

Abstract
Proliferative cell nuclear antigen (PCNA), a conserved plant protein as well as an important replication factor, is induced in response to geminivirus infection in the resting cells of the phloem tissues. The biochemical role of PCNA in rolling circle replication (RCR) of geminivirus DNA has not been explored in detail. The initiation of RCR of the bipartite genome of a geminivirus, Indian mung bean yellow mosaic virus (IMYMV), is mainly controlled by viral protein Rep (or AL1 or AC1). The role of host PCNA in RCR of IMYMV was revealed by studying the physical and functional interactions between recombinant PCNA and recombinant IMYMV Rep. Pea nuclear PCNA as well as recombinant pea PCNA showed binding to recombinant Rep in experiments involving both affinity chromatography and yeast two-hybrid approaches. The contacting amino acid residues of PCNA seemed to be present throughout a wide region of the trimeric protein, while those of Rep appeared to be localized only in the middle part of the protein. The site-specific nicking-closing activity and the ATPase function of IMYMV Rep were impaired by PCNA. These observations lead to interesting speculations about the control of viral RCR and dynamic profiles of protein-protein interactions at the RCR origin of the geminiviruses.
Indian mung bean yellow mosaic virus (IMYMV), a species of the Begomovirus genus in the Geminiviridae family, poses a tremendous threat to legume cultivation, especially in the northern parts of India. A majority of the begomoviruses have single-stranded bipartite genomes, and the begomoviruses are the most abundant species of the geminiviruses (36). Although the geminiviruses form a very diverse family and infect almost all crops with narrow host ranges, the modes of viral DNA replication are essentially the same for all (31). The viral genomes multiply by rolling circle replication (RCR), and all of the geminivirus genomes have a conserved nonamer (TAATATT↓AC) at their unique replication origins (33). Besides the conserved nonamer, the RCR origin of each geminivirus is characterized by iterons and some other cis-acting elements that belong only to a single or closely related species of the geminiviruses. These elements may serve as the landing ground of various transacting factors that are encoded by either the virus or the host and that are important for the initiation of RCR. The most important of these factors is the virus-encoded Rep protein; in some cases, the Rep protein alone can initiate RCR without requiring other accessory viral factors (14).
IMYMV Rep is a site-specific DNA binding, site-specific nicking-closing enzyme and an ATP-activated type I DNA topoisomerase; these biochemical activities are required for the initiation and termination of DNA replication of the IMYMV genome (26, 29). Since the coding capacities of the geminivirus DNA genomes are extremely limited, the replication and transmission processes of these viruses are largely dependent on host factors. In this way, the geminiviruses also serve as important model systems for studies of plant DNA replication and transcription (13). Most of the host factors crucial for RCR initiation and control and viral pathogenesis have been found to interact with the virus-encoded Rep protein. The Rep proteins of many geminiviruses have been found to interact with plant retinoblastoma-like proteins (pRBR) to alter the cell cycle programs of infected nondividing cells (11, 1). The Arabidopsis GRIK, GRIMP, and histone H3 proteins interact with the Rep proteins of tomato golden mosaic virus and cabbage leaf curl virus (20). The host GRAB proteins and replication factor C (RF C) proteins have been found to bind to geminivirus Rep proteins (12, 23). Of these various interactions, the pRBR-Rep association is of extreme importance for reprogramming of the cell cycle of infected cells, a scenario often encountered with the mammalian DNA tumor viruses (17).
A majority of the geminiviruses infect terminally differentiated cells that have exited the cell division cycle and contain very small (or nil) amounts of DNA replication enzymes (4). In such noncycling cells or cells in the resting cell cycle phase (G0), pRBR represses transcription by sequestering E2F and keeps E2F-responsive genes in an effective heterochromatized state by utilizing E2F, histone deacetylase, and chromatin remodeling proteins (40). Following geminivirus infection, the pRBR-Rep complex releases E2F to turn on the E2F-responsive genes. As a result, infected G0 cells are activated to partial S phase (7), thus ensuring viral growth processes (6). At least one important host replication factor, proliferative cell nuclear antigen (PCNA), an accessory to DNA polymerase δ, has been found to be induced by this mechanism following infection by tomato golden mosaic virus (21) and other geminiviruses, including IMYMV (S. K. Mukherjee, unpublished data).
PCNA acts as a sliding clamp for processive DNA synthesis (19) and forms a trimeric ring, as evidenced from its crystal structure (10). The primary amino acid sequences of PCNA across the eukaryotic kingdom are conserved, and the plant PCNA proteins share more than 90% homology among themselves. The function of the protein is entirely dependent on its structure, and many of its deletion derivatives lose functional significance because of the structural perturbations (3). The PCNA ring has two dissimilar surfaces: an outside surface consisting of β sheets and an inside layer of α helices that are rich in basic amino acids. PCNA links the template double-helical DNA topologically with the catalytic help of heteropentameric RF C (28). The inner surface of the PCNA ring encircles DNA, and at the same time, PCNA freely slides along the DNA by virtue of its helical composition. PCNA also increases the processivity of DNA polymerase by engaging in protein-protein interactions with its outer surface while remaining linked to the DNA (18). Because of its typical structure, it also interacts with a plethora of cellular proteins, which belong to three major classes: the DNA replication factors, the DNA repair proteins, and the cell cycle regulatory factors (24). Panning of a random peptide display library (39) and multiple alignments of many PCNA binding proteins have helped reveal two conserved PCNA binding motifs: the PCNA-interacting protein (PIP) box and the KA box (22, 37, 38). The consensus sequence of the PIP box is Q-xx-h-x-x-a-a, where “h ” represents a moderately hydrophobic amino acid (e.g., L, I, or M), “a ” represents a highly hydrophobic residue with an aromatic side chain (e.g., F or Y), and “x ” is any amino acid; proteins such as p21 and FenI harbor this motif. The other motif (the KA box), with the sequence K-A-(A/L/I)-(A/L/Q)-x-x-(L/V), is found in proteins such as cyclin D3 and MSH6. Some PCNA binding proteins, such as DNA polymerase δ and UNG2, contain both motifs, whereas other PCNA partners do not contain these motifs at all. It is noteworthy that IMYMV Rep also does not contain the KA box. PCNA in eukaryotic cells thus plays key roles in regulating several biochemical reactions through the coordination and organization of various interacting partners.
Since PCNA is induced as a result of a geminivirus infection, PCNA might be used for virus-specific DNA replication and growth. In order to explore the virus-specific mode of utilization of PCNA, we examined the ability of PCNA to form complexes with IMYMV Rep. Here we report the region of Rep responsible for binding to PCNA as well as the amino acid residues of PCNA that contact the Rep protein. The possible consequences of complex formation on the biochemical activities of Rep were also investigated. Besides acting as an accessory protein for DNA polymerase δ, PCNA might have a role in limiting the viral DNA copy number.
MATERIALS AND METHODS
DNA constructs and purification of recombinant proteins.
DNA isolated from the full-length clone of Rep-pET28a was used to PCR amplify various deletion mutations in Rep with specific sets of primers containing BamHI and HindIII restriction sites at the N and C termini, respectively (Table 1). The amplified fragments were first cloned in vector pGEMT (Promega) and subsequently recloned in vector pET28a (Novagen) at the same restriction sites. Six-His-tagged recombinant proteins were purified by Ni-nitrilotriacetic acid (NTA)-agarose chromatography.
TABLE 1.
Primers used for IMYMV Rep deletion mutations
| Orientationa | Sequence of primer (5′→3′)b | Mutation |
|---|---|---|
| S | ATG GAT CCA TGC CAA GGG AAG GTC GT | None (wild type) |
| A | TGA AAG CTT TCA ATT CGA GAT CGT CGA | |
| S | CGA AAA ACC AAA GGT CCT CCG ACC TCC ATT CC | Rep (FF75,76SS) |
| A | GCT TTT TGG TTT CCA GGA GGC TGG AGG TAA GG | |
| S | ATG GAT CCA TGC CAA GGG AAG GTC GT | Rep1-183aac |
| A | CTA AGC TTA GGC GAC TCA TAT GCC TG | |
| S | ATG GAT CCA TGC CAA GGG AAG GTC GT | Rep1-133aa |
| A | AGA AGC TTC TAT GCG TCG TTG GCA GAT TG | |
| S | CCG GAT CCG GCA GAT CAG CTA GAG GAG G | Rep120-362aa |
| A | TGA AAG CTT TCA ATT CGA GAT CGT CGA ATT GC | |
| S | AAA GGA TCC TTT ACA TTG GAG TCA TTC GAC | Rep184-362aad |
| A | TGA AAG CTT TCA ATT CGA GAT CGT CGA ATT GC |
S, sense; A, antisense.
All of the primers used for deletion mutations contained a BamHI restriction site (underlined) in the sense primer and a HindIII restriction site (underlined) in the antisense primer preceded by a stop codon.
N terminus.
C terminus.
Point mutants of Rep and PCNA were constructed by using a Quick-change site-directed mutagenesis kit (Stratagene) by following the manufacturer's protocol. PCR amplification was carried out with complementary mutagenesis primers and the full-length wild-type clone of Rep-pET28a or PCNA-pGEX4T-1 as a template, respectively. A list of each set of primers is given in Table 2. The mutations were confirmed by sequencing. Double point mutations in PCNA, which are spatially distal to each other, were made by the addition of single point mutations in a serial manner.
TABLE 2.
Primers used for PCNA mutations
| Orientationa | Sequence of primer (5′→3′)b | Change in:
|
|
|---|---|---|---|
| Amino acid | Nucleotide in the sense strand | ||
| S | GGG AAG TCT GCT GCA GAA GGT TCT AG | K13Q | A to C |
| A | CCC TTC AGA CGA CGT CTT CCA AGA TC | ||
| S | GGG AAG TCT GCT CGA GAA GGT TCT AG | K13R | GA to CG |
| A | CCC TTC AGA CGA GCT CTT CCA AGA TC | ||
| S | GAA GTC TGC TGA AGG AGG TTC TAG AAT C | K14E | A to G |
| A | CTT CAG ACG ACT TCC TCC AAG ATC TTA G | ||
| S | TAG AAT CAA TTA CGG ATC TGG TGA AC | K20T | A to C |
| A | ATC TTA GTT AAT GCC TAG ACC ACT TG | ||
| S | GCG CTA GTG GCG CTG CTG GCC CGA TCT GAG GGT TTT GAG | L52A | CT to GC |
| A | CGC GAT CAC CGC GAC GAC CGG GCT AGA CTC CCA AAA CTC | ||
| S | GCG CTA GTG GCG CTG CTG CGC CGA TCT GAG GGT TTT GAG | L52R | T to G |
| A | CGC GAT CAC CGC GAC GAC GCG GCT AGA CTC CCA AAA CTC | ||
| S | CAC CGT CAC TTT CAT GAG CCC CAC CCA AGA AGA CAA G | Δ(FE103,104) | 6 Nucleotides deleted |
| A | GTG GCA GTG AAA GTA CTC GGG GTG GGT TCT GTT GTT C | ||
| S | GAC AGT GAG CAT CTT GCG ATT CCA GAA GCA GAG G | G127A | G to C |
| A | CTG TCA CTC GTA GAA CGC TAA GGT CTT CGT CTC C | ||
| S | CTT GGG ATT CCA GAA GCA GAG GAG GCC CAT GCT ATT CTT ATA ATG CC | Y133A | TA to GC |
| A | GAA CCC TAA GGT CTT CGT CTC CTC CGG GTA CGA TAA GAA TAT TAC GG | ||
| S | GGT TTT ACC TGC CAC CTA AGA TTG | A252F | G to C |
| A | CCA AAA TGG ACG GTG GAT TCT AAC | ||
| S | TTT TAC CTG GCA GCT AAG ATT GAA G | P253A | C to G |
| A | AAA ATG GAC CGT CGA TTC AAA CTT C | ||
| S | GCA CCT AAG ATT AGA GAG GAT GAA GAG | E256R | GA to AG |
| A | CGT GGA TTC TCC TCT CTC CTA CTT CTC | ||
S, sense; A, antisense.
Underlining indicates the change(s) in the sequence.
Full-length Rep and deletion mutants of Rep were overexpressed in Escherichia coli strain BL-21(DE3) and purified to near homogeneity under nondenaturing conditions as described earlier (26). The activity of the recombinant Rep proteins was checked in vitro by site-specific cleavage (26) and/or ATPase function (see below).
Wild-type PCNA was overexpressed in E. coli BL-21(DE3) from plasmid vectors pET28a and pGEX4T-1 as polyhistidine and glutathione S-transferase (GST) fusion proteins, respectively, and purified to near homogeneity by following the manufacturer's protocol (Pharmacia Biotech). Point mutants of PCNA were purified as GST fusion proteins. The activity of the recombinant PCNA proteins was checked by the ability to stimulate DNA polymerase δ-mediated extension of DNA synthesis (15).
ATPase assay.
The ATPase assay protocol was adapted from that of Fukuda et al. (8). The buffer used for the ATPase reaction contained 20 mM Tris-HCl (pH 8.0), 1 mM MgCl2, 100 mM KCl, 8 mM dithiothreitol (DTT), and 80 μg of bovine serum albumin (BSA)/ml. Briefly, 1 μl (10 μCi) of [γ-32P]ATP (6,000 Ci/mmol) was diluted 50-fold with 5 mM ATP. Dilute radiolabeled ATP (1 μl) was mixed with the desired amounts of protein and incubated at 37°C for 30 min. After the reaction, 1 μl of the reaction mixture was spotted on a polyethyleneimine thin-layer chromatography (TLC) plate (Sigma-Aldrich) and air dried, and chromatography was performed with 0.5 M LiCl and 1 M HCOOH as the running solvents. Following the completion of chromatography, the TLC paper was dried and autoradiographed.
Preparation of a nuclear extract.
A nuclear extract was prepared essentially as described by Dignam et al. (5). Briefly, about 500 g of fresh leaf tissue from 8- to 10-day-old pea seedlings was ground at 4°C in 1 liter of STM buffer (500 mM sucrose, 50 mM Tris-HCl [pH 8.0], 10 mM MgCl2, 0.1% β-mercaptoethanol). The finely ground leaf tissue was passed twice through Mira cloth and centrifuged at 1,000 × g for 10 min at 4°C. The pellet was resuspended in 100 ml of STM buffer containing 2.5% Triton X-100 and kept on ice for 30 min with periodic shaking followed by centrifugation at 3,000 × g for 15 min at 4°C. The above step was repeated three or four times until clear nuclei were obtained. The nuclei were lysed by homogenization in 20 ml of lysis buffer (20 mM HEPES [pH 7.0], 25% glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM phenylmethylsulfonyl fluoride, 0.5 mM DTT, 50 μM leupeptin, 1 μM pepstatin). The homogenate was stirred for 30 min at 4°C with a magnetic stirrer and centrifuged at 25,000 × g for 30 min. The supernatant was dialyzed against 50 mM KCl-50 mM Tris-HCl (pH 8.0)-20% glycerol with three exchanges, followed by centrifugation at 15,000 × g for 25 min at 4°C. The concentrated supernatant (20 to 50 mg/ml) was collected and stored in aliquots at −70°C.
DNA synthesis assay.
The protocol for the DNA synthesis assay was modified from that of Hop et al. (15). About 2 μg of M13mp18 DNA was annealed to a 17-mer universal primer (Stratagene). The annealed DNA was incubated with 10 μg of pea nuclear extract, 200 μM (each) dATP, dGTP, and dTTP, 2 μM dCTP, 6 nM [α-32P]dCTP, and increasing amounts of either wild-type or mutant PCNA in a 50-μl reaction buffer containing 10 mM Tris-HCl (pH 8.0), 50 mM KCl, 1.5 mM MgCl2, 0.01% gelatin, and 0.01% Triton X-100 for 30 min at 37°C. The reaction was stopped by the addition of 1 ml of 5% ice-cold trichloroacetic acid, and the mixture was kept on ice for 1 h. The entire reaction mixture was passed through prewashed GF/C (Millipore) filters in a Manifold system (Millipore). The filters then were washed with 10 volumes of 90% alcohol followed by reaction buffer and were allowed to dry under a lamp. The Cerenkov counts of acid-insoluble radioactivity retained on the filters (DNA synthesis) were read with a multipurpose scintillation counter (Beckman LS 6500).
Pull-down assays.
Purified six-His-tagged Rep proteins were incubated with equal amounts of wild-type or mutant GST-PCNA proteins in binding buffer B (25 mM Tris-HCl [pH 8.0], 75 mM NaCl, 2.5 mM EDTA, 5 mM MgCl2, 2.5 mM DTT, 1% NP-40) at 37°C for 30 min, following which 10 μl of prewashed and binding buffer B-equilibrated glutathione-Sepharose (Pharmacia) resin was added. The mixture was slowly stirred for 30 min at room temperature. The unbound protein fraction was separated from the resin by centrifugation at 3,000 × g for 3 min. The resin containing the bound protein was washed by centrifugation (3,000 × g, 3 min) with increasing concentrations of NaCl (100 to 300 mM) in binding buffer B. An equal volume of 2× sample buffer (100 mM Tris-Cl [pH 6.8], 200 mM DTT, 4% SDS, 0.2% bromophenol blue, and 20% glycerol) was added to this resin, the mixture was boiled for 5 min and centrifuged at 3,000 × g for 3 min, and the supernatant was analyzed by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE). The protein bands were visualized by staining with either silver nitrate or Coomassie blue. The intensities of the bands were estimated by using a Fluor-S Multi-Imager (Bio-Rad) with Quantity 1 (4.1.1) software (Bio-Rad). Each experiment was repeated three times. Similarly, equal amounts of various six-His-tagged Rep deletion mutants were studied for interactions with wild-type GST-PCNA. For oligomerization studies, each of the Rep proteins or Rep protein derivatives was purified with two different N-terminal tags, i.e., six-His and maltose binding protein (MBP), and the interactions between the two forms of fusion proteins were analyzed in pull-down assays with amylose resin (New England Biolabs).
Yeast two-hybrid analysis.
IMYMV Rep DNA was excised from the BamHI and XhoI sites of pET28a and recloned into the BamHI and SalI sites of pGBD-C1 (16). Similarly, either full-length PCNA or point mutants of PCNA were excised from the EcoRI and XhoI sites of pGEX4T-1 and recloned into the EcoRI and SalI sites of pGAD-C1 (16). This cloning effort resulted in the in-frame fusion of the Gal4 DNA binding domain with Rep and the Gal4 activation domain with PCNA. All of the constructs were verified by restriction digestion and sequencing. DNA manipulations were carried out as described by Sambrook et al. (30).
The yeast two-hybrid assay was performed with yeast strain Y190, which was transformed with the appropriate plasmids and grown on synthetic defined plates in the absence of Trp and Leu. Protein interaction analysis was performed by using synthetic defined plates without Leu, Trp, and His. After 3 days at 30°C, individual colonies were streaked and tested in liquid and filter β-galactosidase assays, in a colony-forming assay in the presence of 20 mM 3-amino-1,2,3-triazole (3-AT), and in a diploid His− assay described earlier (34). The filter β-galactosidase assay, which directly reflects the strength of protein interactions, was performed by streaking doubly transformed yeast colonies onto filter paper and allowing the colonies to grow for 2 days in selection medium. The yeast cells within the yeast-impregnated filter were permeabilized by freezing in liquid nitrogen and thawing at room temperature. The filter was placed on a second filter presoaked in 0.1 M phosphate buffer (pH 7.0) containing 300 μg of 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal)/ml and 0.27% β-mercaptoethanol. The filter was allowed to develop a blue color over 48 h.
The liquid β-galactosidase assay was carried out with the substrate chlorophenol red-β-d-galactopyranoside as described previously (2). Relative enzymatic activities were determined for five independent transformants. Data for quantitative assays were corrected for yeast cell number and are expressed as the mean and standard error of the mean (SEM) of triplicate assays. Appropriate positive and negative controls and buffer blanks were used. Host strain Y190 containing pAS2-ORF3 or pACT2-ORF2 was used as a positive control (35) for the yeast two-hybrid analysis.
DNA binding assay.
A reaction mixture consisting of 1 ng of 5′ 32P-labeled IMYMV A origin DNA (216 bp) and either Rep protein containing amino acids (aa) 1 to 362 (Rep1-362aa protein) or Rep1-133aa protein in a binding buffer containing 10 mM Tris-HCl (pH 8), 300 mM NaCl, 5 mM MgCl2, 2.5 mM DTT, 1 mM EDTA, and 3% glycerol was incubated at 37°C for 30 min. An aliquot of the reaction mixture was resolved by 8% PAGE and analyzed by autoradiography. About 20 to 60 ng of unlabeled origin DNA was used as a cold competitor.
In the presence of PCNA, the origin DNA binding activity of Rep1-133aa or Rep1-362aa was analyzed by using nitrocellulose filters (DAWP 02500, 0.65 μm-pore size; Millipore). The 50-μl reaction mixture consisted of 1 ng of 5′ 32P-labeled IMYMV A origin DNA (216 bp), PCNA, and either Rep1-133aa or Rep1-362aa in a binding buffer containing 20 mM Tris-HCl (pH 8.0), 25 mM KCl, 1 mM DTT, 0.1 μg of BSA/μl, 0.02% poly(dI-dC), and 10% dimethyl sulfoxide. After 30 min of incubation at 37°C, the reaction mixture was filtered through prewashed nitrocellulose filters in the Manifold system. The filters then were washed with 10 volumes of binding buffer and were allowed to dry under a lamp. The extent of origin DNA bound by Rep proteins or Rep-PCNA complexes was calculated from the Cerenkov counts retained on the filters, as read with the multipurpose scintillation counter (Beckman LS 6500).
Molecular modeling of tobacco PCNA.
A Rasmol (2.7.2) display of the three-dimensional (3D) model of the tobacco PCNA monomer was obtained by using the ProMod method in Swiss Model, an automated protein modeling server (27) based on the 3D structures of homologous genes deposited in the Brookhaven Protein Databank (9, 32).
RESULTS
IMYMV Rep binds to pea nuclear PCNA.
About 10 μg of recombinant six-His-tagged Rep was immobilized in an Ni-NTA-agarose column through which about 400 μg of pea nuclear extract was chromatographed. The loosely bound proteins were eluted in the presence of 1% Triton X-100, 10 mM imidazole, and 150 mM NaCl. The firmly bound proteins were step eluted with 250 mM imidazole and were subjected to Western analysis with rabbit polyclonal antibodies (diluted 1:10,000) raised against 67-kDa recombinant pea GST-PCNA. To quantitate the amount of bound PCNA, about 50 ng of 67-kDa recombinant GST-PCNA was exogenously added to the eluted nuclear proteins, as shown in lane 2 of Fig. 1. It appeared that about 4 μg of pea nuclear PCNA was bound by 10 μg of immobilized IMYMV Rep. Similar results were obtained with nuclear PCNA derived from Nicotiana tabacum. Since tobacco PCNA was about 96% identical to pea PCNA, the rabbit antibodies raised against recombinant pea GST-PCNA recognized the PCNA proteins from both sources with almost equal efficiencies.
FIG. 1.
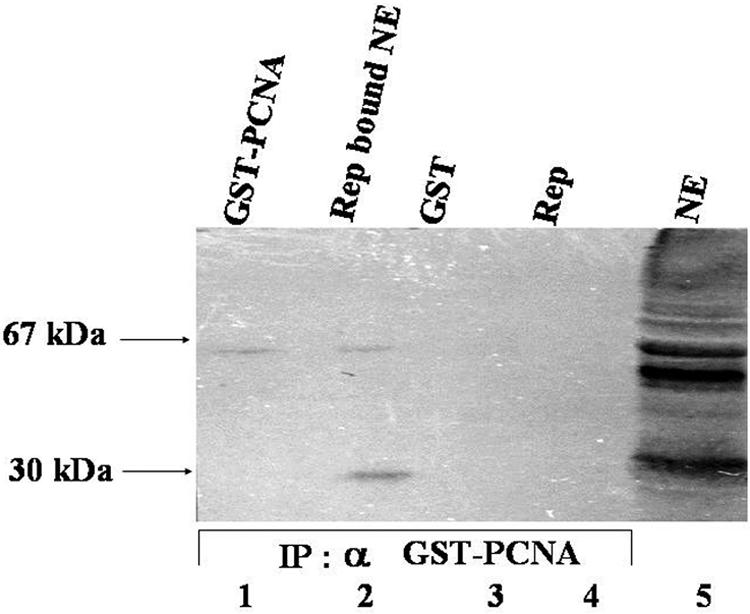
IMYMV Rep binds to pea nuclear PCNA. Lane 1 shows ∼50 ng of GST-PCNA. About 2 μg each of 27-kDa GST (lane 3) and 43-kDa Rep (lane 4) was used to reveal the specificity of the antibodies. Input pea nuclear extract (100 μg), resolved by SDS-10% PAGE and stained with Coomassie blue, is shown in lane 5. NE, nuclear extract; IP, immunoprobe.
Expression of recombinant tobacco PCNA.
In order to further characterize the interactions and map the domains of association, wild-type as well as various mutant tobacco PCNA proteins were produced in the recombinant fashion. Since the recombinant proteins carried N-terminal tags, they were purified by using the appropriate affinity matrix. Figure 2A shows purified GST-tagged tobacco PCNA and six-His-tagged tobacco PCNA in lanes 4 and 5, respectively, and the two site-specific mutant proteins in lanes 6 and 7 as representative examples of the mutant proteins. All of the mutants listed in Table 3 were purified to apparent homogeneity and were tested for their functional roles in DNA synthesis as well as binding to IMYMV Rep. A partially purified nuclear extract containing DNA polymerase δ activity was isolated from pea leaves. The proteins in this extract were capable of copying the M13mp18 single-stranded DNA annealed with a 17-mer oligonuleotide primer. The copying reaction, i.e., DNA synthesis, was enhanced in the presence of GST-tagged wild-type tobacco PCNA in a dose-responsive manner, as shown in Fig. 2B. The K13Q mutant, believed to be defective in associating with DNA polymerase δ, failed to enhance extract-mediated DNA synthesis. The K14E and K20T mutants were also similarly incapacitated in activating DNA synthesis. The L52A and G127A mutants were almost as effective as wild-type PCNA. The influences of all of the other mutants were also tested; the effects of only some of these are shown in Fig. 2B as representative examples.
FIG. 2.

Purification and characterization of PCNA mutants. (A) Expression of recombinant tobacco PCNA. Various protein fractions were analyzed by SDS-10% PAGE and visualized by Coomassie blue staining. Lanes 2, 3, and 1 each contain about 100 μg of protein from lysates of E. coli BL21(DE3) cells that harbored induced GST-PCNA, induced six-His-tagged PCNA, and uninduced protein, respectively. Lanes 4 and 5 contain PCNA proteins apparently purified as GST-tagged and six-His-tagged fusions, respectively. Representative PCNA mutants are shown in lanes 6 and 7. Arrows indicate molecular mass standards. (B) DNA polymerase stimulatory (DNA synthesis) activity of purified PCNA proteins. The different curves represent data obtained with various PCNA proteins. WT, wild type. Error bars indicate SEMs.
TABLE 3.
Description of PCNA mutants
| PCNA | Descriptiona | Interaction with Rep in the following assayb
|
|
|---|---|---|---|
| In vitro pull-down (mean ± SEM) | Yeast two hybrid | ||
| Wild type | 264-aa homotrimeric ring-shaped protein with charged amino acids at ends | 100.0 ± 5.2 | 100.0 |
| K13Q | In DNA Pol δ binding domain; charge alteration | 49.6 ± 13.9 | 43.0 |
| K13R | In DNA Pol δ binding domain; charge conservation; bulky side chain | 100.0 ± 12.0 | 86.0 |
| K14E | In DNA Pol δ binding domain; charge alteration | 84.3 ± 17.9 | 68.0 |
| K20T | In DNA Pol δ binding domain; charge alteration | 65.9 ± 12.1 | |
| L52A | Present on exposed β sheet; likely to mediate protein-protein interactions | 116.2 ± 4.0 | |
| L52R | Present on exposed β sheet; charge imbalance; bulky side chain | 76.5 ± 12.1 | |
| Δ(FE103,104) | Deletion of these two residues, which are on exposed β sheet, may disrupt interactions | 71.5 ± 14.7 | |
| G127A | Present in interdomain connector loop (IDCL), which interacts with other PCNA-interacting proteins; substitution of glycine leads to helix stability | 253.3 ± 14.2 | |
| Y133A | Present in IDCL; bulky side chain is replaced by smaller residue | 100.4 ± 3.1 | |
| A252F | Present in PCNA oligomerization domain; alanine is substituted with aromatic heavy side chain | 79.2 ± 2.8 | 30.0 |
| P253A | Present in PCNA oligomerization domain; charge conservation and structural stability | 82.9 ± 22.8 | 75.0 |
| E256R | Present in PCNA oligomerization domain; charge imbalance and structural change | 79.6 ± 11.9 | |
| K13Q + A252F | Double mutant | 126.2 ± 8.2 | |
| L52R + A252F | Double mutant | 126.0 ± 14.0 | |
Pol, polymerase.
The intensities of bands in the pull-down assay were estimated by using a Fluor-S Multi-Imager with Quantity 1 (4.1.1) software. The percentage of Rep pulled down by GST-PCNA was calculated from the amount of Rep bound by a standard fixed amount of PCNA. The amount of Rep bound by wild-type PCNA was arbitrarily assigned a value of 100%. The levels of interaction in the yeast two-hybrid assay were estimated by using a liquid β-galactosidase (colorimetric) assay. The β-galactosidase activity of wild-type PCNA was arbitrarily assigned a value of 100.
Recombinant tobacco PCNA associates with recombinant Rep.
The binding between recombinant PCNA and six-His-tagged Rep was examined by Ni-NTA-agarose chromatography. About 6 μg of GST-tagged tobacco PCNA was chromatographed through the Ni-NTA-agarose matrix, in which recombinant IMYMV Rep was immobilized. The bound proteins were step eluted with salt followed by 250 mM imidazole. At 400 mM NaCl, some of the bound GST-PCNA proteins were eluted (Fig. 3A, lane 7), and the rest were coeluted with Rep (lane 5). As a control, E. coli proteins (about 100 μg) enriched with GST (20 μg) were chromatographed similarly through a separate Ni-NTA-agarose column in which Rep was immobilized. Lanes 3 and 4 of Fig. 3A show that the control proteins including GST failed to bind to the column.
FIG. 3.

PCNA binds to Rep. (A) In vitro association of recombinant tobacco PCNA with recombinant Rep. Bound PCNA was serially eluted with various concentrations of NaCl (lanes 6 to 8) and finally with 250 mM imidazole (ID) (lane 5). As a control, 100 μg of bacterial extract containing overexpressed GST protein was used. Most of these proteins did not bind to the column (lane 1). With 250 mM imidazole, only immobilized Rep was eluted (lane 4). Proteins in various chromatographic fractions were examined by SDS-12% PAGE and visualized by Coomassie blue staining. Fl, flowthrough; Mr., protein standards. Arrows show the banding patterns of important proteins. (B) Ex vivo association of recombinant tobacco PCNA with recombinant Rep. All experimental results shown are from yeast two-hybrid assays with either single or double transformants containing in-frame fusions of full-length constructs. Yellow boxes (BD) show the vector containing the Gal4 DNA binding domain, and aqua boxes (AD) show the same for the activation domain. Blue boxes show full-length IMYMV Rep. Red boxes show full-length PCNA. His−depicts the growth of yeast clones streaked on synthetic dextrose plates lacking histidine. β-Galactosidase (β-gal) activity was measured on filters and quantitated by a liquid-based assay, the relative activity units of which are shown in brackets. The strength of the positive interactions was determined by the ability of the cotransformants (10−4 dilution) to grow on a His− plate supplemented with 20 mM 3-AT (AT). ORF2 and ORF3 fusion constructs were used as positive controls. Diploids were obtained from genetic crosses of haploids (single transformants) and were tested for His prototrophy (DHis−).
The interaction between tobacco PCNA and IMYMV Rep was also examined with a yeast two-hybrid system. The PCNA and Rep proteins were fused in frame with the activation domain and the DNA binding domain of the Gal4 protein, respectively. Yeast cells harboring both kinds of fusion proteins were able to grow on His− plates. These cells also exhibited β-galactosidase activities (Fig. 3B, sixth row). Since the histidine and β-galactosidase reporter genes were downstream of the Gal4-responsive promoter, the expression of these reporter genes suggested the reconstitution of Gal4 activities due to the association between PCNA and Rep within the yeast cells. In these experiments, the open reading frame 2 (ORF2) and ORF3 proteins of hepatitis E virus were used as a pair of positive control partners (Fig. 3B, seventh row). The growth of cotransformed yeast colonies in the presence of 20 mM 3-AT and the high level of expression of β-galactosidase in these yeast cells indicated the relatively high strength of the interaction between PCNA and Rep. The unrestricted growth of all transformants shown in Fig. 3B was observed on yeast extract, peptone, dextrose plates. Neither plasmids encoding the fusion proteins alone (Fig. 3B, third and fourth rows) nor plasmid vectors in any combination (Fig. 3B, first, second, and fifth rows) were able to induce His3 or LacZ expression in yeast cells.
The specificity of the Rep-PCNA interaction was also confirmed with a yeast genetic approach (34). After genetic crossing of single transformants (haploids), the His3 prototrophy of diploid strains (a/α) was tested. Only diploids containing both constructs (i.e., binding domain fused to Rep and activation domain fused to PCNA) showed a positive phenotype similar to that of the diploid positive control (binding domain fused to ORF3 and activation domain fused to ORF2). From the results of all of the above experiments, we conclude that the Rep and PCNA proteins interacted with each other in the yeast two-hybrid system.
Interactions between IMYMY Rep and tobacco PCNA mutants.
In an attempt to locate the amino acid residues of PCNA responsible for the interaction with IMYMV Rep, a panel of site-specific mutants was constructed as shown in Table 3 and Fig. 4C. Each mutant was expressed in bacteria as the GST-tagged version, and the purified mutant was tested for its interaction with six-His-tagged Rep in a GST pull-down assay. The ratio of the intensities of Rep and mutant PCNA in the bound fraction was taken as a quantitative measure of the interaction between the partners. As a representative sample, the behavior of three mutants is shown in Fig. 4A. The L52R mutant was partially defective in the interaction, while the Y133A mutant was similar to wild-type PCNA. As shown in Table 3, the K13Q mutant was the most defective (yet non-null) in the interaction. The data shown in Fig. 4C and Table 3 suggested that PCNA probably interacts with Rep over a wide surface area and that the interacting residues are distributed on both the inner and the outer surfaces of the PCNA ring (Fig. 4C). For some mutants, such as L52A and K14E, alterations in the levels of the interactions seemed to be rather marginal. When the changes were ≤25%, at least eight repeats of the pull-down experiments were carried out to confirm the reproducibility of the marginal changes. Thus, although the data for the interactions of five of the mutants shown in Fig. 4C reflected marginal effects, the reported changes were statistically significant.
FIG.4.

PCNA mutants defective in interactions with Rep. (A) In vitro association of recombinant tobacco PCNA mutants with recombinant Rep. A silver-stained SDS-10% polyacrylamide gel shows fixed amounts of input proteins for six-His-tagged Rep and GST-PCNA mutants (lanes 1 to 4). Bound complexes of Rep and PCNA were separated and examined by SDS-PAGE. Lane 6 shows six-His-tagged Rep bound to wild-type (WT) GST-PCNA. Lanes 7, 8, and 9 show six-His-tagged Rep bound to GST-PCNA mutants L52A, L52R, and Y133A, respectively. The relative mobilities of standard molecular mass markers are shown in lane 5. (B) In vitro association of recombinant tobacco PCNA double mutants with recombinant Rep. A silver-stained SDS-10% polyacrylamide gel shows equal amounts of input proteins for six-His-tagged Rep and two GST-PCNA double mutants (lanes 1 to 3). Lane 5 shows six-His-tagged Rep bound to wild-type GST-PCNA. Lanes 6 and 7 show six-His-tagged Rep bound to GST-PCNA double mutants K13Q + A252F and L52R + A252F, respectively. The relative mobilities of standard molecular mass markers are shown in lane 4. (C) Locations of tobacco PCNA mutations on the 3D model of the tobacco PCNA monomer. The contacted amino acids are denoted by the single-letter code followed by the coordinate number and the changed amino acid. Values in parentheses indicate increased (↑), decreased (↓), or nearly equal (≈) interactions of mutant PCNA with IMYMV Rep relative to that of wild-type PCNA of tobacco.
In an attempt to identify a noninteracting mutant, two double mutants, K13Q + A252F and L52R + A252F, were isolated and assayed for interactions. Although each of the single site-specific mutants was partly defective in the Rep interaction, both of the double mutants showed enhanced interactions (Fig. 4B, lanes 6 and 7) compared to the single mutants as well as wild-type tobacco PCNA. In each case, mutation at one location surprisingly appeared to suppress the binding defect of the other mutant.
X-ray diffraction data are available for yeast PCNA and human PCNA. With these data, the structure of tobacco PCNA was modeled as shown in Fig. 4C. The putative interacting residues are shown, and changes in the levels of interactions of the mutants compared to the wild type are indicated.
Interactions between wild-type GST-PCNA and various deletion derivatives of Rep.
In order to find the region of Rep responsible for interactions with PCNA, various deletion derivatives of Rep were isolated and examined for interactions. Since the geminiviral Rep proteins have modular structures (13), the isolation of various deletions in IMYMV Rep was preferred for this study. All of the Rep deletion derivatives were N-terminally tagged with six histidines, and wild-type PCNA was tagged with GST. To detect pairwise interactions, GST pull-down assays were carried out as mentioned earlier. The C-terminal Rep fragment (aa 184 to 362) failed to bind PCNA (Fig. 5A, lane 5), but the N-terminal fragment (aa 1 to 183) bound PCNA tightly (lane 4). However, the fragment spanning the first 133 aa failed to bind PCNA (Fig. 5A, lane 3), but deletion of the first 119 aa (i.e., the fragment spanning aa 120 to 362) did not have any deleterious effect on binding (lane 2). Thus, the zone of Rep spanning aa 134 to 183 (or aa 120 to 183) seemed to be involved in binding to PCNA. Interestingly, the Rep peptide 69QTKNQRFF76 resembled the PIP box, which was presumed to be the target site for mediating the interaction with PCNA (34). Accordingly, a site-specific mutant with altered FF sites (i.e., FF75,76SS) within the full Rep protein backbone was designed and purified. Surprisingly, this mutant was as effective as wild-type Rep in binding to PCNA (Fig. 5A, lane 11). Hence, the presumed PIP box did not appear to be functional.
FIG. 5.

Delimitation of the Rep zone for interactions with PCNA. (A) In vitro associations of various deletion derivatives of Rep with recombinant tobacco PCNA. A Coomassie blue-stained SDS-12.5% polyacrylamide gel shows purified input proteins for GST-PCNA and six-His-tagged Rep (lanes 6 to 10). Lane 1 shows the full-length wild-type six-His-tagged Rep protein bound to GST-PCNA. Bound six-His-tagged Rep120-362aa and six-His-tagged Rep1-183aa are shown in lanes 2 and 4, respectively. Six-His-tagged Rep1-133aa and six-His-tagged Rep184-362aa are shown in input lanes (lanes 8 and 10) but not as bound and eluted proteins (lanes 3 and 5). Lanes 11 and 12 show bound (B) and input (I) six-His-tagged Rep1-362aa (FF75,76SS), respectively. Molecular mass standards (in kilodaltons) are shown in lane 13. (B) Various domains and motifs of IMYMV Rep and summary of IMYMV Rep and PCNA interactions. Various domains of Rep and the corresponding coordinates are indicated in the upper panel. The active residue (Y) of nicking-closing activity is shown. The nonfunctional PIP box is shown in an expanded manner; conserved residues are underlined. Interaction characteristics are summarized in the bottom panel; +++, ++, and − indicate the degrees of interactions (+++ and ++, presence; −, absence).
Figure 5B summarizes the modular structure of IMYMV Rep. The PCNA binding sites are indicated, along with other modules. Figure 5B also summarizes the interactions of PCNA with various Rep deletion derivatives. The descriptions of the deletions are provided in Table 4.
TABLE 4.
Description of Rep mutants
| Rep | Description | Interaction with PCNA in vitro (relative value) |
|---|---|---|
| Wild-type six-His-tagged Rep1-362aa | Initiates site-specific viral replication and terminates it by ligating the replication product | 100.0 |
| Six-His-tagged Rep1-362aa (FF75,76SS) | Two phenylalanines in the presumed PCNA signature motif69QTKNQRFFI76 are converted to serines | 100.0 |
| Six-His-tagged Rep1-183aa | N′-terminal half of Rep protein has DNA cleavage, DNA ligation, DNA binding, oligomerization, and REn interaction domains | 43.4 |
| Six-His-tagged Rep184-362aa | C′-terminal half of Rep protein has ATPase activity | 2.24 |
| Six-His-tagged Rep1-133aa | Rep deletion mutant retaining DNA binding activity and partial oligomerization domains | 0.0 |
| Six-His-tagged Rep120-362aa | Rep deletion mutant for DNA binding and cutting | 100.0 |
The Rep1-133aa mutant is functionally active.
In order to reinforce the conclusion regarding the PCNA binding zone of Rep, it was imperative to show that the deletion mutant Rep1-133aa had not lost its biochemical activity following the structural alterations caused by the large deletion. As shown in lanes 3 to 5 of Fig. 6A, the mutant retained its site-specific nicking activity. This observation is quite consistent with the fact that the active residue Y103 is left undisturbed by the deletion mutation. Lanes 3 and 4 of Fig. 6B show that the mutant protein bound to the IMYMV CR region, i.e., the region harboring the origin of replication (26), to cause the electrophoretic shift of the labeled CR region. This binding was also sequence specific, as revealed by competition analysis (data not shown).
FIG. 6.

Intact functional activity of Rep1-133aa. (A) Site-specific origin DNA cleavage activity of Rep1-133aa. An autoradiogram of a 15% polyacrylamide-urea gel shows the 5′ 32P-labeled 26-mer substrate containing the conserved origin region and the 18-mer cleaved products that were formed either with increasing amounts of six-His-tagged Rep1-133aa (lanes 3 to 5) or 5 μg of six-His-tagged Rep1-362aa (lane 2). Lane 1 shows the 5′ 32P-labeled 26-mer substrate alone. (B) Site-specific origin DNA binding activity of Rep1-133aa. An autoradiogram of a 6% polyacrylamide nondenaturing gel shows a shift of the 5′ 32P-labeled 216-bp DNA fragment (lane 1) containing the IMYMV origin region in the presence of either six-His-tagged Rep1-133aa (lanes 3 and 4) or 1 μg of full-length six-His-tagged Rep1-362aa (lane 2). (C) Oligomerization property of Rep1-133aa. Shown in the silver-stained SDS-12.5% polyacrylamide gel are purified input proteins for MBP-Rep and six-His-tagged Rep1-362aa (lanes 5 to 8). Proteins eluted from MBP chromatography are shown in lanes 1 to 4.
To examine whether Rep1-133aa retained oligomerization properties, the binding of six-His-tagged mutant Rep1-133aa to MBP-tagged wild-type Rep was probed in MBP pull-down assays with amylose resins. Lanes 2 to 4 of Fig. 6C show that the six-His-tagged Rep1-133aa deletion derivative retained its property to bind to MBP-tagged Rep1-362aa. Assuming that Rep-Rep interactions are mediated by the oligomerization domain (25), this result may imply that the oligomerization activity of the mutant was not lost. All of these facts taken together prove that the C-terminal deletion in Rep did not cause any drastic functional perturbations in the mutant protein. The mutant protein retained all of the biochemical functions compatible with its structural information but had lost the PCNA binding module.
PCNA inhibits the enyzmatic functions of Rep.
For RCR of the IMYMV DNA genome, the site-specific nicking (26) and ATPase activities of Rep are extremely important. Hence, we wanted to test the modulation of these activities in the presence of PCNA. Figure 7A shows that PCNA inhibited the nicking performance of wild-type Rep but did not influence the similar activity in six-His-tagged Rep1-133aa to a great extent. This observation indicates a direct correlation between the physical binding of protein partners and the functional inactivation of Rep. Rep also shows ATPase activity because of its C-terminal Walker A and Walker B motifs (Fig. 5B). The Rep-mediated release of radioactive phosphate (Pi) from [γ-32P]ATP was monitored on TLC plates as shown in Fig. 7B. PCNA also inhibited ATPase activity in a dose-dependent fashion (Fig. 7B, lanes 2 to 4). Since the Rep 1-133 mutant lacks the Walker A and Walker B motifs, it did not show any ATPase activity, as expected (Fig. 7B, lanes 5 to 8). Thus, on the basis of the observations in Fig. 7A and B, it appeared that PCNA may have a regulatory role in controlling Rep-mediated RCR of the IMYMV genome.
FIG. 7.

Influence of PCNA on Rep activity. (A) Rep-dependent cleavage activity in the presence of PCNA. An autoradiogram of a 15% polyacrylamide-urea gel shows the 5′ 32P-labeled 26-mer substrate containing the conserved origin region (lane 1) and the 18-mer cleaved product with 1 μg of six-His-tagged Rep1-362aa (lane 2) and 1.5 μg of six-His-tagged Rep1-133aa (lane 2, right panel). The amounts of six-His-tagged Rep1-362aa-mediated cleavage products obtained in the presence of increasing amounts (2, 5, and 10 μg) of GST-PCNA are shown in lanes 3 to 5, respectively, of the left panel. Similarly, the amounts of six-His-tagged Rep1-133aa-mediated cleavage products obtained in the presence of 5 and 15 μg of PCNA are shown in lanes 3 and 4, respectively, of the right panel. Lane 5 (right panel) shows that the substrate remained unchanged following incubation with 10 μg of GST-PCNA. (B) Rep-dependent ATPase activity in the presence of GST-PCNA. An autoradiogram of the results of an ATPase assay performed with a TLC sheet shows the release of labeled free phosphate with 1.5 μg of six-His-tagged Rep1-362aa (lanes 1 to 4) or 1.5 μg of six-His-tagged His-Rep1-133aa (lanes 5 to 8). Increasing amounts (0, 1, 2, and 5 μg) of GST-PCNA were added to lanes 1, 2, 3, and 4 and to lanes 5, 6, 7, and 8, respectively. Lane 9 contained the labeled ATP probe only. (C) Origin DNA binding activity of Rep in the presence of PCNA. Panel i shows the percentage of 5′ 32P-labeled IMYMV origin DNA bound by Rep. The bound DNA was retained on the nitrocellulose filter with either six-His-tagged Rep1-362aa or six-His-tagged Rep1-133aa in the presence of increasing amounts of PCNA. Panel ii shows the percentage of 5′ 32P-labeled origin DNA retained on the nitrocellulose filter with no protein, 4 μg of GST-PCNA, 1 μg of six-His-tagged Rep1-362aa, and 0.5 μg of six-His-tagged Rep1-133aa, from left to right, respectively. Error bars indicate SEMs.
However, not all of the biochemical functions of Rep were downregulated by PCNA. The sequence-specific binding of Rep to IMYMV origin DNA was analyzed in DNA binding assays (26). At low concentrations of PCNA (5 to 10 μg/ml), Rep-mediated origin DNA binding was enhanced in the presence of PCNA (Fig. 7C, panel i). However, the enhancement was hardly recognizable when Rep1-133aa was used in the DNA binding assay, reflecting the lack of interaction of Rep1-133aa with PCNA. Figure 7C also revealed that Rep1-133aa showed better binding to the CR region than did wild-type Rep.
DISCUSSION
Since PCNA is induced in response to geminivirus infection, we argued that it would be used for virus-DNA specific replication. To address the issue, we examined the biochemical interaction between recombinant IMYMV Rep and recombinant PCNA of either legume or solanaceous origin. A strong interaction between the protein partners was observed in the in vitro pull-down and yeast two-hybrid assays. A zone in Rep that spanned aa 134 to 183 and was responsible for the interaction with PCNA was identified, whereas the interacting residues of PCNA were spread throughout PCNA. As a result of this interaction, recombinant Rep was incapacitated in its site-specific nicking-closing activity and ATPase function. Since both of these activities are essential for RCR of viral genomes, it is tempting to suggest that PCNA is responsible for control of the viral DNA copy number within infected host plants. The interaction between the host plant PCNA protein and the Rep protein of begomoviruses may be of general significance; i.e., the reported interaction may not reflect any speciality or peculiarity of IMYMV Rep. Castillo et al. demonstrated that the tomato PCNA protein (Le-PCNA) interacted with the Rep protein of tomato yellow leaf curl sardina virus (TYLCSV Rep) when the proteins were coexpressed in Schizosaccharomyces pombe (4). However, that particular study did not address the mapping of the interaction domains or the biochemical effects of the interaction on the viral Rep protein.
There were several reasons for choosing pea or tobacco PCNA in the present investigation. First, PCNA of mung bean origin does not have any special advantage over pea or tobacco PCNA. A comparison of all plant PCNA sequences reveals that the plant PCNA proteins are extremely (>80%) homologous. Second, a partially dimeric construct of IMYMV DNA-A containing the green fluorescent protein reporter gene can replicate well in pea, French bean, or tobacco when the construct is delivered to the plant through agroinoculation (N. Mohammad et al., unpublished data). Such assays on transient replication suggest that pea or tobacco PCNA may be functionally equivalent to mung bean PCNA. Third, the interaction of IMYMV Rep with PCNA of pea (legume) was similar to that between of viral Rep with PCNA of nonlegume origin (tobacco). These facts suggest that the substitution of mung bean PCNA with either pea or tobacco PCNA was not unjustified.
For most studies of the Rep-PCNA interaction, in vitro pull-down assays were used; yeast two-hybrid analyses were carried out only on some occasions. Data obtained by these two types of assays agree reasonably well and suggest a strong interaction between Rep and PCNA. The trimeric PCNA protein is structurally very sensitive, and hence no deletion mutants were used for probing the nature of the interaction. We were surprised at the results obtained with double mutants, which indicated that mutation at one allele could act as a suppressor mutation for the other locus when both are present at spatially distant locations. Hence, we restricted the interaction analysis to only site-specific single amino acid changes in PCNA. However, for Rep, we resorted to a deletion analysis. From studies of naturally available recombinant geminiviruses, a consensus regarding the modular structure of Rep proteins emerged in view of the abundance of recombination between the Rep proteins of various geminiviruses. The concept of the modular nature of the IMYMV Rep protein (Fig. 5B) was also reinforced in the present study, since various deletion mutants with intact functional properties could be generated. These mutants showed a loss of function corresponding to the deleted regions only. The mutant Rep1-133aa failed to interact with PCNA, thus delimiting the interaction zone to between aa 134 and 183 of Rep of IMYMV. However, this mutant retained site-specific DNA binding, nicking-closing activity and oligomerization activity. Since this mutant lost both ATPase activity and binding capacity to PCNA, which are perhaps extremely important for viral replication, it may function in a dominant-negative manner for IMYMV DNA replication. This idea needs to be tested by screening of IMYMV-resistant mung bean plants that could be generated by using Rep1-133aa DNA as the transgene.
RCR of the viral genome is extremely dependent on the site-specific nicking and ATPase activities of the Rep protein. Since PCNA dampened both of these activities, PCNA appeared to be an inhibitor of RCR. While this inhibitory activity may be important for limiting the viral DNA copy number, PCNA would supposedly find a way to perform its usual role as an accessory of DNA polymerase δ for elongation of the viral DNA replication fork. At present, we do not have any evidence suggesting how this happens, but a speculative scenario can be proposed. Rep binds to PCNA (this report) as well as RF C (23); the latter helps load PCNA eventually at the 3′ end of the primer for replication synthesis. From the three-way protein interaction of Rep, PCNA, and RF C, a two-way complex of PCNA and RF C may form later as a result of high affinity between these two components. In this way, Rep can be freed kinetically to perform site-specific nicking at the replication origin. RF C can load PCNA subsequently at the 3′OH end of nicked DNA, which may serve as the primer for DNA polymerases. Thus, in this model, PCNA may play a dual role. During the initial phase of viral DNA replication, when the Rep protein may be relatively more abundant, the Rep-PCNA-RF C complex is likely to form; subsequently, the complex may disassemble to allow for primer formation and elongation. However, in the late phase of viral replication, the PCNA protein concentration may increase markedly and may inhibit the initiation of RCR by Rep, thus controlling the viral DNA copy number inside the infected plant nucleus.
Acknowledgments
We thank S. Tyagi for assistance with yeast two-hybrid work. The assistance of D. Gupta with PCNA modeling is greatly appreciated. S. Chattopadhyay assisted with densitometric scanning.
Partial financial help from DBT, New Delhi, India (to S.K.M.), and CSIR, New Delhi, India (to B.B.), is acknowledged.
REFERENCES
- 1.Ach, R. A., T. Durfee, A. B. Miller, P. Taranto, L. Hanley-Bowdoin, P. C. Zamryski, and W. Gruissem. 1997. RRB1 and RRB2 encode maize retinoblastoma-related proteins that interact with a plant D-type cyclin and geminivirus replication protein. Mol. Cell. Biol. 17:5077-5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bai, C., and S. J. Elledge. 1996. Gene identification using the yeast two-hybrid system. Methods Enzymol. 273:331-347. [DOI] [PubMed] [Google Scholar]
- 3.Brand, S. R., R. M. Bernstein, and M. B. Mathews. 1994. Trimeric structure of human proliferating cell nuclear antigen. Implications for enzymatic function and autoantibody recognition. J. Immunol. 153:3070-3078. [PubMed] [Google Scholar]
- 4.Castillo, A. G., D. Collinet, S. Deret, A. Kashoggi, and E. R. Bejarano. 2003. Dual interaction of plant PCNA with geminivirus replication accessory protein (Ren) and viral replication protein (Rep). Virology 312:381-394. [DOI] [PubMed] [Google Scholar]
- 5.Dignam, J. D., R. M. Lebovitz, and R. G. Roeder. 1983. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11:1475-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Egelkrout, E. M., D. Robertson, and L. Hanley-Bowdoin. 2001. Proliferating cell nuclear antigen transcription is repressed through an E2F consensus element and activated by geminivirus infection in mature leaves. Plant Cell 13:1437-1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Egelkrout, E. M., L. Mariconti, S. B. Settlage, R. Cella, D. Robertson, and L. Hanley-Bowdoin. 2002. Two E2F elements regulate the proliferating cell nuclear antigen promoter differently during leaf development. Plant Cell 14:3225-3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fukuda, K., H. Morioka, S. Imajou, S. Ikeda, E. Ohtsuka, and T. Tsurimoto. 1995. Structure-function relationship of the eukaryotic DNA replication factor, proliferating cell nuclear antigen. J. Biol. Chem. 270:22527-22534. [DOI] [PubMed] [Google Scholar]
- 9.Guex, N., and M. C. Peitsch. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714-2723. [DOI] [PubMed] [Google Scholar]
- 10.Gulbis, J., M. Z. Kelman, J. Hurwitz, M. O'Donnell, and J. Kuriyan. 1996. Structure of the C-terminal region of p21WAF1/CIP1 complexed with human PCNA. Cell 87:297-306. [DOI] [PubMed] [Google Scholar]
- 11.Gutierrez, C. 2000. DNA replication and cell cycle in plants: learning from geminiviruses. EMBO J. 19:792-799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gutierrez, C. 2000. Geminiviruses and the plant cell cycle. Plant Mol. Biol. 43:763-772. [DOI] [PubMed] [Google Scholar]
- 13.Hanley-Bowdoin, L., S. B. Settlage, B. M. Orozco, S. Nagar, and D. Robertson. 2000. Geminiviruses: models for plant DNA replication, transcription, and cell cycle regulation. Crit. Rev. Biochem. Mol. Biol. 35:105-140. [PubMed] [Google Scholar]
- 14.Hong, Y., J. Stanley, and R. Wezel. 2002. Novel system for the simultaneous analysis of geminivirus DNA replication and plant interactions in Nicotiana benthamiana. J. Virol. 77:13315-13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hop, D. V., A. Gaikwad, B. S. Yadav, M. K. Reddy, S. Sopory, and S. K. Mukherjee. 1999. Suppression of pea nuclear topoisomerase I enzyme activity by pea PCNA. Plant J. 19:153-162. [DOI] [PubMed] [Google Scholar]
- 16.James, P., J. Halladay, and E. A. Craig. 1996. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 144:1425-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jansen-Durr, P. 1996. How viral oncogenes make the cell cycle. Trends Genet. 12:270-275. [DOI] [PubMed] [Google Scholar]
- 18.Kelman, Z., and M. O'Donnell. 1995. Structural and functional similarities of prokaryotic and eukaryotic DNA polymerase sliding clamps. Nucleic Acids Res. 23:3613-3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kelman, Z. 1997. PCNA: structure, functions and interactions. Oncogene 14:629-640. [DOI] [PubMed] [Google Scholar]
- 20.Kong, L. J., and L. Hanley-Bowdoin. 2002. A geminivirus replication protein interacts with a protein kinase and a motor protein that display different expression patterns during plant development and infection. Plant Cell 14:1817-1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kong, L. J., B. M. Orozco, J. L. Roe, S. Nagar, S. Ou, H. S. Feiler, T. Durfee, A. B. Miller, W. Gruissem, D. Robertson, and L. Hanley-Bowdoin. 2000. A geminivirus replication protein interacts with the retinoblastoma protein through a novel domain to determine symptoms and tissue specificity of infection in plants. EMBO J. 19:3485-3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lane, M. E., K. Sauer, K. Wallace, Y. N. Jan, C. F. Lehner, and H. Vaessin. 1996. Dacapo, a cyclin-dependent kinase inhibitor, stops cell proliferation during Drosophila development. Cell 87:1225-1235. [DOI] [PubMed] [Google Scholar]
- 23.Luque, A., A. P. Sanz-Burgos, E. Ramirez-Parra, M. M. Castellano, and C. Gutierrez. 2002. Interaction of geminivirus Rep protein with replication factor C and its potential role during geminivirus DNA replication. Virology 302:83-94. [DOI] [PubMed] [Google Scholar]
- 24.Maga, G., and U. Hubscher. 2003. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J. Cell Sci. 116:3051-3060. [DOI] [PubMed] [Google Scholar]
- 25.Orozco, B. M., L. J. Kong, L. A. Batts, S. Elledge, and L. Hanely-Bowdoin. 2000. The multifunctional character of replication protein is reflected by its complex oligomerization properties. J. Biol. Chem. 275:6114-6122. [DOI] [PubMed] [Google Scholar]
- 26.Pant, V., D. Gupta, N. R. Choudhury, V. G. Malathi, A. Varma, and S. K. Mukherjee. 2001. Molecular characterization of the Rep protein of the blackgram isolate of Indian mung bean yellow mosaic virus. J. Gen. Virol. 82:2559-2567. [DOI] [PubMed] [Google Scholar]
- 27.Peitsch, M. C. 1995. Protein modeling by E-mail. Bio/Technology 13:658-660. [Google Scholar]
- 28.Podust, L. M., V. N. Podust, J. M. Sogo, and U. Hubscher. 1995. Mammalian DNA polymerase auxiliary proteins: analysis of replication factor C-catalyzed proliferating cell nuclear antigen loading onto circular double-stranded DNA. Mol. Cell. Biol. 15:3072-3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raghavan, V., P. S. Malik, N. R. Choudhury, and S. K. Mukherjee. 2004. The DNA-A component of a plant geminivirus (Indian mung bean yellow mosaic virus) replicates in budding yeast cells. J. Virol. 78:2405-2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sambrook, J., E. F. Fritsch, and T. Maniatis (ed.). 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 31.Saunders, K., A. Lucy, and J. Stanley. 1991. DNA forms of the geminivirus African cassava mosaic virus consistent with a rolling circle mechanism of replication. Nucleic Acids Res. 19:2325-2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schwede, T., J. Kopp, N. Guex, and M. C. Peitsch. 2003. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31:3381-3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stenger, D. C., G. N. Revington, M. C. Stevenson, and D. M. Bisaro. 1991. Replicational release of geminivirus genomes from tandemly repeated copies: evidence for rolling-circle replication of a plant viral DNA. Proc. Natl. Acad. Sci. USA 88:8029-8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tyagi, S., and S. K. Lal. 2000. Combined transformation and genetic technique verification of protein-protein interactions in the yeast two-hybrid system. Biochem. Biophys. Res. Commun. 277:589-593. [DOI] [PubMed] [Google Scholar]
- 35.Tyagi, S., H. Korkaya, M. Zafrullah, S. Jameel, and S. K. Lal. 2002. The phosphorylated form of the ORF3 protein of hepatitis E virus interacts with its non-glycosylated form of the major capsid protein, ORF2. J. Biol. Chem. 277:22759-22767. [DOI] [PubMed] [Google Scholar]
- 36.Varma, A., and V. G. Malathi. 2003. Emerging threats of gemininvirses: a serious threat to crop production. Ann. Appl. Biol. 142:145-164. [Google Scholar]
- 37.Warbrick, E., W. Heatherington, D. P. Lane, and D. M. Glover. 1998. PCNA binding proteins in Drosophila melanogaster: the analysis of a conserved PCNA binding domain. Nucleic Acids Res. 26:3925-3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Warbrick, E. 2000. The puzzle of PCNA's many partners. Bioessays 22:997-1006. [DOI] [PubMed] [Google Scholar]
- 39.Xu, H., P. Zhang, L. Liu, and M. Y. Lee. 2001. A novel PCNA-binding motif identified by the panning of a random peptide display library. Biochemistry 40:4512-4520. [DOI] [PubMed] [Google Scholar]
- 40.Zhang, H. S., and D. C. Dean. 2001. Rb-mediated chromatin structure regulation and transcriptional repression. Oncogene 20:3134-3138. [DOI] [PubMed] [Google Scholar]