gel extraction kit (QIAGEN). PCR was carried out by using the Triple Master system (Eppendorf AG, Hamburg, Germany) in a GeneAmp PCR System 2700 (Applied Biosystems, Foster City, Calif.). When required, PCR products were cloned with pGEM-T Easy (Promega) according to the manufacturer's instructions. Oligonucleotides were synthesized, and DNA sequences were determined by the dideoxy chain termination method (40) at the Tufts University Core Facility.
Construction and characterization of a dapB deletion in Pf0-1.
The region of the Pf0-1 genome specifying DapB was identified from the draft genome sequence (http://genome.jgi-psf.org/draft_microbes/psefl/psefl.home.html) by a Blast search using dapB of P. aeruginosa PA01 as the query sequence. As in other fluorescent pseudomonads, the locus had probable dnaJ, dapB, and carA genes. The dapB gene was deleted by amplifying sequences flanking the open reading frame (ORF) and ligating them together, thus removing dapB. PCR primer pair DapD1 (5′-GGAAGATCTGTTCAAGGAGG) and DapD2 (5′-ccGGATCCATCGCCGAAGCGTTTCAC) was designed to amplify 991 bp upstream (separated from the dapB ORF by 15 bp), while DapD3 (5′-ccGGATCCGATGTTCTGAAGCCGGCG) and DapD4 (5′-GTCTGTGCCGGAACGACAGTG) were specific for 924 bp downstream of dapB. The reverse primer for the upstream sequence and the forward primer for the downstream sequence both had a BamHI site included at the 5′ end (underlined). Amplicons were digested with BamHI, ligated together, and used as the template in a second PCR with primers DapD1 and DapD4, resulting in the in vitro generation of a dapB deletion. The deletion amplicon was cloned in pGEM-T Easy, and then cloned in the NotI site of the suicide plasmid pSR47s by utilizing the NotI sites flanking the cloning site in pGEM-T Easy. The resulting plasmid (pSRΔdap) was transferred to Pf0-1 by conjugation, and the dapB region in Pf0-1 was replaced with the deletion by allele exchange (see below). Sucrose-tolerant recombinant colonies (i.e., those that had lost the suicide plasmid) were tested for DAP auxotrophy by replica plating onto PMM with and without DAP and lysine supplements. DAP auxotrophy confirmed the successful deletion of dapB. PCR with DapD1 and DapD4 amplified a product that was consistent in size with the deletion of dapB and could be cleaved by BamHI, resulting in DNA fragments of the same size as the upstream and downstream PCR products. The absence of dapB was further confirmed by the inability to amplify a sequence within the dapB ORF from the deletion strain, which was amplifiable from the parental strain (data not shown).
The deletion of dapB rendered Pf0-1 dependent on exogenous DAP and lysine. For a strain to be of use in an IVET screening, gradual death, or at least an inability to grow in the environment of interest, is essential. Pf0-1Δdap was unable to proliferate in soil, and the population slowly declined over 7 days. Over the same time period, the wild-type Pf0-1 population increased by 4 orders of magnitude (data not shown).
Construction of an IVET vector and library.
The promoter-trapping plasmid was constructed by inserting a promoterless dapB gene into the universal IVET construct pUIC3 (32). pUIC3 has a number of features that make it ideal for construction of an IVET reporter plasmid. First, it has promoterless lacZY genes with restriction sites located 5′ of the lacZ Shine-Dalgarno sequence to allow cloning of further promoterless reporter genes. Second, a unique BglII restriction site is located upstream of the sites used for cloning reporter genes, allowing the cloning of random genomic DNA fragments upstream of the reporter genes. Finally, pUIC3 is dependent on the Pir protein for replication, so it is not maintained as an autonomous replicon in strains that do not produce Pir. We used primers DapSPF (5′-ggACTAGTCTTCTTCGGCGATCTGTAAGG) and DapSPR (5′-ggACTAGTTCAATGCAGGCCGAGCACGTC) to amplify the dapB ORF, along with the probable Shine-Dalgarno sequence at the 5′ end, from Pf0-1. SpeI restriction sites (underlined) incorporated into the 5′ end of the primers allowed cloning of the dapB′ amplicon into the unique SpeI site of pUIC3, upstream of the promoterless lacZY genes. Thus, pIVETdap has a promoterless dapB-lacZY transcriptional fusion.
Four fusion libraries were constructed with pIVETdap. The first of these was constructed by cloning BglII-digested total DNA into the corresponding site of pIVETdap. The other three libraries were constructed by isolating 1.5- to 2.5-kb fragments of Pf0-1 DNA, partially digested with Sau3A, from an agarose gel and ligating these into the BglII site of pIVETdap. After the transformation of E. coli S17-1 with the libraries, 20 random colonies were chosen and the plasmid DNA was prepared. Restriction digestion indicated that 90% of the isolates had inserts, and a range of insert sizes had been cloned, supporting the description as a random genomic library (data not shown).
Selection of soil-induced promoters.
Four genomic libraries were constructed in pIVETdap, and each was transferred to Pf0-1Δdap by conjugation. Since pIVETdap cannot replicate in Pf0-1, we concluded that colonies that were recovered had library clones integrated into the Pf0-1 genome by recombination between the plasmid-borne and chromosomal copies of the cloned fragment. Pooled transconjugant colonies arising from each library were used to inoculate two separate soil samples (see below for soil procedures). Each inoculum consisted of cells from approximately 1,500 transconjugant colonies. After 7 days of selection in soil, bacteria were recovered and plated onto PMM supplemented with DAP, lysine, tetracycline, and X-Gal. Bacteria that survived selection produced colonies; white ones were chosen for further use.
Soil growth and survival assay.
Strains were grown for 20 h in PMM broth with appropriate antibiotics and growth supplements. The soil used was a gamma-irradiated fine sandy loam, which has been described previously (9). Bacteria were diluted to an approximate population of 2 × 104 CFU/ml in sterile distilled H2O, and 1 ml was mixed with 5 g of sterile soil (achieving approximately 50% water-holding capacity). After 30 min, a 1-g sample was removed to allow the initial recoverable CFU/g to be determined. Subsequent samples were taken when required. CFU/g of soil was determined by adding 1 ml of sterile H2O to 1 g of soil and vortexing vigorously for 1 min. The soil suspension was allowed to settle briefly so that any large particles fell out of suspension. The soil suspension was then serially diluted 10-fold and plated on selective media to allow enumeration of viable, culturable bacteria by colony counting on PMM plates with appropriate supplements to support growth.
Construction of plasmids for knockout of iiv2, iiv3, and iiv7.
A wild-type DNA fragment was amplified from each locus by using Pf0-1 total DNA as a template, with the oligos iiv2F (5′-CGAGCAACTGATTCACTCCA) and iiv2R (5′-CGGCGATATTTTGCATAGGT); iiv3F (5′-GACGATCGGGTGTTCACTTT) and iiv3R (5′-AGTTCTCTTCGGCAGTGGAA); and iiv7F (5′-ATGAATATCCTTGTTGTCG) and iiv7R (5′-TCACGCACCTGCGTCTCG). Each product was then cloned with pGEM-T Easy. Gene disruptions were generated by making partial deletions and inserting a Smr omega cassette as follows. The iiv2 clone (pGEMiiv2) was digested with MscI (and dephosphorylated with calf intestinal alkaline phosphatase [CIAP]), for which there are recognition sites at positions 2997 and 3976 of the 5,535-bp iiv2 ORF. A Smr cassette was liberated from pHRP317 with EcoRV and SmaI, gel purified, and ligated to the MscI-cut pGEMiiv2, generating the plasmid pGEMiiv2Sm in which a Smr cassette replaces 973 bp of iiv2 sequence between two MscI sites. The iiv3 clone (pGEMiiv3) was digested with BamHI, which cuts the 1,560-bp iiv3 ORF sequence at base pair positions 744 and 1176, and treated with CIAP. A Smr cassette with BamHI ends was cut from pHRP315, gel purified, and ligated to the BamHI-digested pGEMiiv3, resulting in pGEMiiv3Sm. This plasmid has a 426-bp deletion between two BamHI sites in the iiv3 coding sequence, which is replaced by the Smr cassette. The sequence amplified in the iiv7 clone (pGEMiiv7) contained the ORFs for the iiv7 response regulator and sensor kinase (see Results). It was digested with ClaI (by using DNA prepared from the dam-negative strain GM2163), end-filled with Klenow fragment, and treated with CIAP. ClaI cuts the cloned sequence at 398 and 1,009 bp. A Smr cassette cut from pHRP317 with EcoRV and SmaI was ligated to the free ends of the digested pGEMiiv7, creating pGEMiiv7Sm. This new plasmid lacks the last 287 bp of the 672-bp response regulator gene and the first 340 bp of the 1,416-bp sensor kinase gene and has a Smr cassette in place of the deleted sequence. Finally, each of the iiv::Sm sequences in pGEM-T Easy clones was released by digestion with NotI and ligated with NotI-digested pSR47s (23), resulting in the suicide constructs pSRiiv2Sm, pSRiiv3Sm, and pSRiiv7Sm.
Allele exchange mutagenesis.
Mutated alleles cloned into the suicide plasmid pSR47s (see above) were transferred to P. fluorescens Pf0-1 by conjugation, by using E. coli S17-1 as the donor strain. Transconjugants resistant to antibiotic markers carried by the plasmid (Kmr) and, in the case of iiv mutations, to the mutated allele (Smr), were selected. Since pSR47s cannot replicate in P. fluorescens, the growth of transconjugants indicated that the entire vector carrying the disrupted allele had integrated into the chromosome by recombination. The recombinant bacteria were grown overnight in LB broth containing ampicillin and plated on LB agar supplemented with ampicillin, streptomycin (for iiv knockouts), and 5% sucrose. Growth of iiv knockout candidates indicated that the mutated allele was present (Smr) and that the plasmid vector had been lost since pSR47s has the sacB gene, which confers sucrose sensitivity. In the case of constructing the dapB deletion, candidates were screened for DAP auxotrophy by replica plating. That the wild-type allele had been replaced by the mutated one was verified by PCR with total DNA from the recombinant strains as the template.
Computational and statistical analyses.
Database searches were carried out by using the Blastn, Blastx, and Blastp programs, accessed from http://www.ncbi.nlm.nih.gov/BLAST/ (1). Open reading frames were found by using ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html). Promoter searches were carried out by using SoftBerry software. Codon usage was examined by comparison to the P. fluorescens codon usage table (http://www.kazusa.or.jp/codon/cgi-bin/showcodon.cgi?species=Pseudomonas+fluorescens+[gbbct]), by using the Graphical Codon Usage Analyzer (http://gcua.schoedl.de/).
The growth of bacterial strains in soil was analyzed by using a two-tailed t test, assuming unequal variance between data sets. Analysis was carried out by using Microsoft Excel.
RESULTS AND DISCUSSION
Selection and sequencing of soil-induced promoters.
Growth of the Pf0-1 IVET library-bearing strains in soil (see Materials and Methods) led to the recovery of active promoter fusions to dapB′. Prior to soil inoculation, 31% of transconjugants were blue on X-Gal. However, greater than 99% of recovered bacteria gave rise to blue colonies, indicating that growth in soil had indeed enriched the population for active fusions, leading to the expression of lacZY. Among the recovered colonies, those that were white or very pale blue (<1%) possessed putative soil-induced promoters since they indicated low expression of lacZY in laboratory medium. These candidates were tested for dependence upon DAP and lysine. In total, 26 strains that survived 7 days in soil but had DAP and lysine dependency in laboratory medium were isolated.
Each strain was reintroduced separately into soil to confirm its ability to proliferate compared to the parental Pf0-1Δdap strain (Fig. 1). Each strain was able to grow in soil significantly better than the ΔdapB mutant, which was consistently unable to multiply, supporting the suggestion that these isolates have soil-induced promoters fused to the promoterless dapB. However, none of these strains were fully complemented to wild-type levels by the promoter fused to dapB, which may indicate that this screening was useful in recovering promoters with moderate (less than native dapB) expression levels.
FIG. 1.

Growth of 22 individual IVET fusion strains in soil (4 of the original 26 were duplicates). (a) Positive fusion strains. (b) Cryptic fusion strains. In each case, soil was inoculated with bacteria at a density of between 103 and 104 CFU/g of soil. Bacteria were enumerated by colony counting after 1 and 3 days of growth. The initial titer was established by recovery of bacteria from 1 g of soil 30 min after inoculation. Data are the means of results from three or four independent experiments. After 3 days in soil, all fusion strains had a significantly higher population (indicated by asterisks) than Pf0-1ΔdapB, while there was no significant difference between initial population sizes (t test; P < 0.05). Error bars indicate standard deviations.
Plasmids harboring IVET fusions were recovered from their hosts by using conjugation, as described previously (33). The primer IVET3 (5′-GCCTCGACCAGAATCTTGC) was designed to anneal near the start of dapB reading toward the start codon and was used to determine the DNA sequences fused to dapB. Among the 26 fusions, we identified 22 different sequences; four sequences (iiv7, iiv9, iiv14, and iiv20) were isolated twice. Although originally from the same library, the duplicate fusion strains were isolated from separate soil selection experiments, supporting their being genuine soil-induced fusions. The 22 different sequences obtained fell into two distinct groups. The first group, which we call positive fusions, has similarity to known or predicted genes in the correct orientation to drive the expression of dapB (Table 2). The second group showed similarity to known or predicted genes on the DNA strand in the opposite direction of dapB (Table 3). It has been suggested that the sequences in the second group might harbor previously unrecognized promoters fused to dapB, and therefore these have been called cryptic fusions (41).
TABLE 2.
Analysis of positive fusions
| Fusion | Pfluc | COG | Best hitd | PAO1 match (% identity, % positive substitutions)e |
|---|---|---|---|---|
| Nutrient utilization | ||||
| iiv3 | Pflu5341 | COG0318, acyl-CoA synthetases (AMP-forming)/AMP-acid ligases | Putative ligase; Bordetella parapertussis; E = e−138 | PA3300 (29, 45) |
| iiv11 | Pflu5302 | COG0726, predicted xylanase/chitin deacetylase | Putative saccharide deacetylase; Sinorhizobium meliloti; E = 2e−29 | PA1517 (26, 40) |
| Transport | ||||
| iiv6b | Pflu4867 | COG3633, Na+/serine symporter | Sodium-dicarboxylate symporter family; P. putida KT2440; E = 0.0 | PA2042 (80, 87) |
| iiv15 | Pflu4596 | COG1457, purine-cytosine permease and related proteins | Cytosine/purine/uracil/thiamine/allantoin permease, P. putida KT2440; E = e−174 | PA5099 (35, 53) |
| iiv18 | Pflu5167 | COG3451, type IV secretory pathway, VirB4 components | Conserved hypothetical protein with VirB4 domain; P. putida; E = 0.0 | |
| iiv20a | Pflu0451 | COG0477, permeases of the major facilitator superfamily | Permeases of the major facilitator superfamily; P. syringae pv. syringae; B728a; E = 0.0 | PA0229 (75, 84) |
| Regulation | ||||
| iiv7a | Pflu4942 | COG0642, signal transduction histidine kinase | Signal transduction histidine kinase; Burkholderia fungorum; E = e−121 | PA4886 (45, 62) |
| iiv9a | Pflu2333 | COG1167, MocR family transcriptional regulators | COG1167: transcriptional regulator; P. syringae pv. syringae B728a; E = 0.0 | PA1654 (80, 88) |
| Detoxification and metabolism | ||||
| iiv17 | Pflu4220 | COG0604, NADPH quinone reductase and related Zn-dependent oxidoreductases | Putative oxidoreductase; Streptomyces avermitilis MA-4680; E = e−108 | PA3567 (31, 48) |
| iiv21 | Pflu1840 | COG0315, molybdenum cofactor biosynthesis enzyme | Molybdopterin biosynthetic protein C; P. aeruginosa; E = 5e−60 | PA3918 (75, 86) |
| Hypothetical | ||||
| iiv2 | Pflu5100 | None | None | None |
| iiv24 | None | None | Hypothetical protein; P. putida KT2440; E = 5e−19 | None |
This fusion was isolated twice in the IVET screen for soil-induced promoters.
The fusion is likely to contain the promoter for Pflu4867. None of this ORF is present in the fusion.
Pflu numbers are the NCBI designations for genes identified in the NCBI annotation of the Pf0-1 genome.
Database searches were carried out by using Blastx, with the Pflu sequences as queries. Best hit excluding matches to Pf0-1 is shown.
Closest match in the P. aeruginosa PA01 genome sequence (http://www.pseudomonas.com).
TABLE 3.
Analysis of “cryptic fusions”
| Fusion | ORF length (bp)b | Codon usage%c | Predicted promoterd | Gene on opposite strande |
|---|---|---|---|---|
| iiv1 | 627 | 15.77 | Yes | Pflu5418; COG0604: NADPH quinone reductase and related Zn-dependent oxidoreductases |
| iiv4 | 1,569 | 16.98 | No | Pflu1118; COG1902: NADH flavin oxidoreductases, Old Yellow |
| iiv5 | 1,038 | 15.67 | Yes | Pflu1358; COG1012: NAD-dependent aldehyde dehydrogenases |
| iiv8 | 1,701 | 16.39 | No | Pflu2278; COG0855: polyphosphate kinase |
| iiv12 | 1,308 | 17.7 | Yes | Pflu2679; COG2081: predicted flavoprotein |
| iiv13 | 213 | 27.47 | No | Pflu5361; COG0583: transcriptional regulator (LysR type) |
| iiv14a | 1,020 | 14.81 | Yes | Pflu0506; COG1289: predicted membrane protein. Similar to predicted fusaric acid resistance proteins |
| iiv19 | 1,908 | 17.7 | Yes | Pflu5051; COG0119: isopropylmalate/homocitrate/citramalate synthases |
| iiv23 | 321 | 20.78 | No | Pflu3174; COG0584: glycerophosphoryl diester phosphodiesterase |
| iiv25 | 267 | 23.39 | No | Pflu3727; COG0189: glutathione synthase/ribosomal protein S6 modification enzyme (glutaminyl transferase) |
This fusion was isolated twice in the IVET screening for soil-induced promoters.
Length of the cryptic gene ORF identified from IVET fusions.
Mean differences when compared to the P. fluorescens codon usage table, http://www.kazusa.or.jp/codon/cgi-bin/showcodon.cgi?species=Pseudomonas+fluorescens+[gbbct].
Putative promoters upstream of the iiv ORF predicted by using SoftBerry software.
Gene found on the opposite DNA strand relative to the cryptic genes, based upon NCBI annotation of P. fluorescens Pf0-1 genome sequence (NZ_AAAT00000000).
Analysis of DNA sequences fused to dapB′ in positive IVET fusions. (i) Genes related to nutrition.
Possible roles in the acquisition and utilization of nutrients are revealed by six positive fusions (iiv3, iiv11, iiv6, iiv15, iiv18, and iiv20; see Table 2), suggesting that the soil used has a number of useful compounds that can support growth but that these are not found in standard laboratory media. We suspect that many biosynthetic genes would also be expressed in soil but would have been excluded from this study because of the use of a minimal medium for selection. The predicted product of iiv3 is similar to FadD acid-coenzyme A (CoA) ligases, and has 35% identity (51% positive) with FadD of E. coli (GenBank accession number L02649). Cloning and expression of E. coli fadD demonstrated that the product was responsible for the acyl-CoA synthetase activity, which esterifies fatty acids into metabolically active CoA thioesters (2). This reaction makes the fatty acid available for β-oxidation and thus a useable carbon and energy source. In iiv11, the Pflu5302 gene, predicted to encode a xylanase/chitin deacetylase, is found. Interestingly, upstream of the gene is another predicted xylanase/chitin deacetylase gene (Pflu5303), the 3′ end of which overlaps the 5′ end of Pflu5302 by 8 bp. It is likely that these two xylanase/chitin deacetylase genes are transcribed together, possibly from a promoter upstream of Pflu5303. The induction of iiv11 may reflect the presence of xylans and/or chitin, of plant or insect origin, respectively, whose degradation would yield a useful carbon source.
Four potential transporters were upregulated during growth in soil (iiv6, iiv15, iiv18, and iiv20). The iiv6 sequence is predicted to specify a Na+/serine symporter. Unlike the other positive fusions, the sequence juxtaposed to dapB in iiv6 was not part of a gene annotated in the Pf0-1 draft genome sequence. Rather, the DNA cloned in the fusion ended 194 bp upstream of the predicted Na+/serine symporter (Pflu4867). An examination of the 1,486 bp of intergenic sequence upstream of the Pflu4867 gene with SoftBerry software suggests the presence of two predicted promoters, both in the correct orientation to drive expression of Pflu4867 and both within the region contained in the iiv6 fusion. The predicted transcriptional start site from the promoter nearest to the symporter ORF starts 259 bp upstream of the ORF.
iiv15 is predicted to specify a cytosine/purine/uracil/thiamine/allantoin permease (Pflu4595). An NCBI conserved-domain search using the position-specific scoring matrix (PSSM) indicates that Pflu4595 aligns with CodB (7). However, Pflu4595 appears to be about 90 amino acids shorter than the other members of that group.
A gene that could encode a protein with export or import function is found in iiv18, a member of the clustered orthologous group 3451 (COG3451) type IV secretory pathway, VirB4 components group. NCBI conserved-domain searching indicates that there is some significant alignment with a VirB4 consensus, but that this alignment is only between amino acid residues 326 and 799, which represents just 52.3% of the protein. Using Blastp, we found that the nearest match in public databases is with a conserved hypothetical protein with a VirB4 domain (CAE92911). Blastp searches with the amino acid sequences before and after the VirB4 consensus failed to give further insight into potential functions. VirB4 is an ATPase, and VirB4-like proteins are part of the plasmid conjugation machinery and the type IV secretion apparatus. In addition, competence (DNA import) in Helicobacter pylori requires homologues of a number of conjugation proteins, including VirB4.
It is interesting that the genes found in fusions iiv15 and iiv18 may both, conceivably, function in the transport of nucleotides or DNA. The increased expression of codB in nitrogen-limited conditions (25) suggests that import of nitrogen-containing compounds, such as cytosine, purines, uracil, thiamine, and/or allantoin, may supply a nitrogen source. Although generally considered with respect to acquiring new genetic information, importing DNA also provides a good nitrogen and carbon source, e.g., for E. coli growing in starvation cultures (10).
The iiv20 sequence is predicted to specify a member of the major facilitator superfamily of permeases. The iiv20 sequence has similarity with the sequence encoded by PcaT, which is thought to function as a system by which starved cells can scavenge β-ketoadipate from the environment (31). In Pf0-1 and Pseudomonas putida, pcaT is found among other genes specifying factors of importance in the protocatechuate branch of the β-ketoadipate pathway. Therefore, the trapping of pcaT could relate to the induction of an upstream gene that is important for another part of the β-ketoadipate pathway.
While the induction of genes involved in nutrient acquisition and utilization solves a basic need for nutrient sources, an additional role in environmental sensing cannot be excluded. O'Toole et al. proposed that the catabolite repression control protein (Crc) in P. aeruginosa integrates nutritional cues as part of a pathway to regulate biofilm development (28). In P. fluorescens WCS365, biofilm formation proceeds via a number of convergent pathways responding to environmental (nutrient) factors (29). In Pseudomonas aureofaciens, low concentrations of inorganic phosphate lead to the expression of the Pho regulon, which is involved in scavenging phosphate from the environment and repression of biofilm formation (24).
(ii) Regulatory genes (iiv7 and iiv9).
In iiv7, the sequence belongs in the signal transduction histidine kinase (COG0642) family. The predicted translation product shows similarity to heavy metal sensor histidine kinases, such as CzcS (which controls cobalt, zinc, and cadmium resistance) of Ralstonia solanacearum (gi 17430958). In support of this prediction, the sequence upstream (Pflu4943) is similar to CzcR, the response regulator partner for CzcS. Despite the possible role in metal resistance, efforts to induce expression of iiv7 in laboratory media by exposure to sublethal concentrations of copper, zinc, and cobalt were unsuccessful (data not shown). This failure may indicate a specific inducer in the soil environment.
The gene identified in iiv9 is predicted to encode a member of the GntR family of regulators, which contain a DNA-binding helix-turn-helix domain and an aminotransferase domain. This family of regulators was first described by Haydon and Guest (13) and has been further refined into subfamilies (37). The sequence found in iiv9 most closely fits with the MocR subfamily defined by Rigali et al. (37).
(iii) Genes reflecting detoxification and metabolism.
Although we hypothesize that soil represents a harsh environment in comparison to laboratory media, only one fusion with a possible role in the direct resistance to toxic compounds was identified. The iiv17 construct has a dapB fusion with a putative NADPH quinone reductase (COG0604). Enzymes of this family catalyze the one-electron reduction of some quinones, which may detoxify quinones that might have an adverse effect on survival.
The predicted product for the gene identified at the iiv21 locus shows strong similarity with predicted molybdenum cofactor biosynthesis protein C (MoaC) from P. aeruginosa (PA3918; 75% identity) and Pseudomonas syringae (accession number ZP_00125576; 72% identity). However, the similarity is apparent only between residues 98 and 254. iiv21 appears to specify a protein that is 93 amino acids longer than many other members of the COG0315 group. An exception is a predicted protein from Azotobacter vinelandii (accession number ZP_00089475), which is of a similar size as the iiv21 gene. In E. coli, MoaC is involved in the synthesis of a molybdopterin precursor, which is ultimately converted to molybdopterin guanine dinucleotide (35). The molybdenum cofactor can be found in at least nine enterobacterial anaerobic enzymes (12).
(iv) Hypothetical genes.
The iiv2 sequence does not match any sequence in GenBank; the 1,844-amino acid predicted translation product has no significant (E < e−10) matches to sequences in public databases, and no conserved domains are evident (NCBI conserved domain search). The iiv24 sequence is annotated as hypothetical by the Joint Genome Institute but does not appear in the NCBI annotation of the Pf0-1 genome. The predicted translation product (113 amino acids) has 61% identity with another hypothetical protein from P. putida KT2440 between amino acids 33 and 109 but no matches that span the full-length sequence. Using the SignalP server (http://www.cbs.dtu.dk/services/SignalP-2.0/) (26), we found that the first 20 amino acids are predicted to be a signal peptide, cleaved between amino acids 20 and 21. Neither the iiv2 nor iiv24 predicted proteins can be matched with a COG. The novel nature of these sequences supports the contention that growth in nonlaboratory environments requires functions not previously found by studies with laboratory media.
Cryptic IVET fusions.
The second major class of fusions (10 out of 22) isolated in this study were those in which the sequence trapped in the same orientation as dapB′ had no similarity to known genes or proteins (Blastn and Blastx searches). This group of trapped sequences had considerable overlap with previously described or predicted genes on the opposite DNA strand (Table 3 and Fig. 2). We have termed these cryptic fusions, because the expressed sequences that they reveal have not been identified during genome annotation efforts (41). The draft genome sequence of Pf0-1 revealed that for each cryptic fusion, an ORF can be found in the direction of the trapped promoter (Fig. 1b). The association of active promoters with ORFs indicates that the cryptic fusions have the promoters for hitherto unknown genes specifying novel proteins. Candidate promoters were detected by SoftBerry software upstream of the iiv1, iiv5, iiv12, iiv14, and iiv19 ORFs. Thus, our analysis suggests that there may be protein-coding genes present on both DNA strands. When compared to the P. fluorescens codon usage table, the mean differences of the ORFs ranged from 14.81 to 27.47% (Table 3). Although these values are considerably different from what might be expected for P. fluorescens, an examination of the codon usage of RecA, DapB, GlnA, and FlgB from Pf0-1 revealed mean differences of 14.33, 11.84, 14.69, and 16.8%, respectively (41). Thus, the seemingly high mean differences might be attributed to variations between Pf0-1 and other P. fluorescens strains.
FIG. 2.

Genetic organization of ORFs identified in cryptic fusions, relative to the annotated genes that they overlap. Pflu numbers were assigned during the automated annotation of the Pf0-1 genome by the NCBI Microbial Genomes Annotation Project. Each ORF of the cryptic fusion and its opposing gene is shown with numbers to indicate the start and stop of the ORF in Pf0-1. Asterisks indicate the point at which the cryptic ORF was fused to dapB′ in the IVET fusion strains (iiv1-bp 571; iiv4-bp 1265; iiv5-bp 1034; iiv8-bp 895; iiv12-bp 1201; iiv13-bp 15; iiv14-bp 346; iiv19-bp 1042; iiv23-bp 33; iiv25-bp 237). iiv5 was described by Silby et al. (41).
Interestingly, the sequence trapped in the cryptic fusion iiv4 is antisense to a predicted fadH gene (encoding 2,4-dienoyl-CoA reductase) on the opposite strand, which has been shown to be required for the β-oxidation of unsaturated fatty acids with double bonds extending from even-numbered carbon atoms (46). The induction of such a sequence raises the possibility of RNA-mediated regulation of fatty acid utilization in soils.
Our identification of cryptic genes could indicate that the P. fluorescens genome, and perhaps other bacterial genomes, have more genes than currently thought, which would naturally imply a greater coding density in the genome sequence. Possible functions include the transcript acting as an antisense regulator of the opposite gene. In prokaryotes, antisense regulation is involved in controlling plasmid replication and bacteriophage gene expression (for a review, see reference 44), and a growing number of regulatory RNA molecules are being identified in prokaryotes (for examples, see references 14 and 22). Alternatively, the activation of the cryptic gene could shut down transcription of the opposite gene due to competition for the template. The extent to which this largely stochastic situation might be effective could depend upon the relative promoter strengths in different environments, and thus environmental fluctuations would indirectly control relative transcription at such loci.
We are not the first to report the recovery of such fusions from IVET screenings. Indeed, the first report on IVET (19) mentions a fusion in the rfb operon which when transcribed would be antisense to rfb gene transcripts; and other examples exist (4, 32, 45). However, we are the first to emphasize the significance of these fusions and to use an existing whole genome sequence to examine them further.
Role for iiv genes in proliferation in soil.
The importance of proteins specified by iiv sequences for growth and/or survival in soil was tested by constructing mutants in which iiv sequences representing different classes of genes were partially deleted. We disrupted the genes found in iiv2 (hypothetical protein), iiv3 (FadD), and iiv7 (two-component sensor), replacing part of each ORF with a streptomycin resistance gene cassette (see Materials and Methods for the construction). The ability of the recombinants to grow in soil was assessed by using our standard soil growth assay.
Each iiv::Sm mutant showed a significant defect in soil growth after 1 day (P < 0.05) relative to Pf0-2x (Fig. 3). Pf0-2x is a Pf0-1 derivative harboring Smr in the adnA gene (38), and was used to rule out reduced fitness linked to the Smr cassette. AdnA is important for long-term persistence and is spread in natural soils under field conditions (21), but under conditions similar to those employed here, the lack of AdnA in Pf0-2x did not influence growth (9). The population of the iiv mutants increased less than 100-fold, compared to Pf0-2x, which increased over 1,000-fold after 1 day. Interestingly, after 3 days in soil, the three iiv::Sm mutants had established populations similar to Pf0-1 and Pf0-2x. Mutations in these three iiv genes clearly have an impact on soil fitness, presumably impairing early establishment of the population. This finding was not associated with a discernible growth defect in cultures of iiv::Sm mutants in vitro. Although the growth characteristics shown in Fig. 3 resemble those in Fig. 1A, the slow growth of fusion strains (Fig. 1) correlates with slow growth in laboratory media (data not shown) and is due to the fact that growth of fusion strains depends on the expression of dapB by heterologous promoters. In contrast, dapB in the iiv mutants is driven by its native promoter, and the growth defect is a consequence of mutations in iiv genes.
FIG. 3.
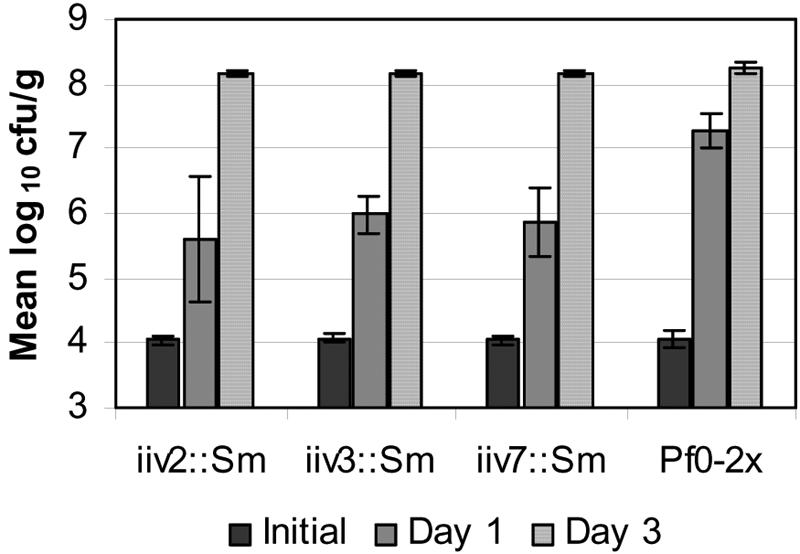
Growth of P. fluorescens Pf0-1 strains bearing null mutations in iiv genes compared to Pf0-2x. Data presented are the means of results from three or four independent replicates. Initial CFU/g titers were not significantly different. Each iiv mutant had a significantly lower titer than Pf0-2x after 1 day in soil (P < 0.05). No significant differences were seen among strains after 3 days of soil growth. iiv mutations had no effect on growth rate in culture (doubling times in PMM culture: Pf0-1, 71 min; iiv2Sm, 69 min; iiv3Sm, 73 min; and iiv7Sm, 71.5 min). Error bars indicate standard deviations.
Most IVET-type screens to date have been focused on elucidating the mechanisms of bacterial pathogenesis in mammals (for examples, see references 4, 16, 20, and 45). More recently, IVET approaches have been used to examine the processes of plant pathogenesis (3), colonization of a plant pathogenic fungus by Pseudomonas putida (17), and rhizosphere colonization by Pseudomonas spp. (11, 32, 36). This IVET study has focused on genes induced during growth in soil. Of interest, we have found some overlap between soil-induced and rhizosphere-induced genes. Rainey (32) isolated a fusion (rhi-3) containing sequences similar to copR and copS, which are involved in resistance to copper. Upstream of iiv7, a putative sensor kinase from a two-component regulatory pair, is a sequence predicted to encode a protein that is 61% identical to CopR (GenBank accession number L05176) and has 63% identity with CzcR (GenBank accession number Q44006), a regulator of resistance to cobalt, zinc, and cadmium. iiv7 is likely to specify the functional equivalent of rhi-3 and CopS or CzcS. A second overlap appears between iiv20 and the cii-10 fusion reported by Rediers et al. (36). In cii-10, genes with a high degree of similarity to pcaB and pcaC were found. The products of these genes are important in the protocatechuate branch of the β-ketoadipate pathway for the degradation of aromatic compounds, which is the same system in which PcaT (iiv20) functions. In another example, Gal et al. identified a putative acyl CoA dehydrogenase (COG1960) in the fusion rhi-27 (11), whereas the sequence in fusion iiv3 is two genes downstream from a COG1960-family acyl CoA dehydrogenase gene which is likely to be functionally equivalent to rhi-27. Finally, both iiv15 and the rhi-73 fusion isolated by Gal et al. (11) specify predicted COG1457 nucleotide permeases. These similarities in recovered sequences between the rhizosphere studies and our soil work suggest that those shared gene fusions may be ones more generally responsive to the soil surrounding the rhizosphere than to the rhizosphere per se. In contrast, there is very little overlap between the small number of fusions recovered here and those identified by using IVET, differential display, and signature-tagged mutagenesis to examine gene expression in model pathogenesis systems (18).
This study, the third which utilized the requirement for DAP, uncovered two groups of fusions which are affected by soil contact. The first group has similarity to known or predicted genes, while the second does not. The second group comprises 10 cryptic genes and points to a novel genome organization that has so far been overlooked in genome annotation efforts.
ADDENDUM IN PROOF
The NCBI annotation of the P. fluorescens pf0-1 genome sequence has been updated to reflect newly released data. With this update, Pflu numbers have been changed. We anticipate that Pflu numbers may change again before the genome sequence is completed. Anyone interested in the P. fluorescens sequences described herein should contact the authors for the most recent accession numbers.
Acknowledgments
We thank Laura McMurry for reading the manuscript.
This study was supported by Department of Energy grant DE-FG02-97ER62493.
REFERENCES
- 1.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. [DOI] [PubMed] [Google Scholar]
- 2.Black, P. N., C. C. DiRusso, A. K. Metzger, and T. L. Heimert. 1992. Cloning, sequencing, and expression of the fadD gene of Escherichia coli encoding acyl coenzyme A synthetase. J. Biol. Chem. 267:25513-25520. [PubMed] [Google Scholar]
- 3.Boch, J., V. Joardar, L. Gao, T. L. Robertson, M. Lim, and B. N. Kunkel. 2002. Identification of Pseudomonas syringae pv. tomato genes induced during infection of Arabidopsis thaliana. Mol. Microbiol. 44:73-88. [DOI] [PubMed] [Google Scholar]
- 4.Camilli, A., and J. J. Mekalanos. 1995. Use of recombinase gene fusions to identify Vibrio cholerae genes induced during infection. Mol. Microbiol. 18:671-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Casaz, P., A. Happel, J. Keithan, D. L. Read, S. R. Strain, and S. B. Levy. 2001. The Pseudomonas fluorescens transcription activator AdnA is required for adhesion and motility. Microbiology 147:355-361. [DOI] [PubMed] [Google Scholar]
- 6.Compeau, G., B. J. Al-Achi, E. Platsouka, and S. B. Levy. 1988. Survival of rifampin-resistant mutants of Pseudomonas fluorescens and Pseudomonas putida in soil systems. Appl. Environ. Microbiol. 54:2432-2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Danielsen, S., M. Kilstrup, K. Barilla, B. Jochimsen, and J. Neuhard. 1992. Characterization of the Escherichia coli codBA operon encoding cytosine permease and cytosine deaminase. Mol. Microbiol. 6:1335-1344. [DOI] [PubMed] [Google Scholar]
- 8.DeFlaun, M. F., B. M. Marshall, E.-P. Kulle, and S. B. Levy. 1994. Tn5 insertion mutants of Pseudomonas fluorescens defective in adhesion to soil and seeds. Appl. Environ. Microbiol. 60:2637-2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeFlaun, M. F., A. S. Tanzer, A. L. McAteer, B. Marshall, and S. B. Levy. 1990. Development of an adhesion assay and characterization of an adhesion-deficient mutant of Pseudomonas fluorescens. Appl. Environ. Microbiol. 56:112-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Finkel, S. E., and R. Kolter. 2001. DNA as a nutrient: novel role for bacterial competence gene homologs. J. Bacteriol. 183:6288-6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gal, M., G. M. Preston, R. C. Massey, A. J. Spiers, and P. B. Rainey. 2003. Genes encoding a cellulosic polymer contribute toward the ecological success of Pseudomonas fluorescens SBW25 on plant surfaces. Mol. Ecol. 12:3109-3121. [DOI] [PubMed] [Google Scholar]
- 12.Gennis, R. B., and V. Stewart. 1996. Respiration, p. 217-261. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella, 2nd ed., vol. 1. ASM Press, Washington, D.C. [Google Scholar]
- 13.Haydon, D. J., and J. R. Guest. 1991. A new family of bacterial regulatory proteins. FEMS Microbiol. Lett. 79:291-296. [DOI] [PubMed] [Google Scholar]
- 14.Johansson, J., and P. Cossart. 2003. RNA-mediated control of virulence gene expression in bacterial pathogens. Trends Microbiol. 11:280-285. [DOI] [PubMed] [Google Scholar]
- 15.Kirner, S., S. Krauss, G. Sury, S. T. Lam, J. M. Ligon, and K. H. van Pee. 1996. The non-haem chloroperoxidase from Pseudomonas fluorescens and its relationship to pyrrolnitrin biosynthesis. Microbiology 142:2129-2135. [DOI] [PubMed] [Google Scholar]
- 16.Lai, Y.-C., H.-L. Peng, and H.-Y. Chang. 2001. Identification of genes induced in vivo during Klebsiella pneumoniae CG43 infection. Infect. Immun. 69:7140-7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee, S.-W., and D. A. Cooksey. 2000. Genes expressed in Pseudomonas putida during colonization of a plant-pathogenic fungus. Appl. Environ. Microbiol. 66:2764-2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahan, M. J., D. M. Heithoff, R. L. Sinsheimer, and D. A. Low. 2000. Assessment of bacterial pathogenesis by analysis of gene expression in the host. Annu. Rev. Genet. 34:139-164. [DOI] [PubMed] [Google Scholar]
- 19.Mahan, M. J., J. M. Slauch, and J. J. Mekalanos. 1993. Selection of bacterial virulence genes that are specifically induced in host tissues. Science 259:686-688. [DOI] [PubMed] [Google Scholar]
- 20.Mahan, M. J., J. W. Tobias, J. M. Slauch, P. C. Hanna, R. J. Collier, and J. J. Mekalanos. 1995. Antibiotic-based selection for bacterial genes that are specifically induced during infection of a host. Proc. Natl. Acad. Sci. USA 92:669-673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marshall, B., E. A. Robleto, R. Wetzler, P. Kulle, P. Casaz, and S. B. Levy. 2001. The adnA transcriptional factor affects persistence and spread of Pseudomonas fluorescens under natural field conditions. Appl. Environ. Microbiol. 67:852-857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Masse, E., N. Majdalani, and S. Gottesman. 2003. Regulatory roles for small RNAs in bacteria. Curr. Opin. Microbiol. 6:120-124. [DOI] [PubMed] [Google Scholar]
- 23.Matthews, M., and C. R. Roy. 2000. Identification and subcellular localization of the Legionella pneumophila IcmX protein: a factor essential for establishment of a replicative organelle in eukaryotic host cells. Infect. Immun. 68:3971-3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Monds, R. D., M. W. Silby, and H. K. Mahanty. 2001. Expression of the Pho regulon negatively regulates biofilm formation by Pseudomonas aureofaciens PA147-2. Mol. Microbiol. 42:415-426. [DOI] [PubMed] [Google Scholar]
- 25.Muse, W. B., C. J. Rosario, and R. A. Bender. 2003. Nitrogen regulation of the codBA (cytosine deaminase) operon from Escherichia coli by the nitrogen assimilation control protein, NAC. J. Bacteriol. 185:2920-2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nielsen, H., J. Engelbrecht, S. Brunak, and G. von Heijne. 1997. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 10:1-6. [DOI] [PubMed] [Google Scholar]
- 27.Osbourn, A. E., C. E. Barber, and M. J. Daniels. 1987. Identification of plant-induced genes of the bacterial pathogen Xanthomonas campestris pathovar campestris using a promoter-probe plasmid. EMBO J. 6:23-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O'Toole, G. A., K. A. Gibbs, P. W. Hager, P. V. Phibbs, Jr., and R. Kolter. 2000. The global carbon metabolism regulator Crc is a component of a signal transduction pathway required for biofilm development by Pseudomonas aeruginosa. J. Bacteriol. 182:425-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O'Toole, G. A., and R. Kolter. 1998. Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol. Microbiol. 28:449-461. [DOI] [PubMed] [Google Scholar]
- 30.Parales, R. E., and C. S. Harwood. 1993. Construction and use of a new broad-host-range lacZ transcriptional fusion vector, pHRP309, for gram− bacteria. Gene 133:23-30. [DOI] [PubMed] [Google Scholar]
- 31.Parke, D., D. A. D'Argenio, and L. N. Ornston. 2000. Bacteria are not what they eat: that is why they are so diverse. J. Bacteriol. 182:257-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rainey, P. B. 1999. Adaptation of Pseudomonas fluorescens to the plant rhizosphere. Environ. Microbiol. 1:243-257. [DOI] [PubMed] [Google Scholar]
- 33.Rainey, P. B., D. M. Heithoff, and M. J. Mahan. 1997. Single-step conjugative cloning of bacterial gene fusions involved in microbe-host interactions. Mol. Gen. Genet. 256:84-87. [DOI] [PubMed] [Google Scholar]
- 34.Rainey, P. B., and G. M. Preston. 2000. In vivo expression technology strategies: valuable tools for biotechnology. Curr. Opin. Biotechnol. 11:440-444. [DOI] [PubMed] [Google Scholar]
- 35.Rajagopalan, K. V. 1996. Biosynthesis of the molybdenum cofactor, p. 674-679. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella, 2nd ed., vol. 1. ASM Press, Washington, D.C. [Google Scholar]
- 36.Rediers, H., V. Bonnecarrère, P. B. Rainey, K. Hamonts, J. Vanderleyden, and R. De Mot. 2003. Development and application of a dapB-based in vivo expression technology system to study colonization of rice by the endophytic nitrogen-fixing bacterium Pseudomonas stutzeri A15. Appl. Environ. Microbiol. 69:6864-6874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rigali, S., A. Derouaux, F. Giannotta, and J. Dusart. 2002. Subdivision of the helix-turn-helix GntR family of bacterial regulators in the FadR, HutC, MocR, and YtrA subfamilies. J. Biol. Chem. 277:12507-12515. [DOI] [PubMed] [Google Scholar]
- 38.Robleto, E. A., I. Lopez-Hernandez, M. W. Silby, and S. B. Levy. 2003. Genetic analysis of the AdnA regulon in Pseudomonas fluorescens: nonessential role of flagella in adhesion to sand and biofilm formation. J. Bacteriol. 185:453-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 40.Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 74:5463-5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silby, M. W., P. B. Rainey, and S. B. Levy. 2004. IVET experiments in Pseudomonas fluorescens reveal cryptic promoters at loci associated with recognizable overlapping genes. Microbiology 150:518-520. [DOI] [PubMed] [Google Scholar]
- 42.Simon, R., U. Priefer, and A. Puhler. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram-negative bacteria. Bio/Technology 1:784-791. [Google Scholar]
- 43.Stover, C. K., X. Q. Pham, A. L. Erwin, S. D. Mizoguchi, P. Warrener, M. J. Hickey, F. S. Brinkman, W. O. Hufnagle, D. J. Kowalik, M. Lagrou, R. L. Garber, L. Goltry, E. Tolentino, S. Westbrock-Wadman, Y. Yuan, L. L. Brody, S. N. Coulter, K. R. Folger, A. Kas, K. Larbig, R. Lim, K. Smith, D. Spencer, G. K. Wong, Z. Wu, I. T. Paulsen, J. Reizer, M. H. Saier, R. E. Hancock, S. Lory, and M. V. Olson. 2000. Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature 406:959-964. [DOI] [PubMed] [Google Scholar]
- 44.Wagner, E. G., and R. W. Simons. 1994. Antisense RNA control in bacteria, phages, and plasmids. Annu. Rev. Microbiol. 48:713-742. [DOI] [PubMed] [Google Scholar]
- 45.Wang, J., A. Mushegian, S. Lory, and S. Jin. 1996. Large-scale isolation of candidate virulence genes of Pseudomonas aeruginosa by in vivo selection. Proc. Natl. Acad. Sci. USA 93:10434-10439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.You, S. Y., S. Cosloy, and H. Schulz. 1989. Evidence for the essential function of 2,4-dienoyl-coenzyme A reductase in the beta-oxidation of unsaturated fatty acids in vivo. Isolation and characterization of an Escherichia coli mutant with a defective 2,4-dienoyl-coenzyme A reductase. J. Biol. Chem. 264:16489-16495. [PubMed] [Google Scholar]