

Abstract
Cyclic Immunofluorescence (CycIF) is a public domain method for performing highly multiplexed immunofluorescence imaging using a conventional epifluorescence microscope. It uses simple reagents and existing antibodies to construct images with up to 30 channels by sequential 4–6 channel imaging followed by fluorophore inactivation. Three variant methods are described, the most generally useful of which involves staining fixed cells with antibodies directly conjugated to Alexa Fluor dyes and imaging in four colors, inactivating fluorophores using a mild base in the presence of hydrogen peroxide and light and then performing another round of staining and imaging. Cell morphology is preserved through multiple rounds of CycIF and signal to noise ratios appear to increase. Unlike antibody-stripping methods, CycIF is gentle and optimized for monolayers of cultured cells. A second protocol involves indirect immunofluorescence and a third enables chemical inactivation of genetically encoded fluorescent proteins, allowing multiplex immunofluorescence to be combined with live-cell analysis of cells expressing fluorescent reporter proteins.
Keywords: CycIF, immunofluorescence, high-content imaging, multiplexing, systems biology, Signal transduction, multiplex single-cell imaging
INTRODUCTION
The recent development of methods for very high-plex (20–100 channel) flow cytometry and single-cell sequencing has emphasized the value of making many simultaneous measurements on each cell in a population (Gaudet & Miller-Jensen, 2016). Multiplex single-cell data reveals correlations between measurements from which it is possible to infer causal relationships, reconstruct developmental pathways and infer some aspects of kinetics.
In principle, immunofluorescence microscopy is well suited to multiplex single-cell analysis. It is a simple, well-established means for determining morphology and measuring the levels, states of modification and localization of intracellular and extracellular proteins in cultured cells and tissues. Immunofluorescence is also compatible with RNA and DNA in situ hybridization (ISH) and other methods for assaying nucleic acids. Finally, high-content imaging is well suited to high-throughput screening of small molecules, siRNA and CRISPR guides that induce specific molecular and phenotypic changes (Zanella, Lorens, & Link, 2010). Using conventional microscopes and reagents, immunofluorescence imaging is typically limited to 4–6 channels, above which cross-talk between fluorophores becomes a problem. The use of fluorescent protein reporters further limits this because such proteins typically have broad excitation spectra and small Stokes shifts. Multiple methods have been developed for increasing the multiplicity of immunofluorescence imaging, most of which involve specialized instrumentation, such as multi-spectral microscopes (Stack, Wang, Roman, & Hoyt, 2014) or reagents (Frei et al., 2016). In contrast, CycIF is a simple and open-source method which uses standard reagents and conventional microscopes.
STRATEGIC PLANNING
Choosing dye-antibody conjugates
Several variants of CycIF are possible involving either direct immunofluorescence with dye-conjugated antibodies or indirect immunofluorescence with secondary antibodies (Figure 1). Direct detection is preferred because the multiplexity of indirect immunofluorescence is constrained by the isotypes and species reactivity of secondary antibodies: unless protease-mediated antibody stripping is employed, only one primary species/isotype is possible per experiment (e.g., one rabbit, one goat, one mouse primary antibody etc.). Antibody stripping is a harsh procedure with several disadvantages: (1) protease digestion degrades antigens and damages cell structures, particularly with cells grown as monolayers. (2) antibodies with different isotypes or host species are digested at different rates, and (3) protease-stripping methods need to be optimized for each conjugate, a laborious procedure.
Figure 1. Three ways to achieve highly multiplexed read-outs in single cells.
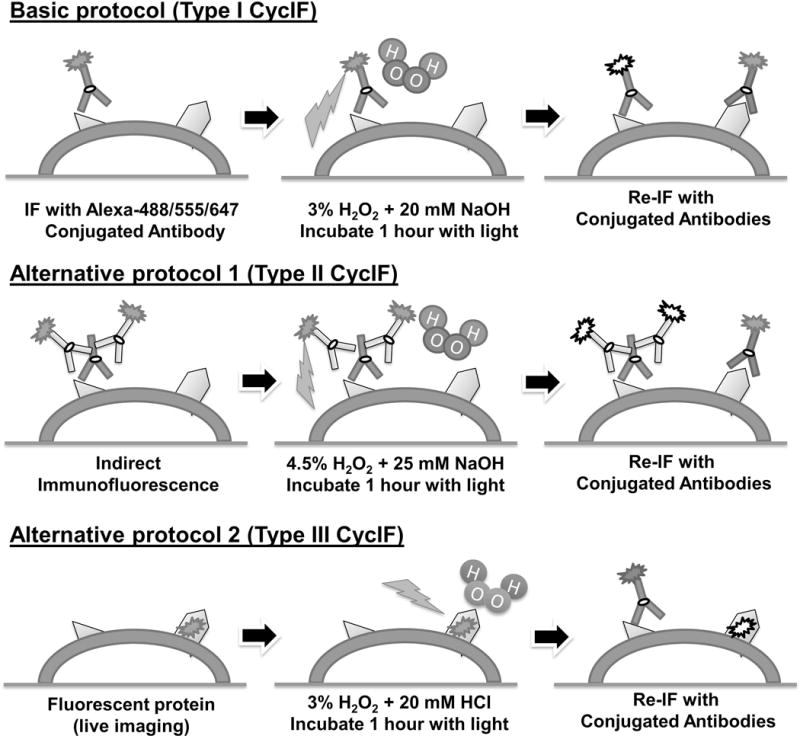
See text for details.
A drawback of fluorophore-conjugated antibodies is that signals are generally weaker than those generated by indirect immunofluorescence (in which multiple secondary antibodies can bind the primary antibody) making the detection of low abundance epitopes more difficult. The process of crosslinking dyes to antibodies (typically using lysine-reactive esters) also has the potential to modify and damage antigen combining domains. Nonetheless we have had good luck with commercially available antibodies conjugated to Alexa Fluor® 488/555/647 fluorophores, which are substantially brighter and more photo-stable than antibodies conjugated to FITC/Cy3/Cy5 dyes (Panchuk-Voloshina et al., 1999). Many vendors provide Alexa Fluor conjugated antibodies that have been tested for immunofluorescence or immunohistochemistry applications. The use of commercial reagents also facilitates reproducible application of CycIF across different laboratories. Commercial antibodies validated for CycIF applications are listed in Table 1 and a website hosted by the HMS LINCS Center maintains an up-to-date list (http://lincs.hms.harvard.edu/lin-NatCommun-2015/).
Table 1.
Antibodies tested in the CycIF protocol
| Alexa-488/FITC conjugated | Alexa-555/TRITC conjugated | Alexa-647 conjugated |
|---|---|---|
|
*** p-ERK1/2 T202/Y204 (CST #4344, Lot #12): 1:200 |
*** p-Rb S807/S811 (CST #8957, Lot #1): 1:400 |
*** p21 Waf1/Cip1 (CST #8587, Lot #3): 1:200 |
|
*** EGFR (CST #5616, Lot #4): 1:400 |
*** p-Histone H3 S10 (CST #3475, Lot #2): 1:800 |
*** p-S6 S235/S236 (CST #4851, Lot #22): 1:400 |
|
*** Lamin A/C (CST #8617, Lot #2): 1:400 |
***β-Actin (CST #8046, Lot #1): 1:200 |
*** beta-Tubulin (CST #3624, Lot #4): 1:200 |
|
*** p-S6 S240/S244 (CST #5018, Lot #4): 1:800 |
*** VEGFR2 (CST #12872, Lot #1): 1:400 |
*** beta-Catenin (CST #4627, Lot #5): 1:400 |
|
*** PCNA (CST #8580, Lot #1): 1:400 |
*** Vimentin (CST #9855, Lot #1): 1:200 |
*** mTOR (CST #5048, Lot #2): 1:300 |
|
*** Ki-67 (CST #11882, Lot #4): 1:400 |
*** p-S6 S235/S236 (CST #3985, Lot #4): 1:300 |
*** pan-Akt (CST #5186, Lot #3): 1:400 |
|
*** Cyclin D1 (AB #AB190194, Lot #GR199456-1): 1:400 |
*** p-AuroraABC (CST #13464, Lot #1): 1:200 |
*** p65 NFkB (AB #AB190589, Lot #GR199457-1): 1:800 |
|
*** Bax (BIO #633603, Lot #B169774): 1:400 |
*** S6 (CST #6989, Lot #2): 1:200 |
*** p27 (AB #AB194234, Lot #GR200274-1): 1:400 |
|
** EpCAM (CST #5198, Lot #9): 1:100 |
** pan-Keratin (CST #3478, Lot #4): 1:200 |
p75 NGF Receptor (AB #AB195180, Lot #GR203573-1): 1:400 |
|
** c-JUN (AB #AB193780, Lot #GR203494-1): 1:400 |
** p-Histone H2A.X S139 (CST #8228, Lot #3): 1:200 |
*** p-H2.AX S139 (CST #9270, Lot #15): 1:400 |
|
** E-Cadherin (CST #3199, Lot #11): 1:200 |
** LC3A/B (CST #13173, Lot #1): 1:200 |
*** Vimentin (CST #9856, Lot #7): 1:800 |
|
* p-c-JUN (CST #12714, Lot #6) |
** ActinRed 555 (Invitrogen #R371112, Lot #1646656) |
*** CD45 (BIO #304020, Lot #B1810139): 1:400 |
|
* p-CREB (CST #9187, Lot #6) |
** cPARP (CST #6894, Lot #1): 1:200 |
*** p-H2.AX S139 (BIO #613407, Lot #B199199): 1:400 |
|
* p-HSP27 (CST #12172, Lot #1) |
** p21 Waf1/Cip1 (CST #8493, Lot: #2): 1:200 |
** p-Tyrosine (CST #9415, Lot #8): 1:100 |
|
* Cyclin B1 (SC #SC-752, Lot #K1008) |
* Sox2 (CST #5179, Lot #4) |
** Her2 (BIO #324412, Lot #B179768): 1:200 |
|
* cdc2/CDK1 (SC #SC-54, Lot #G0606) |
* Oct-4A (CST #4439, Lot #1) |
* FOXO3a (AB #AB196539, Lot #GR202407-1) |
|
* cMyc (SC #SC-40, Lot #B2813) |
* Bcl-2 (BIO #658705, Lot #B180139) |
Has not worked in any cell line or condition tested to date
Works in specific cells/conditions,
Works across all cell lines and conditions tested to date
CST = Cell Signaling Technology AB = Abcam BIO = BioLegend SC = Santa Cruz Biotechnology
In addition, many Alexa Fluor® based subcellular labeling products (e.g., NucRed 647 and ActinRed 555) are available from ThermoFisher/Invitrogen and are fully compatible with the chemical inactivation procedure used in CycIF. These are particularly useful for staining specific organelles and membranes.
Planning the sequence in which to use antibodies in multiple CycIF cycles
Increasing the number of channels in each round of CycIF is advantageous. However, with the microscopes we have, crosstalk is minimized and detection of weak signals optimized if each round of imaging is limited to four channels, one of which is occupied by the Hoechst dye present in every CycIF cycle. Users of this protocol might want to investigate this for themselves, since different microscopes and filter sets will have different degrees of spectral discrimination.
In CycIF one channel in each cycle is dedicated to a dye (typically Hoechst 33342 or DAPI) that stains all cells; this is used to monitor cell loss and register successive images. Specific antigens are then detected using antibodies conjugated to one of three Alexa 488/555/647 fluorophores. Each Alexa Fluor dye has a different intrinsic brightness and the achievable signal to noise ratio in each channel is also dependent on instrument-specific emission/excitation filters and properties of the light source. Alexa Fluor® 488 is the brightest (quantum yield: 0.92), Alexa Fluor® 647 the second brightest (quantum yield: 0.33) and Alexa Fluor® 555 the least bright (quantum yield: 0.1); all Alexa fluorophores outperform Cyanine-based fluorophores. In CycIF, Alexa Fluor® 488 is generally used for antibodies that give the weakest signals, most commonly phospho-specific antibodies, and Alexa Fluor® 555 is used for antibodies that generate the brightest signals. However, some biological samples have relatively high background fluorescence at shorter wavelengths, which reduces signal-to-noise ratios for Alexa Fluor® 488-conjugated antibodies. In contrast, auto-fluorescence at 647 nm is usually very low which could be served as an optional channel/fluorophore for weak signals as well.
Even well-designed filter sets result in some cross-talk between Alexa dyes and this must be kept in mind when combining antibodies. In general, it is not a good idea to combine antibodies that will stain very strongly with those that stain weakly, especially when patterns of staining (the localization of antigens) is similar. Fluorophore inactivation is nearly complete for Alexa 488/555/647 dyes (Lin, Fallahi-Sichani, & Sorger, 2015) making cross-talk between cycles less of a concern than cross-talk within each cycle (users new to the method should confirm this for themselves; we always image cells after fluorophore inactivation). As a general rule, we combine antibodies with similar signal strengths in each CycIF cycle. We find that antigen loss is minimal and that signal/noise ratio appears to increase with cycle number, perhaps because the sample is increasingly well blocked. Nonetheless we tend to start with low-signal antibodies (e.g., antibodies against phospho-proteins) in early cycles and then proceed to higher-signal antibodies (e.g. antibodies against cytoskeleton or abundant proteins) in subsequent cycles. When very precise registration of signals is required, it may be necessary to exchange dyes on a pair of antibodies and check for spectral shifts arising from optical aberrations and slight sample distortion as the number of CycIF cycles increases. A typical order of use for antibodies in a six-round CycIF experiment is listed in Table 2.
Table 2.
An example of 6-round CycIF experiment using both indirect and direct IF
| Round 1 | Round 2 | Round 3 | Round 4 | Round 5 | Round 6 | |
|---|---|---|---|---|---|---|
| Alexa 488 | Foxo3a(R)1 | p-ERK | CycD1 | p-S6(240) | Bax | PCNA |
| Alexa 555 | Actin-5552 | p-RB | p-Aurora | p-H3 | pan-S6 | Keratin |
| Alexa 647 | p53(m)3 | p21 | p27 | p-S6(235) | γH2ax | AKT |
Rabbit Foxo3a antibody plus Alexa-488 goat anti-rabbit IgG
ActinRed™ 555 ReadyProbes® Reagent from ThermoFisher/Invitrogen
Mouse p53 antibody plus Alexa-647 goat anti-mouse IgG
Tips for preventing cell loss during CycIF
The primary constraint on the number of CycIF cycles that can be performed involves cell loss during washing steps. When the procedure is working well, we observe detachment of less than 1–2% per cycle from cover slips or multi-well plates per CycIF rounds. Achieving this, requires prolonged fixation with a combination of paraformaldehyde (PFA) and methanol and gentle washes. It is also helpful to pre-coat plates with poly-lysine or collagen. The total duration of an experiment from the first to the last cycle should be minimized: when stored at 4°C in PBS, fixed cells gradually detach over time. Some users have reported “peeling” of cells from plates during the washing steps and we have traced this to excessive agitation. An automated plate washer, such as BioTek EL406, is highly recommended for gentle washing and dispensing (see below).
Special considerations for the first and last CycIF cycles
It is often advantageous to use indirect immunofluorescence in the first cycle of CycIF. In this case, secondary antibodies conjugated to Alexa dyes are inactivated following the usual CycIF procedure and subsequent cycles are performed using directly conjugated antibodies (see Alternate Protocol 1, Fig. 1). Non-antibody reagents such as fluorescently-labelled Phalloidin to stain actin, Annexin V to stain apoptotic cells and Mitotracker to measure mitochondrial function can also be incorporated into the CycIF procedure. Those dyes and fluorophores that cannot be efficiently inactivated should be used in the last CycIF cycle. It is important to note, however, that the integrity of some subcellular structures and organelles is compromised by fixation and/or peroxidation. For example, Mitotracker CMXRos (Invitrogen, Cat: M-7512) is sensitive to oxidation and must be used prior to fixation. In general, empirical testing is required to determine the compatibility of dyes and fluorophores with the first, middle and last CycIF cycles.
Incorporation of fluorescent reporter proteins
The rapid growth of live-cell reporters based on fluorescent proteins has opened many new avenues for single-cell research (Hoppe, Coutu, & Schroeder, 2014). In Alternate Protocol 2 a method is described for chemically inactivating fluorescent proteins that makes samples compatible with conventional CycIF (Fig. 2). By analyzing cells that express fluorescent reporters with CycIF it is possible to combine time-resolved live-cell imaging with highly multiplexed fixed-cell microscopy.
Figure 2. Experimental workflow with time estimations.
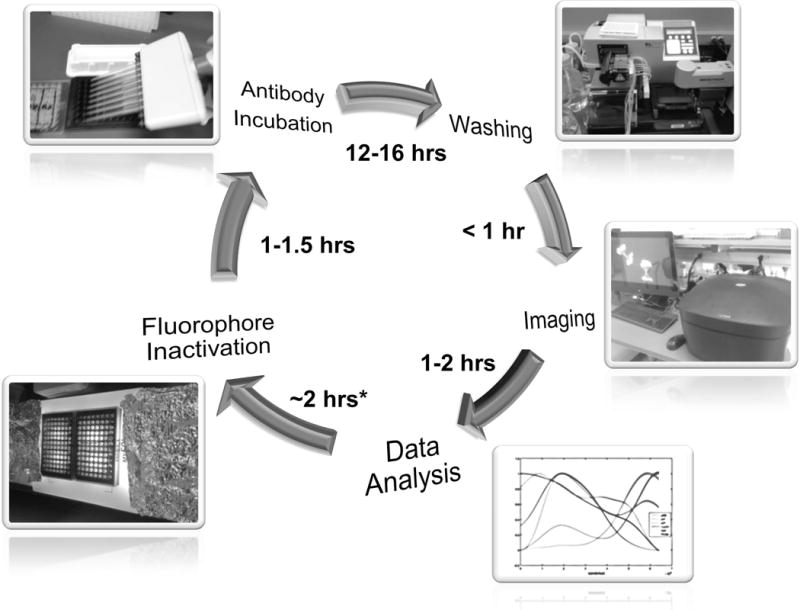
Illustrated here is a typical CycIF cycle. Start from the antibody incubation step and follow by washing, imaging and preliminary data analysis. Fluorophore inactivation is performed after the confirmation of image/data quality. One full cycle can be completed in 24 hours.
Choice of cell line and culture conditions
The protocols described below have been optimized for adherent cells grown in 96-well plates. To date, a wide variety of cell lines have been tested including, MCF7, MCF10A, COLO858, WM115, WM1552C, LOXIMVI, RPE1, human primary fibroblasts and human iPSCs. Cultured cells are generally seeded at 20–50% confluency and fixed and stained 2 to 3 days after seeding, which results in 70–90% final confluency. When cultures are over- or under- confluent increased detachment of cells is observed during CycIF. Optimal conditions for growing cells, time required for cells to achieve confluency and optimal conditions for performing biological assays are cell-line dependent and must be determined empirically. Further details are discussed in the Troubleshooting section. To minimize cell loss washes should be performed using a Biotek EL406 washer/dispenser using the protocol described below or, alternatively, a multichannel pipette with gentle technique.
Basic Protocol 1
Type I CycIF: Using antibodies directly conjugated to Alexa fluorophores
Materials
Antibodies against antigens of interest that have been directly conjugated to Alexa Fluor dyes; many such antibodies are available and those tested with CycIF to date are listed in Table 1.
16 % Paraformaldehyde/PFA (Electron Microscopy Sciences, Cat:15710): diluted 1:4 in PBS to make 4% PFA.
20× Phosphate-buffered Saline/PBS (Santa Cruz Biotech, Cat:SC-362299): Dilute 1:20 in milli-Q water to make 1× solution.
Methanol 99.9% (Fisher Scientific, Cat: AC61009), store in −20°C
Odyssey blocking buffer (LICOR Odyssey, Cat:927-40125)
Hoechst 33342 10 mg/mL (Invitrogen, Cat: H3570)
Hydrogen peroxide/H2O2 solution 30 wt. % in H2O (Sigma-Aldrich, Cat: 216763)
Sodium hydroxide/NaOH 1M solution, (Sigma-Aldrich, S5881)
Corning 96 well plates; clear-bottom; black (Sigma-Aldrich, Cat: CLS3603)
Biotek EL406 Washer/dispenser or multichannel pipetter
Microscope for imaging multi-well plates (e.g. GE Cytell Cell Imaging System).
-
Fixation: Fix cells in 96-well plates with 140 μl 4% PFA at room temperature for 30 min.
Extensive fixation is required. For loosely adherent cells the fixation time may be extended up to 1 hr.
-
Wash four times with 250 μl PBS per wash.
All washes should be as gentle as possible. A plate washer is recommended. A protocol for use of a Bio-Tek EL406 automated plate washer/dispenser is as follows:.
The following protocol is used for wash steps associated with CycIF in 96-well microplates.
- <Protocol begin>
- Step Details: W-Wash
- Pre-dispense before washing: No
- Bottom Wash: No
- Number of Wash Cycles: 4 Aspirate per cycle
- Vacuum Filtration: False
- Travel Rate: 3 CW 7.3 mm/sec
- Delay: 0 msec
- Z Offset: 35 steps (4.45 mm above carrier)
- X Offset: −40 steps (1.83 mm left of center)
- Y Offset: 0 steps (center of well)
- Secondary Aspirate: No
- Dispense per cycle
- Buffer: A Volume: 250 μL/well
- Flow Rate: 2
- Z Offset: 110 steps (13.97 mm above carrier)
- X Offset: −25 steps (1.14 mm left of center)
- Y Offset: 0 steps (center of well)
- Pre-dispense: not available
- Delay start of Vacuum until
- Volume dispensed: 300 μL/well
- Shake/Soak after dispense:
- Pre-dispense between cycles: No
- Final Aspirate: No
<Protocol end>
-
3
Permeabilization: Add 140 μl ice-cold methanol and allow plates to sit at room temperature for 10 min, then wash four times with 250 μl PBS per wash.
-
4
Blocking: Incubate the cells with 50 μl Odyssey blocking buffer at room temperature for 1 hr.
-
5
Staining: Dilute antibodies in 50 μl Odyssey buffer, and add to cells. Incubate at 4°C overnight, and then wash four times with 250 μl PBS per wash.
The optimal dilutions of antibodies tested for CycIF are provided in Table 1. The detailed guideline for antibody dilution in immunofluorescence can be found elsewhere but generally requires empirical testing (Donaldson, 2002; Hoffman, Le, & Sita, 2008).
-
6
Hoechst staining: Dilute Hoechst 33342 (1mg/ml stock) 1:5000 in 140 μl PBS and incubate with cells for 15 min at room temperature. Then wash four times with 250 μl PBS per wash. Leave 10–20 μl of PBS in each well after the last wash step.
-
7
Imaging: Use a microscope that is compatible with multi-well plates; the Cytell imaging system from GE Healthcare is a particularly simple and low-cost system that works well.
The Cytell system is equipped with optimal filter sets for 4-channel immunofluorescence (Hoechst, FITC/Alexa 488, Cy3/Alexa 555, and Cy5/Alexa 647), minimizing crosstalk between fluorophores. On-camera binning can be used to increase signals from weak antibodies or low-abundant antigens.
-
8
Fluorophore inactivation: After imaging, remove PBS and add 140 μl fluor-inactivation solution (3% H2O2 in PBS plus 20 mM NaOH). Incubate at room temperature for 1 hr under a tabletop lamp with a standard LED or fluorescent bulb. Wash four times with 250 μl PBS per wash, leaving 10–20 μl of PBS in each well after the last wash step.
After washing, cells can be stored at 4°C before proceeding, but it is best to proceed with the next round of imaging as soon as possible to minimize cell loss.
Use of a lamp is optional, but it may increase the efficiency of fluorophore inactivation by 10–20%, depending on the light source. A fluorescent or LED white bulb has been tested for performance in the CycIF protocol. Further details about lamps tested are described in the Reagents, Supplies and Instruments section.
-
9
Start next CycIF cycle: After fluorophore inactivation, image plates again to ensure that fluorescence has returned to background levels and then start the next round of CycIF (step 5: staining).
In some cases, re-blocking (starting CycIF from step 4) will reduce background from non-specific antibody binding, but this extra step can also increase cell loss. Thus, re-blocking is normally omitted unless background staining is unacceptably high.
Image registration
To align images acquired in successive rounds of CycIF, a microscope with good control over plate alignment and stage position is required, as is software image registration. Since only a subset of the information in each image is retained during registration, stitching together images from overlapping fields prior to registration allows for a larger fraction of cells to be retained in the final registered image.
Hoechst images acquired during each CycIF round are used as references to generate registration information based on a rigid body transformation algorithm. The registration information is then applied to all channels from that CycIF round to generate a hyperstack of images. Image registration is performed using the ImageJ scripts (can be downloaded from the LINCS website: http://lincs.hms.harvard.edu/lin-NatCommun-2015/) and plugins StackReg: http://bigwww.epfl.ch/thevenaz/stackreg/ and MultiStackReg: http://bradbusse.net/downloads.html).
Alternate Protocol 1
Type II CycIF: Cyclic immunofluorescence with both non-conjugated antibodies and conjugated antibodies
This protocol involves indirect immunofluorescence and can be used in the first CycIF cycle after which directly labelled antibodies are used according to the Basic Protocol (Steps 5–9). A different species of unlabeled primary antibody and a corresponding Alexa Fluor® 488/555/647-labelled secondary antibody is required for each channel.
Materials
Antibodies against antigens of interest.
Secondary antibodies conjugated to Alexa Fluor dyes (e.g. ThermoFisher/Invitrogen Goat anti-Rabbit IgG Alexa Fluor 488 cat: #A11008)
All other materials are the same as in Basic Protocol 1
-
Step 1–4
Same as Basic Protocol 1
-
Step 5a
Primary antibody staining: Dilute antibodies in 50 μl Odyssey buffer, and add to the cells. Incubate at 4°C overnight, and then wash 4 times with 250 μl PBS per wash.
Optimal dilutions of antibodies tested for CycIF are provided in Table 1. The detailed guideline for antibody dilution in immunofluorescence can be found elsewhere (Donaldson, 2002; Hoffman et al., 2008).
-
Step 5b
Secondary antibody staining: Dilute Alexa conjugated secondary antibodies (1:1000~1:2000) in 50 μl Odyssey buffer, and add to the cells. Incubate at room temperature for 1 hr, and then wash 4 times with 250 μl PBS per wash.
Dilutions of secondary antibodies are determined empirically, with the highest dilution (lowest concentration) of antibody giving acceptable staining intensity regarded as optimal.
-
Step 6 & 7
(Hoechst staining and Imaging). Same as basic protocol.
-
Step 8
Fluorophore inactivation: After imaging, remove PBS and add 140 μl fluor-inactivation solution (4.5% H2O2 in PBS plus 25 mM NaOH). Incubate at room temperature for 1–1.5 hr, then wash four times with 250 μl PBS per wash, leaving 10–20 μl of PBS in each well after the last wash step.
Secondary antibodies conjugated with Alexa dyes usually produce stronger signals due to the formation of antibody sandwiches and take longer to inactivate. Before proceeding to the next step, make sure the inactivation is complete by imaging plates.
-
Step 8
Start next CycIF cycle: After fluorophore inactivation, start the second round of CycIF from step 4 (blocking) or step 5 (staining). Only fluorophore-conjugated antibodies should be used from this point to avoid cross-reactivity with primary antibodies still bound to the sample.
In some cases, re-blocking (starting CycIF from step 4) will reduce background from nonspecific antibody binding, but this extra step can also increase cell loss. Thus, re-blocking is normally omitted unless background staining is unacceptably high.
Alternate Protocol 2
Type III CycIF: Cyclic immunofluorescence for cells expressing fluorescent proteins (FPs)
This protocol is performed after live-cell imaging of cells expressing fluorescent proteins and prepares them for analysis by CycIF.
Materials
Hydrogen Chloride/HCl 1M solution, (dilute from 12 M solution; Sigma-Aldrich, cat. #258148)
All other materials are the same as in Basic Protocol for Type I CycIF
-
Step 1 to 7
Same as Basic Protocol 1.
-
Step 8a
Inactivation of fluorescent proteins: After imaging, remove PBS and add 140 μl acidic fluorophore inactivation solution (3% H2O2 in PBS plus 20 mM HCl). Incubate at room temperature for 1–1.5 hr, then wash four times with 250 μl PBS per wash, leaving 10–20 μl of PBS in each well after the last wash step.
For Turquoise/ECFP fusion proteins, increase the inactivation time to 2 hr (or bleach a second time with fresh inactivation solution).
Step 8a is performed only prior to the first CycIF cycle.
-
Step 8b
Fluorophore inactivation for Alexa Dyes: Remove PBS and add 140 μl basic fluorophore-inactivation solution (3% H2O2 in PBS plus 20 mM NaOH). Incubate at room temperature for 1–1.5 hr, then wash four times with 250 μl PBS per wash, leaving 10–20 μl of PBS in each well after the last wash step.
Step 8b is performed after imaging in each CycIF cycle (started from the first antibody staining).
-
Step 9
Start CycIF: After fluorophore inactivation, image plates to ensure fluorescence is at background levels and then start CycIF from step 4 (blocking).
REAGENTS, SUPPLIES AND INSTRUMENTS
Corning 96-well Plates, Clear Bottom, black (Sigma-Aldrich, Cat: CLS3603)
4% paraformaldehyde/PFA (diluted from the 16% stock from Electron Microscopy Services, cat. #15710)
Methanol 99.9% (Fisher Scientific, Cat: AC61009)
Hoechst 33342 (ThermoFisher/Invitrogen, cat. #H3570)
Odyssey blocking buffer (LI-COR Biosciences, cat. #927-40001)
20× Phosphate-buffered Saline/PBS (Santa Cruz Biotech, cat. #SC-362299):
30% hydrogen peroxide (Sigma-Aldrich, cat. #H1009)
1 M NaOH (dissolve from pellets; Sigma-Aldrich, cat. #S5881)
2 M HCl (dilute from 12 M solution; Sigma-Aldrich, cat. #258148)
Basic fluorophore-inactivation solution for Alexa dyes: 3% H2O2 (1/10 dilution from 30% stock), 20 mM NaOH (1/50 dilution from 1 M stock) in PBS
Basic fluorophore-inactivation solution for indirect IF: use up to 4.5% H2O2 and 25 mM NaOH in PBS.
Acidic fluorophore-inactivation for GFP/YFP/mCherry fusion proteins: 3% H2O2 (1/10 dilution from 30% stock), 20 mM HCl (1/100 dilution from 2M stock) in PBS
Biotek EL406 Washer/dispenser
Plate-based high content imaging system (such as GE Cytell Cell Imaging System).
Lamp to promote fluorophore inactivation. A simple table lamp equipped with Lithona 211E71 LED light (UCLD 24 WH, Color temperature: 3000k with CRI:83) has proven acceptable. The peak wavelength of the light source is at 630 nm with second peak at 540 nm and third peak at 450 nm (Fig. 3).
Figure 3.
The light device for fluorophore inactivation.
COMMENTARY
Background Information
The use of antibody stripping procedures to increase the multiplicity of antibody-based staining of tissue samples is not a new concept in immunohistochemistry. The heating and microwave steps used in antigen retrieval protocols are also frequently used to remove antibodies from formalin-fixed paraffin-embedded (FFPE) tissue samples (Robertson, Savage, Reis-Filho, & Isacke, 2008). However, such procedures are not directly applicable to cells grown as monolayers in conventional tissue culture dishes because heating or microwaving destroys such samples. A variety of methods for multiplex imaging of cultured cells have been developed (Angelo et al., 2014; Schweller et al., 2012; Zrazhevskiy & Gao, 2013), but most require special reagents such as quantum dots, oligonucleotide- or isotope- conjugated antibodies or special instrumentation such as hyperspectral microscopes, microfluidic devices and mass cytometry which are not widely available and have their own limitations. In contrast, cyclic immunofluorescence (CycIF) uses instruments, reagents and methods that are affordable and widely available. Further, immunofluorescence (and therefore CycIF) is a well-established method to obtain quantitative information on protein abundance and localization, cell morphology and microenvironment. We describe three variants of CycIF: Basic Protocol 1 (Type I CycIF) uses fluorophore-conjugated antibodies and is the most generally useful variant; Alternate Protocol 1 (Type II CycIF) uses indirect immunofluorescence during the first cycle of CycIF and is useful in cases in which directly-labelled antibodies are not available or signals are weak; and Alternate Protocol 2 (Type III CycIF) allows cells that express fluorescent proteins to be analyzed by CycIF.
Chemical inactivation of Alexa dyes in CycIF involves base-catalyzed oxidation. The dye molecules are irreversibly inactivated by oxidation leaving behind primary antibodies that are no longer fluorescent. The Riordan group at Stanford University and the GE Global Research Center have reported the use of similar chemistry for multiplexed immunofluorescence in tissue samples, although their approaches are performed using less stable cyanine dyes (Cy3 and Cy5) (Gerdes et al., 2013; Riordan, Varma, West, & Brown, 2015). Given the similarities between the two approaches it should be possible to apply CycIF to FFPE-embedded tissues and also use a variety of other types of dyes (Cy3, Cy5, etc.) with the same chemical process. We have tested Type I and II CycIF with TRIC or FITC-conjugated antibodies and obtained satisfactory results. Although the CycIF protocol presented here is mainly focused on cultured cells in 96/384-well plates, the method with modifications can be applied to tissue/FFPE samples (Lin et al, in preparation). In addition, we continue to update and optimize the CycIF method and protocols and publish updated methods at http://lincs.hms.harvard.edu/lin-NatCommun-2015/. We anticipate that it will ultimately prove advantageous to combine simple methods such as CycIF with more complex protocols such as DNA PAINT (Jungmann et al., 2014) when some signals are very weak.
Image processing and segmentation
A wide variety of image segmentation and analysis approaches are applicable to CycIF data. In the current protocol we describe a simple means for calculating overall staining intensities from each channel on a per-cell basis. Image segmentation is performed using ImageJ with scripts provided on our website (http://lincs.hms.harvard.edu/lin-NatCommun-2015/). Hoechst images from the last round of CycIF are converted to nuclear masks and regions of interest (ROIs) and ROIs then applied to images in the data channels (Alexa 488/555/647 in the case of four-color CycIF). Mean fluorescence intensities are calculated for complete cells and nuclear masks converted into ring-shape ROIs for quantifying cytoplasmic intensity. Raw intensity data generated by ImageJ are saved in a comma-separated values (CSV) format and processed using Matlab scripts for further data analysis. All ImageJ and Matlab scripts are available at http://lincs.hms.harvard.edu/lin-NatCommun-2015/.
High-dimensional data analysis
The high-dimensional single-cell data generated by CycIF and related methods poses visualization and analysis challenges. We have used general-purpose visualization tools such t-Distributed Stochastic Neighbor Embedding (t-SNE) as well as methods developed for CyTOF data, such as viSNE and Wanderlust algorithms (Amir et al., 2013; Bendall et al., 2014; Lin et al., 2015) (Fig. 4). Principal component analysis (PCA) and cross-correlation analysis can be performed using built-in Matlab functions and additional scripts can be obtained from the HMS LINCS website (http://lincs.hms.harvard.edu/lin-NatCommun-2015/) The viSNE and Wanderlust algorithms are components of the CYT package available from the Pe’er lab webpage (http://www.c2b2.columbia.edu/danapeerlab/html/software.html) (Amir et al., 2013; Bendall et al., 2014). For Wanderlust, the raw intensity data files need to be normalized using the Wanderlust script with default parameters (L number = 30; K number = 5; Number of landmarks = 20; Number of graphs = 25; Distance Metric = Cosine). A substantial need nonetheless exists for new algorithms to analyze high-dimensional morphological features captured by CycIF; datasets for testing such algorithms have recently been published (Honarnejad et al., in preparation).
Figure 4. Analysis of high-dimensional CycIF data.
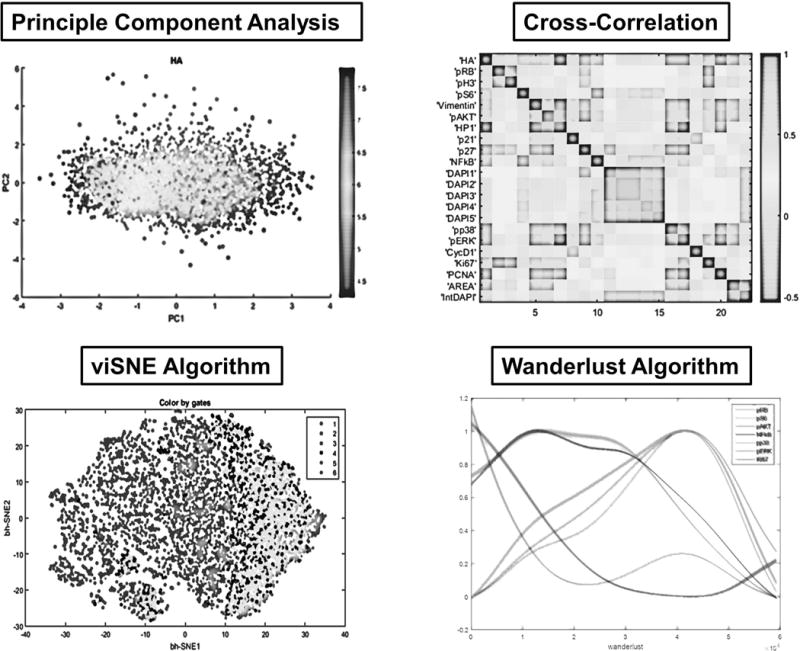
Shown here are four methods for high-dimensional data analysis of CycIF data. Principal Component Analysis (PCA) and viSNE algorithms are generally used for dimensionality reduction and visualization of CycIF data (Amir et al., 2013; Lin et al., 2015). The cross-correlation method is used for probing the relationships between different signals measured by CycIF. The Wanderlust algorithm is a graph-based trajectory detection method, mapping the high-dimensional single-cell CycIF data to a one-dimensional path based on most likely transitional cell states (Bendall et al., 2014; Lin et al., 2015).
Critical Parameters
Achieving consistent results using CycIF protocols involves several key factors. First, samples/cells must be fixed extensively to prevent dissociation through washing. We suggest using a combination of paraformaldehyde and methanol. For some antigens that cannot tolerate PFA, we find that acetone/methanol fixation can yield better results when imaging, especially for the cytoskeletons. Second, CycIF imaging requires a microscope stage with the ability to remount samples multiple times and achieve good registration; multiplexed single-cell measurements in CycIF rely on revisiting the same position through multiple staining cycles (software-based image registration is easier if images are roughly aligned by the instrument). Third, good cameras are important because immunofluorescence with directly conjugated antibodies generates weaker signals than immunofluorescence with secondary antibodies. Fourth, good Alexa dye-conjugated antibodies are important. We have tested antibodies from multiple vendors, including Cell Signaling Technology, Abcam, and BioLegend with good success but have found that the vendor-provided information on antibody dilution or buffer application is often suboptimal. Re-validation and optimization for CycIF procedures seems to be essential, as previously described (Hoffman et al., 2008). A continuously updated list of validated antibodies is available at (http://lincs.hms.harvard.edu/lin-NatCommun-2015/).
Troubleshooting
Insufficient fluorophore inactivation
In general, a one-hour incubation in base-hydrogen peroxide mixture is sufficient for Alexa Fluor® 488/555/647 dyes to bleach below pre-staining fluorescence levels. However, longer incubation times can be used to eliminate residual fluorescence. In the case of strong fluorescent signals resulting, for example, from indirect immunofluorescence (primary/secondary antibody complex), increasing the H2O2 concentration from 3% to 4.5% can also be helpful. Photobleaching using white light improves the efficiency and the speed of fluorophore inactivation (Figure 2).
Uneven staining
It is important to avoid uneven immunostaining due to incomplete fixation or permeabilization, excessively short antibody incubation, and poor washing. Fixation is discussed above. For antibody incubation, it is important to increase the volume of antibody solution in proportion to the area of the sample. We use at least 50 μl of antibody solution per well in a 96-well plate. Washing steps should be performed with at least 250 μl per well per wash using an automatic washer/dispenser, if possible. Manual wash with gentle aspiration/dispense is also possible but care must be taken not to dislodge cells by flowing liquid rapidly across wells.
Cell loss
Cell loss is a potential problem in CycIF and similar methods but with good technique it can be kept to 1–2% per cycle. Fixation, gentle washes and cell density are key variables. Very low or high cell density both appear to increase cell loss. Use of collagen or poly-lysine coating can counteract this to some extent. Once cells are permeabilized, cells become more sensitive to washing, so samples should not be stored for extended periods of time, even at 4°C.
Low signals
Directly conjugated antibodies used in Type I CycIF generate less intense signals than primary-secondary sandwiches. Antibodies used at a lower dilution can sometimes work, but can also lead to higher background. When signals from direct immunofluorescence are too weak, indirect immunofluorescence staining in the first cycle of CycIF is suggested. We continue to perform experiments with different types of fluorophores including those based on polymers; these are potentially very bright but not yet widely conjugated to antibodies.
Anticipated Results
For most of the adherent cells we have tested, five rounds of CycIF can be performed without difficulty, allowing 15 antibody signals to be analyzed; Figure 5 illustrates such a result in retinal pigment epithelium (RPE) cells. For strongly adherent cells such as MCF10A, we have performed 10 CycIF cycles.
Figure 5. Example images for a five-round CycIF experiment.
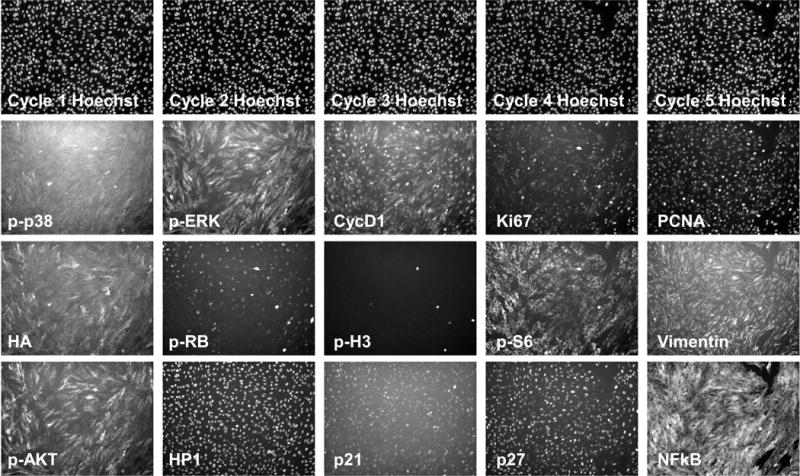
Retinal pigment epithelium (RPE) cells were cultured and fixed in 96-well plates. Five rounds of CycIF staining were performed and representative images are shown with indicated antibodies.
Time Considerations
Since a typical round of CycIF can be performed within one day (including overnight antibody incubation plus imaging and inactivation time), a five-round CycIF experiment takes about one week but with multi-well plates, processing hundreds of samples in parallel is straightforward. To maintain consistent signals and fields of view (which is important for image registration) imaging of all cycles must be performed on the same instrument. In our experience the primary bottleneck in CycIF is therefore imaging with subsequent data analysis an additional time-consuming step. However, automation and better algorithms promise to make CycIF faster.
Acknowledgments
We thank E. Williams for creating the CycIF web-page and editing this protocol. This work was funded by NIH LINCS grant U54-HL127365 to PKS; M.F. is funded by NCI grant K99CA194163 and J.Y.C. by the Jane Coffin Childs Memorial Fund.
LITERATURE CITED
- Amir ED, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Pe’er D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nature Biotechnology. 2013;31(6):545–52. doi: 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelo M, Bendall SC, Finck R, Hale MB, Hitzman C, Borowsky AD, Nolan GP. Multiplexed ion beam imaging of human breast tumors. Nature Medicine. 2014;20(4):436–442. doi: 10.1038/nm.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall SC, Davis KL, Amir EAD, Tadmor MD, Simonds EF, Chen TJ, Pe’er D. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell. 2014;157(3):714–25. doi: 10.1016/j.cell.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson JG. Immunofluorescence staining. Current Protocols in Immunology/Edited by John E Coligan … [et Al], Chapter 21. 2002 doi: 10.1002/0471142735.im2103s48. Unit 21.3. [DOI] [PubMed] [Google Scholar]
- Frei AP, Bava FA, Zunder ER, Hsieh EWY, Chen SY, Nolan GP, Gherardini PF. Highly multiplexed simultaneous detection of RNAs and proteins in single cells. Nature Methods. 2016;13(3):269–275. doi: 10.1038/nmeth.3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet S, Miller-Jensen K. Redefining Signaling Pathways with an Expanding Single-Cell Toolbox. Trends in Biotechnology. 2016;34(6):458–469. doi: 10.1016/j.tibtech.2016.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes MJ, Sevinsky CJ, Sood A, Adak S, Bello MO, Bordwell A, Ginty F. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(29):11982–7. doi: 10.1073/pnas.1300136110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman GE, Le WW, Sita LV. The importance of titrating antibodies for immunocytochemical methods. Current Protocols in Neuroscience/Editorial Board, Jacqueline N Crawley … [et Al], Chapter 2. 2008 doi: 10.1002/0471142301.ns0212s45. Unit 2.12. [DOI] [PubMed] [Google Scholar]
- Hoppe PS, Coutu DL, Schroeder T. Single-cell technologies sharpen up mammalian stem cell research. Nature Cell Biology. 2014;16(10):919–27. doi: 10.1038/ncb3042. [DOI] [PubMed] [Google Scholar]
- Jungmann R, Avendaño MS, Woehrstein JB, Dai M, Shih WM, Yin P. Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and Exchange-PAINT. Nature Methods. 2014;11(3):313–318. doi: 10.1038/nmeth.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JR, Fallahi-Sichani M, Sorger PK. Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nature Communications. 2015;6:8390. doi: 10.1038/ncomms9390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchuk-Voloshina N, Haugland RP, Bishop-Stewart J, Bhalgat MK, Millard PJ, Mao F, Leung WY. Alexa dyes, a series of new fluorescent dyes that yield exceptionally bright, photostable conjugates. The Journal of Histochemistry and Cytochemistry: Official Journal of the Histochemistry Society. 1999;47(9):1179–88. doi: 10.1177/002215549904700910. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/10449539. [DOI] [PubMed] [Google Scholar]
- Riordan DP, Varma S, West RB, Brown PO. Automated Analysis and Classification of Histological Tissue Features by Multi-Dimensional Microscopic Molecular Profiling. PloS One. 2015;10(7):e0128975. doi: 10.1371/journal.pone.0128975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson D, Savage K, Reis-Filho JS, Isacke CM. Multiple immunofluorescence labelling of formalin-fixed paraffin-embedded (FFPE) tissue. BMC Cell Biology. 2008;9:13. doi: 10.1186/1471-2121-9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweller RM, Zimak J, Duose DY, Qutub AA, Hittelman WN, Diehl MR. Multiplexed in situ immunofluorescence using dynamic DNA complexes. Angewandte Chemie (International Ed in English) 2012;51(37):9292–6. doi: 10.1002/anie.201204304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack EC, Wang C, Roman KA, Hoyt CC. Multiplexed immunohistochemistry, imaging, and quantitation: A review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods. 2014;70(1):46–58. doi: 10.1016/j.ymeth.2014.08.016. [DOI] [PubMed] [Google Scholar]
- Zanella F, Lorens JB, Link W. High content screening: seeing is believing. Trends in Biotechnology. 2010;28(5):237–45. doi: 10.1016/j.tibtech.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Zrazhevskiy P, Gao X. Quantum dot imaging platform for single-cell molecular profiling. Nature Communications. 2013;4:1619. doi: 10.1038/ncomms2635. [DOI] [PMC free article] [PubMed] [Google Scholar]