

Abstract
The generation of site-specific bioconjugates of proteins is highly desired for a number of biophysical and nanotechnological applications. To this end, many strategies have been developed that allow the specific modification of certain canonical amino acids and, more recently, non-canonical functional groups. P450 enzymes are heme-dependent monooxygenases involved in xenobiotic metabolism and in the biosynthesis of a variety of secondary metabolites. We became interested in the site-specific modification of these enzymes, CYP3A4 in particular, through our studies of their in vitro biocatalytic properties and our desire to exploit their remarkable ability to oxidize unactivated C-H bonds in a regio- and stereospecific manner. Obtained via a partial cysteine-depletion approach, a functional triple mutant of CYP3A4 (C98S/C239S/C468G) is reported here which is singly modified at C64 by maleimide-containing groups. While cysteine-labelling of the wild-type enzyme abolished >90% of its enzymatic activity, this mutant retained ≥75% of the activity of the unmodified wild-type enzyme with 9 of the 18 maleimides that were tested. These included both fluorescent and solid-supported maleimides. The loss of activity observed after labelling with some maleimides is attributed to direct enzyme inhibition rather than to steric effects. We also demonstrate the functional immobilization of this mutant on maleimide-functionalized agarose resins and silica microspheres.
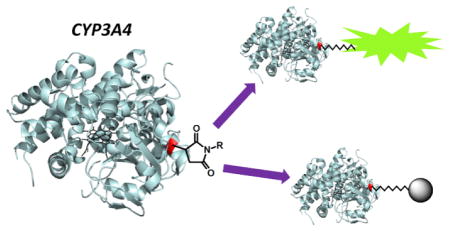
Graphical Abstract
INTRODUCTION
In recent years, the desire to generate covalent protein conjugates has exploded with the rise of more sophisticated biochemical and biophysical analytical techniques, and applications such as single-molecule spectroscopy (SM fluorescence and SM force experiments)1, the fabrication of protein microarrays, biosensor and bioreactors, and the in vivo visualization and tacking of biomolecules. The difficulty remains in obtaining conjugates that retain the activity, functionality and stability found in the native systems. Because of the great diversity of proteins, meeting all these demands is often quite challenging and involves a great deal of trial and error. It is widely acknowledged however that employing site-specific bioconjugation strategies significantly improves the probability of success2,3. By restricting bioconjugation to a single and precise position, one can more easily avoid modifications near residues known to be important for function and stability. An added advantage of generating such conjugates is the resulting uniformity in the population to be studied. This is especially important, for example, in SM experiments in which one wishes to detect biologically significant heterogeneities within a given population.
During our various investigations geared towards the use of human cytochrome P450 enzymes (CYPs or P450s) as in vitro biocatalysts, it was envisaged that the successful generation of functional and site-specific covalent conjugates of these enzymes could potentially open up opportunities to study and apply these catalysts in new ways. P450s form a family of widespread heme-dependent monooxygenases known mainly for their role in xenobiotic metabolism and their notable ability to regio- and stereoselectively oxidize unactivated C-H bonds. Unfortunately, our ability to study and make use of these properties in vitro has been limited by the low activity, the low stability, the requirement for expensive cofactors and the poor product predictability of P450s. Our attention in addressing these issues has been focused mainly on CYP3A44,5,6,7 because of its highly promiscuous yet predictable behaviour, which boosts this P450 enzyme’s potential as a useful and versatile biocatalyst.
A variety of bioconjugation methods that either target specific amino acids (usually cysteine or lysine) or functional groups that are not found in the canonical amino acids (e.g. alkynes, azides, alkenes, phosphines, thioesters, ketones, aldehydes and anilines), termed “bioorthogonal”, have been reported8. The former often requires the use of mutagenesis in order to achieve complete site-specificity, while the latter requires the incorporation of non-native functional groups as unnatural amino acids9,10, or protein fusions either with modified inteins11 or enzymatically modifiable peptide tags8. Unfortunately, most of the bioorthogonal reactions suffer from slow, second-order kinetics and therefore often require high reagent concentrations, harsh conditions (i.e. high temperatures and extreme pHs) and long reaction times that would result in loss of P450 activity. A few improvements in this area have been reported recently however, namely for the CuI-catalyzed azide-alkyne cycloaddition reaction12, the hydrazone-ligation13 and the oxidative coupling of anilines to o-aminophenols14. Considering our observations on the stability of CYP3A4 at room temperature (Figure 1), we thought it prudent to attempt bioconjugation using cysteine-specific maleimide chemistry which can be achieved at room temperature and neutral pH, and proceed to completion within a few hours. There is also precedent in generating functional, partially cysteine-depleted mutants of CYP3A4. Reactions of a quadruple mutant (C98S/C239S/C377S/C468A) with cysteine-reactive fluorescent probes have generated C64-specific labelled CYP3A4 used to study the mechanism of α-naphthoflavone binding by FRET15. Here we present our efforts, by way of this same partial cysteine-depletion strategy, in generating and characterizing site-specific cysteine-modified bioconjugates of CYP3A4 with a variety of maleimide tags, both soluble and solid-supported. Others have reported the immobilization of CYP3A4 on solid matrices such as gold electrodes allowing electrochemically driven drug metabolism16, monolithic chromatographic supports in the development of immobilized enzyme reactors (IMERs)17, glass slides for analysis by total internal reflection microscopy (TIRFM)18, or silver nanoparticles in the development of a localized surface plasmon resonance (LSPR) spectroscopy-based biosensor19. Various other surfaces have also been employed in the development of CYP3A4-based amperometric20,21,22 and oxygen-sensing23 drug metabolism biosensors. However, in all the above cases, CYP3A4 was immobilized in a random manner, either covalently, or non-covalently in biological membranes (microsomal or E. coli-derived) or in synthetic membranes such as Nanodiscs18,24. We report here what is, to our knowledge, the first functional immobilization of CYP3A4 in a covalent and oriented fashion, through site-specific modification of a single cysteine residue on its surface.
Figure 1.

Thermodynamic stability of CYP3A4 at 25°C in 0.1 M potassium phosphate buffer, pH 7.4. The activity was assayed with testosterone as the substrate and cumene hydroperoxide (CHP) as the cofactor4. All values represent an average of three separate experiments each performed in duplicates or triplicates ± 1 standard deviation.
EXPERIMENTAL PROCEDURES
Site-directed mutagenesis
Cysteine mutants of CYP3A4 were generated using the QuikChange Multi Site-Directed Mutagenesis Kit (Stratagene, Agilent) and pSE3A4His encoding N-terminally truncated and tetrahistidine-tagged CYP3A4 wild-type cDNA as the template25. The following PCR primers were used: for C64S, 5′-GGCTTTTGTATGTTTGACATGGAAAGTCATAAAAAGTATGGAAAAGTG-3′; for C98S, 5′-CAAAACAGTGCTAGTGAAAGAAAGTTATTCTGTCTTCACAAACCG-3′; for C239S, 5′-TCCCAATTCTTGAAGTATTAAATATCAGTGTGTTTCCAAGAGAAGT-3′; for C337S, 5′-CTATGAGACTTGAGAGGGTCAGCAAAAAAGATGTTGAGATC-3′ and for C468G, 5′-GAACTTCTCCTTCAAACCTGGTAAAGAAACACAGATCCC-3′. The following mutants were constructed: C98S (mutant 1), C98S/C377S/C468G (mutant 2), C98S/C239S/C468G (mutant 3) and C64S/C98S (mutant 4).
Expression of wild-type and mutant CYP3A4
Protein expression was adapted from a protocol described previously4. Briefly, Escherichia coli DH5α competent cells were transformed with the desired plasmid. A starter culture was generated by inoculation of LB medium (100 mL) containing ampicillin (100 μg/mL) and allowed to grow overnight at 37°C and 250 RPM. Culture flasks (2 L) containing TB medium (750 mL) supplemented with ampicillin (50 μg/mL) were inoculated each with the overnight starter culture (10 mL). The flasks were shaken at 250 RPM and 37°C until an OD600 of 0.6 was reached. Protein expression was then induced by the addition of IPTG (1 mM) and δ-aminolevulinic acid (80 mg/L). Incubation was continued for 48 h at 30°C and 190 RPM. The cells were harvested at 4000 × g and 4°C for 15 min.
Purification of wild-type and mutant CYP3A4
All the following steps were performed at 4°C. The pellet obtained from 5 × 750 mL cultures was resuspended in Buffer A (150–200 mL, 0.1 M potassium phosphate, or KPi, at pH 7.4, containing 10% glycerol) supplemented with phenylmethylsulfonyl fluoride (PMSF, 2 mM). The following protease inhibitors were also added: leupeptin (1.6 μg/mL), aprotinin (1 μg/mL), bestatin (0.8 μg/mL) and pepstatin A (0.7 μg/mL), along with lysozyme (10 mg per g of cells) and CHAPS (1% w/v). The resulting mixture was stirred slowly for 30 min and then sonicated at 60% cycle duty and power 8 for 4 cycles of 30 s each. The lysate was centrifuged for 1 h at 54,000 × g. The supernatant was supplemented with imidazole (5 mM) and KCl (0.5 M) before being loaded onto a column containing Ni-NTA agarose resin (5 mL, Qiagen) pre-equilibrated with Buffer A. The flow rate was adjusted to ca. 1 mL/min. The resin was washed with 10 column volumes of Buffer A containing KCl (0.5 M), CHAPS (0.5 % w/v) and imidazole (5 mM), followed by 10 column volumes of Buffer A containing imidazole (20 mM). The protein was eluted with Buffer A containing more imidazole (200 mM). The fractions containing the protein of interest were combined and dialyzed against Buffer A. The protein was further purified by ion-exchange using Macro-Prep High S Support resin (Bio-Rad). Thus, the protein was loaded onto a column containing the said resin (15 mL) pre-equilibrated with Buffer A. The column was next washed with Buffer A (50 mL), followed by elution with Buffer A containing KCl (0.5 M). Finally, the fractions of interest were combined, dialyzed against Buffer A, and stored at −80°C. Protein concentration was measured using the Bradford method (Pierce, 23236) while the concentration of P450 was calculated from its reduced-CO spectrum26.
Expression and purification of Cytochrome P450 reductase (CPR)
CPR was expressed and purified as described elsewhere4. Protein concentration was measured by the Bradford method (Pierce, 23236). The concentration of holo-CPR was estimated from its flavin content using an extinction coefficient of 21.4 mM−1•cm−1 at 456 nm27.
Cysteine-specific protein labelling with maleimides
Maleimide labels were purchased or generated from the corresponding amine as described below. Stock solutions (20 mM) of the maleimides were prepared in anhydrous DMSO. In a small glass vial, wild-type or mutant CYP3A4 (typically ~10 μM, based on Bradford assay) was combined with 9 equivalents of TCEP (10 mM stock in Milli-Q water) in KPi buffer (0.1 M at pH 7.4), and incubated at 25°C and 250 RPM for 20 min. Then, 100 equivalents of the desired maleimide label were added, and the reaction was incubated for 2 h under the same conditions. Note, equivalents are reported relative to the concentration of CYP3A4. The reaction was quenched by the addition of DTT (2 mM from a 0.1 M stock solution in Milli-Q water) and incubation for 5–10 min. Control reactions were also performed in which pure anhydrous DMSO was added instead of the maleimide label with and without the 2 h incubation period.
Generation of maleimide labels from their corresponding amines
Maleimides 1b, 1c, 2c, 3b, and 3c were prepared as follows (see supplemental information for full product characterization). All other maleimides were purchased and used as is. In a typical reaction, heterobifunctional crosslinker 15b or 15c (6–23 nmol) was combined with 1.05 equivalents of 1a, 2a, or 3a in anhydrous DMSO (final concentration of ~45 mM) and stirred at 25°C. The reaction was monitored by ESI-MS and was usually complete within 4 h after which the crosslinker (15b or 15c) had been consumed and the major peak corresponded to the desired amide bond formation product between the amine and the NHS-ester. A small amount (10–15 %) of double addition product (conjugate addition of the amine to the maleimide) was also observed. Purification was usually not necessary, but was possible using chromatography (see supporting information). The products were stored at −20°C. The final maleimide concentration was quantified spectrophotometrically from the difference in the amount of 4-thiopyridone product formed when 2-mercaptoethanol reacts with 4,4′-dithiodipyridine in the presence and absence of the maleimide reaction mixture (maleimides react with 2-mercaptoethanol but not with 4,4′-dithiodipyridine) 28. Briefly, an aliquot (5 μL) of the reaction mixture in DMSO was diluted into KPi buffer (195 μL of 0.1 M at pH 7.4). A sample of this diluted stock (2–5 μL) was combined with 2-mercaptoethanol (10 μL from a 1.5 mM stock solution prepared fresh in Milli-Q water) and KPi buffer (0.1 M at pH 7.4), to a final volume of 450 μL. Control reactions containing only 2-mercaptoethanol were also prepared. These were incubated 10 min at room temperature after which a saturated solution of 4,4′-dithiodipyridine (50 μL in Milli-Q water) was added before incubation for another 15 min. Absorbance was read at 324 nm against a blank containing only 4,4′-dithiodipyridine. The concentration of the 4-thiopyridone formed from the reaction between 2-mercaptoethanol and 4,4′-dithiodipyridine in the presence and absence of the maleimide was calculated using ε224nm = 19,800 M−1•cm−1. The difference gave the maleimide concentration in the reaction mixture.
Assessing the effect of quenched maleimide labels on enzymatic activity
Samples were prepared in the same manner described above for the cysteine-specific labelling except that the maleimides were quenched with DTT prior to mixing with enzyme. The enzymatic activity was then assayed immediately using testosterone as the substrate and CPR/NADPH as the cofactors (end-point assay, see below), omitting the 2 h incubation period usually required for complete labelling. SDS-PAGE analysis followed by fluorescence visualization of the gels confirmed that CYP3A4 remained essentially label-free when the maleimides were pre-quenched in this manner.
Estimating the labelling yield of CYP3A4 mutant 3 with the maleimide DyLight 549
CYP3A4 mutant 3 was labelled with 10 or 100 equivalents of DyLight 549 maleimide as described above. The excess label was removed by size-exclusion using Zeba Spin Desalting Columns (3 × 0.5 mL, Pierce). The absorbance spectrum of the flow through containing the labelled protein was obtained and the degree of labelling was calculated as per the manufacturer’s instructions using ε280nm = 40,340 M−1•cm−1 (ExPASy ProtParam tool) to determine the protein concentration.
Sample preparation for single-molecule photobleaching analysis
An aliquot of DyLight 549 maleimide-labelled wild-type or mutant CYP3A4 (see above) was combined with SDS sample buffer (4 μL of 0.13 M Tris pH 6.8, 20% glycerol, 4% SDS, 1.25% bromophenol blue, 1.4 M 2-mercaptoethanol) and boiled for 1 min. The sample was then loaded onto a precast 12.5% polyacrylamide gel and subjected to SDS-PAGE (with PhastSystem, Pharmacia) in order to ensure complete removal of excess label. The fluorescent band of appropriate molecular weight (ca. 57 kDa) was excised and subjected to syringe-maceration-extraction29 in order to extract the labelled protein. Coverslips were cleaned in Piranha solution following a procedure reported earlier and used without any further modification of the surface30. Predrilled polycarbonate films with an adhesive gasket (Grace Bio-Labs, Bend, OR) were assembled on top of the cleaned coverslips yielding a chamber with a total volume of 10 μL. Inlet and outlet ports (Nanoport, Upchurch Scientific, Oak Harbor, WA) were glued on top of the chambers30,31. In order to increase the dye photostability, all experiments were run under a constant flow of an oxygen scavenger solution consisting of 2-mercaptoethanol (1% v/v), β-D(+)glucose (3% w/v), glucose oxidase (0.1 mg/mL), and catalase (0.02 mg/mL)30,32,33,. All experiments were conducted at room temperature (22–23°C). Purified labeled proteins were diluted (ca. 1000 fold) and then injected into a clean coverslip until a satisfactory surface density was observed.
Quantification of labelling via single-molecule photobleaching analysis: Single Molecule Imaging
Total internal reflection fluorescence microscopy (TIRF) was utilized to quantify the number of fluorescent dyes per single protein. The setup consisted of an inverted microscope (Olympus IX71) equipped with a laser-based TIRFM illumination module (IX2-RFAEVA-2, Olympus) coupled to a diode-pumped solid-state green laser with 532 nm output (CrystaLaser). The beam position was adjusted using the illuminator to attain total internal reflection through a 60×, N.A. 1.45, oil-immersion objective (Olympus U PLAN SAPO). Fluorescence emission was collected through the objective and images were captured with an electron multiplied back illuminated charged coupled device (EMCCD) camera (CascadeII:512, Photometrics, Roper Scientific). The camera was controlled using Image-Pro Plus 5.1 (Media Cybernetics), capturing 8-bit 512 × 512 pixel images with an exposure time of 200 ms per frame, a conversion gain of 3, and a multiplication gain of 4095. Excitation was carried out at a full power setting (25 mW) with a power output of 6.6 mW measured at the objective.
Quantification of labelling via single-molecule photobleaching analysis: Intensity analysis
A Matlab algorithm was utilized which first maps the ca. hundreds of single molecule spots and following background subtraction it then extracts intensity-time trajectories from the stack of images for each individual molecule. Upon inspecting individual trajectories, photobleaching events were observed as steps. The number of fluorophores per protein was next calculated from the number of photobleaching steps recorded.
In-gel BrCN digestion of labelled protein
An aliquot of the dye-labelled sample (20 μL) was combined with SDS sample buffer (4 μL, see “Sample preparation for single-molecule photobleaching analysis” above). Exceptionally, for dansyl-labelled (1b) samples, an aliquot (500 μL) was first concentrated (to 20 μL) with a centrifugal filter (10 kDa MWCO, Millipore) before combination with SDS sample buffer (4 μL). Samples were boiled for 1 min and separated by SDS-PAGE using a classical vertical separation system. The appropriate bands were excised, washed and treated with cyanogen bromide34. The resulting peptides were analysed by tricine-SDS-PAGE35. Optimal resolution was achieved with a 1.7 mm thick gel when a 10% spacer gel was included between the 4% stacking gel above and the 16%/6 M urea separating gel below. Gels were imaged with a Typhoon Trio + scanner using the Green-excited mode (λex = 532 nm) and the 580 nm band-pass filter (580 BP 30, which transmits light between 565 and 595 nm with a transmission peak centered on 580 nm).
Preparation of maleimide-functionalized resins
Maleimide-functionalized resins were generated through modification at the terminal amine of CarboxyLink resin (4a, Pierce) with 15b or 15c. Aliquots of the resin (1 mL) were washed with KPi buffer (3 × 2 mL, 0.1 M at pH 7.4) through centrifugation/resuspension cycles resulting ultimately in removal of as much supernatant as possible. The crosslinkers (15b or 15c, 22 μmol) were dissolved in anhydrous DMSO (0.5 mL), diluted with the same KPi buffer (0.5 mL) and immediately added to the resin. After brief vortexing, the resin was incubated at room temperature and ~100 RPM for 1.5 h to give 4b or 4c. Reaction completion was confirmed with a ninhydrin test36 as follows. A small aliquot of the resin was transferred to a glass vial and the supernatant was removed. Two drops each of pyridine, 4:1 phenol:ethanol, and 4% (w/v) ninhydrin in EtOH were added to the resin which was then heated to 100°C for 2 min. No colour change indicated that all free amines on the resin had reacted with the crosslinkers. Unmodified resin turned deep blue under the same conditions. The resin was then washed with buffer (5 × 2 mL) to remove excess crosslinker. The degree of maleimide loading was quantified from mixtures of resin aliquots (20 μL, supernatant removed) and 4,4′-dithiodipyridine as described above in “Generation of maleimide labels from their corresponding amines”. The degree of loading was generally ~12 nmol maleimide/20 μL resin.
Immobilization of CYP3A4 mutant 3 onto maleimide functionalized resins
CYP3A4 (~1 nmol based on Bradford assay, ~0.18 nmol active P45026, from a stock in Buffer A) was combined with TCEP (9 nmol, 0.9 μL from a 10 mM stock in Milli-Q water). The volume was adjusted to 100 μL with KPi buffer (0.1 M at pH 7.4) before incubation at 25°C and 250 RPM for 20 min. This TCEP-reduced enzyme solution was then added to an aliquot of resin (200 μL, supernatant removed, contained ca. 120 nmol maleimide) and incubated for 2 h. The enzyme activity was measured both in the supernatant and on the resin. To evaluate the immobilization yield, an aliquot of the supernatant collected after the immobilization reaction was assayed for P450 activity using BFC as the substrate (initial rate measurement, see below). The immobilization yield was calculated from the difference in P450 activity found in the supernatant versus the control containing unmodified resin 4a mixed with free mutant 3 enzyme. This was also compared to the standard enzyme activity of mutant 3 in solution to rule out non-specific interaction of the enzyme with the resin. The activity of the enzyme immobilized on the resin was measured using the testosterone assay (end-point assay, see below) after washing the resin with KPi buffer (0.1 M at pH 7.4).
Preparation of maleimide-functionalized silica microspheres
To an aliquot (427 μL) of silica microspheres (0.1 μm, colloidal, Polysciences Inc.) was added 3-(aminopropyl)trimethoxysilane (2 μL). The mixture was vortexed briefly and incubated at room temperature for 5 min. The reaction was then quenched with the addition of HCl (conc., 8 μL). The resulting aminosilanized beads (14a) were then washed with KPi buffer (3 × 1 mL, 0.1 M at pH 7.4) using centrifugation and resuspension cycles. The beads were finally resuspended in the same buffer (500 μL). The beads were then mixed with crosslinker 15c (22 μmol in 500 μL DMSO) and incubated at 25°C and 250 RPM for 1.5 h. Reaction completion was confirmed using the ninhydrin test (see “Preparation of maleimide-functionalized resins” above). The beads were washed again to remove excess crosslinker 15c and resuspended in buffer (500 μL). The degree of maleimide loading was quantified from mixtures of bead aliquots (20 μL washed, supernatant included) and 4,4′-dithiodipyridine as described in “Generation of maleimide labels from their corresponding amines” above. The degree of loading was generally ~4.3 nmol maleimide/20 μL beads. Finally, the microspheres were concentrated by centrifugation and resuspension in KPi buffer (200 μL).
Immobilization of CYP3A4 mutant 3 onto maleimide-functionalized silica microspheres
An aliquot of TCEP-reduced CYP3A4 mutant 3 (43 μL, see “Immobilization of CYP3A4 mutant 3 onto maleimide functionalized resins” above) was combined with the maleimide-functionalized microsphere suspension prepared as above (100 μL, contained ca. ~54 nmol maleimide) and incubated at 25°C and 250 RPM for 2 h. Controls were also performed in which the beads were quenched with DTT (2 mM from a 0.1 M stock solution in Milli-Q water) prior to combining with mutant 3, or in which no beads were included. At the end of the incubation period, the reactions were quenched with DTT and the enzyme activity in the supernatant was assayed with BFC (initial rate measurement, see below) in order to estimate the immobilization yield. The immobilization yield was calculated from the difference in P450 activity found in the supernatant versus the control containing pre-quenched resin. The beads were washed (2 × 1 mL) and resuspended in KPi buffer (final volume ~150 μL, 0.1 M at pH 7.4) before the activity of the immobilized enzyme was assayed using either testosterone or BFC (end-point assays, see below).
CYP3A4 activity assays with testosterone
End-point CYP3A4 activity was assayed from the conversion of testosterone to 6β-hydroxytestosterone. To assay the activity of CYP3A4 in solution, each enzymatic reaction contained P450 (0.4 μM), testosterone (50 μM) and either CPR (1.6 μM)/NADPH (2 mM), or cumene hydroperoxide (CHP, 100 μM) in KPi buffer (0.1 M at pH 7.4) in a final volume of 75 μL. For agarose resin-immobilized P450, each reaction contained P450 (ca. ~0.4 μM, 70 μL resin, supernatant removed), testosterone (667 μM) and either CPR (1.6 μM) and NADPH (1 mM) or CHP (1 mM) in KPi buffer (0.1 M at pH 7.4) in a final volume of 150 μL. Similarly, for microsphere-immobilized P450, each reaction contained P450 (ca. 0.4 μM, 150 μL microsphere suspension), testosterone (667 μM) and CHP (1 mM) in KPi buffer (0.1 M at pH 7.4) in a final volume of 300 μL. The buffer, testosterone, P450 and CPR (if required) were combined and pre-incubated for 5 minutes at 37°C. The reactions were initiated by the addition of NADPH or CHP, and the reaction was allowed to proceed for 1 h at 37°C with shaking at 250 RPM. At the end of the reaction, the tubes were spiked with internal standard cortexolone (5 μL from a 200 μM stock solution in methanol) and extracted with ethyl acetate (0.5 mL). The extracts were evaporated and re-dissolved in acetonitrile (75 μL for the resin or 150 μL for the microspheres) before HPLC analysis. Separation and quantification of the product (relative to the internal standard cortexolone) was achieved using a 250 × 4.60 mm Synergi 4 μm Hydro-RP 80 Å column in line with a UV detector (set to monitor at 244 nm) using mobile phases A (Milli-Q water) and B (acetonitrile) at a flow rate of 0.5 mL/min. Elution consisted of an initial isocratic step at 50% phase B for 5 min followed by an increase to 70% phase B over the next 7 min and finally to 95% phase B over the final 8 min. The retention times were generally 7.1 min for the product 6β-hydroxytestosterone, 10.1 min for the internal standard cortexolone and 15.8 min for the substrate testosterone.
Determination of initial rates for CYP3A4 using the substrate 7-benzyloxy-4-trifluoromethylcoumarin (BFC)
To assay initial enzyme activity rates, the debenzylation of BFC to the fluorescent product 7-hydroxy-4-trifluoromethylcoumarin (HFC) was monitored over time with a microtiter plate fluorimeter (SpectraMax Gemini XS). CYP3A4 (~50 nM), CPR (150 nM) and BFC (30 μM) were first combined in KPi buffer (0.1 M at pH 7.4, used to adjust the final reaction volume to 150 μL) and pre-incubated for 5 minutes at 37°C. The reaction was initiated with the addition of NADPH (1 mM). Product formation was monitored over time with an excitation wavelength of 410 nm and an emission wavelength of 530 nm.
End-point activity determination for CYP3A4 using the substrate 7-benzyloxy-4-trifluoromethylcoumarin (BFC)
End-point activity of microsphere-immobilized CYP3A4 mutant 3 was also assayed with BFC. Each enzymatic reaction contained P450 (ca. ~0.4 μM from a 150 μL microsphere suspension), BFC (67 μM) and CHP (1 mM) in KPi buffer (0.1 M at pH 7.4) in a final volume of 300 μL. The buffer, BFC and P450 were combined and pre-incubated for 5 minutes at 37°C. The reactions were initiated by the addition of CHP, and the reaction was allowed to proceed for 1 h at 37°C with shaking at 250 RPM. At the end of the reaction, the tubes were spiked with the internal standard 7-hydroxycoumarin (5 μL from a 200 μM stock solution in ethyl acetate) and extracted with ethyl acetate (0.5 mL). The extracts were evaporated and re-dissolved in acetonitrile (150 μL) before HPLC analysis. Separation and quantification of the product (relative to the internal standard 7-hydroxycoumarin) was achieved using a 250 × 4.60 mm Synergi 4 μm Hydro-RP 80 Å column in line with a UV detector (set to monitor at 335 nm) using mobile phases A (Milli-Q water) and B (acetonitrile) at a flow rate of 0.5 mL/min. Elution consisted of a linear gradient from 50% phase B to 95% phase B over 20 min followed by an isocratic step at 95% phase B for 5 min. The retention times were 10.1 min for the product HFC, 6.5 min for the internal standard 7-hydroxycoumarin and 20.8 min for the substrate BFC.
RESULTS AND DISCUSSION
Analysis of fluorescently labelled wild-type CYP3A4
Information about the number and identity of CYP3A4 cysteine residues available for reactions with maleimides is desirable to achieve regioselective conjugation of CYP3A4. The primary sequence of CYP3A4 lists 7 cysteine residues (Figure 2). Upon careful inspection of the crystal structure of CYP3A4, one notes that C442 is coordinated to the heme prosthetic group and buried at the center of the protein, making it inaccessible to maleimide-modification when CYP3A4 is in its correctly folded holo-form. After chemical reaction of wild-type CYP3A4 with maleimide-containing molecules 1b and 3b (Scheme 1, Figure 3), MS analysis gave ambiguous results with poor reproducibility (data not shown). Failure of MS to provide the desired information is likely due to the low stability of CYP3A4 and its notorious tendency to form aggregates.
Figure 2.

A) Primary sequence of human CYP3A4 used here (referred to as the wild-type). Theoretical Cys-containing BrCN digestion peptides are underlined, and their approximate molecular weight when labelled with 10 is shown with the sequence number of the cysteine residue included in each peptide. B) Crystal structure of CYP3A4 (PDB: 1TQN) with cysteine residues highlighted in red.
Scheme 1.

A) General reaction used to label wild-type and mutant CYP3A4 enzymes with maleimides 1-13. B) General approach to preparing the non-commercial maleimide labels. See Figure 2 for detailed structures.
Figure 3.

Structures for molecules 1-9 and 14. The structures for commercial compounds 10-13 are not disclosed by the vendor. The larger sphere represents CarboxyLink resin in 4a-4c. The smaller sphere represents the silica microsphere in 14a and 14c.
A combination of methods was used here to characterize the CYP3A4 conjugates. These include single-molecule fluorescence spectroscopy, electrophoresis, UV and fluorescence spectroscopy, and enzymatic assays. For detailed characterization of the conjugates, wild-type CYP3A4 was reacted with an excess of fluorescent maleimide DyLight 549 (10, structure not disclosed by vendor) as shown in Scheme 1A. A sample of CYP3A4 labelled with 10 was imaged with a single molecule total internal reflection fluorescence microscopy setup (TIRF, see experimental section for details). Analysis of the images acquired allowed us to count the number of fluorescent moieties present per labelled protein molecule (Figure 4). Results indicated that modification with 10 gave predominantly singly modified CYP3A4 (76.9%), with a significant proportion being doubly and triply modified (18.6% and 4.2% respectively). On the down side, this same conjugate only retained ~5% of its original enzymatic activity when assayed with testosterone as the substrate and cytochrome P450 reductase (CPR)/NADPH as the cofactors (Figure 5, wild-type in blue). Replacing the native cofactors with cumene hydroperoxide (CHP) marginally improved the retained activity to ~10%. This deleterious effect on the activity hints at C98 being the main site of modification, based on recent studies indicating that large aromatic substitutions at this residue result in loss of activity37. The C98W and C98F mutations were indeed reported to result in a conformational shift that led to a subsequent loss in catalytic activity, due in part to a reduction in the ability of CYP3A4 to properly interact with its reductase CPR. The C98S CYP3A4 mutant on the other hand was reported to display unchanged activity37.
Figure 4.

Single-molecule photobleaching of variant CYP3A4 enzymes labelled with DyLight 549 maleimide (10). A) Representative example of a wide-field single molecule total internal reflection fluorescence (TIRF) microscope image obtained for fluorescently labelled CYP3A4 upon excitation at 532 nm. B) Representative intensity-time trajectories observed when the protein has one fluorophore/protein molecule (one step), two fluorophores/protein molecule (two steps) and three fluorophores/protein molecule (three steps). These trajectories were tallied for: CYP3A4 wild-type, n = 2212 (C); mutant 1, n = 1778 (D); and mutant 3, n = 703 (E). Pie colours used: blue = one step, red = two steps, purple = three steps, green = four steps.
Figure 5.

Relative activity of CYP3A4 wild-type (in blue, left side) and mutant 3 (in purple, right side) after labelling with maleimides 1-14. Percent activity is reported relative to that of non-labelled wild-type CYP3A4. All values are the average of duplicates or triplicates ± 1 standard deviation.
These results do not eliminate the possibility of different singly labelled regioisomers being formed concurrently. To gain a better handle on the identity of the residues being modified, we turned to an approach combining SDS-PAGE and BrCN digestion of fluorescently labelled P450 samples. BrCN is known to cleave proteins selectively after methionine residues. Sequence and molecular weight of the cysteine-containing peptides expected from BrCN digestion of CYP3A4 are shown on Figure 2. All of the expected digestion peptides contain a maximum of one cysteine residue. Digestion and peptide analysis of 10-labelled wild-type CYP3A4 revealed that at least 5 of the cysteine positions were accessible for modification (Figure 6). Indeed, SDS-PAGE of the digested protein revealed 5 peptide bands exhibiting fluorescence emission between 565 and 595 nm (580 nm band-pass filter) when excited at 532 nm (green-excited mode), as suggested by vendor for DyLight 549 (10). The bands corresponding to the C239- and C377-containing peptides are significantly fainter than the other ones, possibly suggesting minor reactivity at these sites.
Figure 6.

Tricine-SDS-PAGE analysis of BrCN digestion peptides of CYP3A4 variants labelled with DyLight 549 maleimide 10. The image was acquired with a Typhoon Trio + scanner using the Green-excited mode (λex = 532 nm), with a 580 nm band-pass filter (580 BP 30, transmits light between 565 and 595 nm with a transmission peak centered on 580 nm). Mutations include: C98S for mt 1, C98S/C377S/C468G for mt 2, C98S/C239S/C468G for mt 3, and C64S/C98S for mt 4.
Design of cysteine-depleted CYP3A4 mutants for site-specific cysteine modification
As alluded to above, a C98S substitution was desirable in order to prevent conjugation at C98 which is expected to incur a loss in activity. It was also desirable to minimize the number of mutations in order to increase the likelihood of obtaining fully active and stable mutants. Thus, mutant 1 contained only one mutation, C98S. Mutant 2 (C98S/C377S/C468G) and mutant 3 (C98S/C239S/C468G) were also generated to confirm the sites of modifications (especially since some peptides co-migrate on the gel), and in the hopes of producing variants that would be singly modified by maleimides.
The photobleaching profile of mutant 1 labelled with 10 was very similar to that of labelled wild-type enzyme (Figure 4). The key difference was revealed by gel analysis of the digested protein (Figure 6) which shows a decrease in the fluorescence intensity of the band with a molecular weight corresponding to the C98-containing peptide. Because of their very similar predicted molecular weights (4.0 kDa and 3.8 kDa respectively), the C64- and C98-containing peptides are not resolved under the electrophoretic conditions used. Since C98 was mutated to Ser in mutant 1, we do not expect the 98th-residue-containing peptide to have reacted with maleimide 10, thus reducing the fluorescence of the band at ~3.9 kDa. The remainder of fluorescence at this MW implies that C64 is modified in mutant 1, and both C64 and C98 are labelled in the wild-type enzyme. Taken together these results suggest that under our conditions, most of the cysteine residues can be modified, although only up to 2 (or 3, but rarely) are labelled at a time. This is consistent with previous reports suggesting that more than 2 cysteine-modifications per CYP3A4 protein resulted in protein denaturation15.
As for mutants 2 (C98S/C377S/C468G) and 3 (C98S/C239S/C468G), however, our data suggest that both mutants are modified primarily at C64 (Figure 6). They cannot be labelled at the co-migrating peptide containing C98 because of the C98S mutation. In the case of mutant 2, a minor band corresponding to labelling at C239 and a significant band suggesting modification at C442 are observed. The latter is likely due to partial denaturation of the enzyme resulting in exposure of the heme-ligated cysteine residue (C442). Since this band is not observed with mutant 3, a detrimental effect of C377S is inferred. On the other hand, we were delighted to see one major band with labelled mutant 3, suggesting very specific modification by 10 at the C64 position. For reasons that we do not understand, C377 was not modified despite sequence analysis confirming that it had not been mutated. This is likely due to a conformational difference found in mutant 3 rendering C377 unavailable for reactions with maleimides.
Corroborating results were also obtained by UV-Vis spectroscopy indicating the presence of ~0.8 DyLight groups (fluorophore of 10) per protein molecule under saturating conditions (Figure 7), and by photobleaching analysis which indicated that 96.9% of the mutant 3 conjugates were singly labelled (Figure 6). According to the analysis of the gel, the remaining 3–4% is likely to result from a small proportion of the protein being denatured and exposing C442 for labelling. Moreover, this high regioselectivity was consistently observed with all other fluorescent maleimide-conjugates of mutant 3 that were characterized, namely those of 1b, 5d, 8d, 11, 12 and 13 (data not shown).
Figure 7.

Absorbance spectrum of CYP3A4 mutant 3 labelled with DyLight 549 Maleimide (10). In blue, 10 maleimide equivalents were used resulting in ca. ~0.4 labels/protein molecule. In purple, 100 maleimide equivalents were used resulting in ca. ~0.8 labels/protein molecule. An ε280 nm of 40,340 M−1•cm−1 was used to calculate the concentration of CYP3A4.
The identity of the main residue being modified in mutant 3 was further verified with mutant 4, consisting of C64S and C98S mutations. As seen in Figure 6, characterization of 10-labelled mutant 4 shows that the band corresponding to the C64-containing peptide does not fluoresce anymore. This confirms that mutant 3 is modified almost exclusively at C64.
In addition, we were gratified to find that mutant 3 is as stable and even more active (150 ± 30%) than wild-type CYP3A4. And whereas labelled wild-type CYP3A4 lost >90% of its activity, 10-labelled mutant 3 remained at least as active, if not more, than unlabeled wild-type CYP3A4 (120 ± 20%).
Effect of varying the maleimide label on enzyme activity
Next we wanted to expand our scope of maleimides used to label the CYP3A4 mutant 3. This mutant was therefore reacted with a variety of structurally diverse maleimides, either commercially available or synthesized by our group (Scheme 1, Figure 3). Unlike the wild-type enzyme, mutant 3 retained some degree of activity upon labelling with most maleimides that were tested (Figure 5, labelled mutant in purple). DyLight maleimides 10-13 were especially interesting, producing mutant 3 conjugates with 80–120% of the activity of non-modified wild-type. On the other hand, addition of maleimides 1b-c, 2c-d, and 8d proved quite noxious to enzymatic activity. To assess the role that the linker portion (as opposed to the fluorophore portion) plays in the observed loss of activity, maleimides 3b and 3c were prepared and coupled to mutant 3. As seen in Figure 5, when modified with these maleimides, mutant 3 retains most of its activity. This suggests that the fluorophore portions are likely the main culprits causing the loss of activity observed upon labelling of mutant 3. It was envisaged that the fluorophores themselves might inhibit the activity of CYP3A4. This hypothesis was verified by monitoring the enzyme activity of unlabeled wild-type and mutant enzyme in the presence of the free fluorophores. As depicted in Figure 8, in general, the degree of inhibition observed in the presence of free fluorophores correlates with the loss of activity seen upon labelling (Figure 5). We can further explain the effect of modifying mutant 3 on its activity by observing changes in its reduced-CO spectrum. As seen in Figure 9, labelling with maleimides 2d and 9d caused an observable loss in intensity of the band at 450 nm and an increase in absorbance at 420 nm. This is characteristic of a switch to the non-functional P420 form of the enzyme. These results allow us to rule out steric effects and to attribute the loss of activity observed with selected labels to varying degrees of enzyme inhibition by the fluorophores themselves. In the end, it would seem prudent to test a variety of molecular structures when attempting to label CYP3A4.
Figure 8.

Effect of free fluorophores (dark purple: 100 μM, light purple: 1 mM) on the enzymatic activity of non-labelled CYP3A4 mutant 3. The data is normalized against the activity of non-labelled mutant 3 in the absence of free fluorophore. All values are the average of duplicates or triplicates ± 1 standard deviation.
Figure 9.

Effect of labelling with maleimides 9d (A) and 2d (B) on the CO-difference spectrum of CYP3A4 mutant 3 compared to the non-labelled control (C). Each plot compares the CO-difference spectrum before (in blue) and after (in purple) labelling. The CO-difference spectra were acquired in the manner reported by Omura and Sato26. Note that 2d absorbs maximally in the region around 500 nm.
Immobilization of CYP3A4 mutant 3 onto maleimide-functionalized CarboxyLink resin
As an extension of our work with soluble, fluorescent labels, we sought to modify mutant 3 with solid-supports derivatized with maleimide groups. Thus commercial CarboxyLink resin (4a) was modified to display a maleimide group at the end of a chain of varying length (see 4b and 4c). Maleimide coverage of the resin was calculated to be ~0.6 nmol/μL resin. This derivatization was followed by reaction with CYP3A4 mutant 3. No attempts were made to immobilize the wild-type enzyme in light of the fact that attachment of the linker alone (3b or 3c) to wild-type CYP3A4 suffices to bring the enzymatic activity below 5% (Figure 5). Attachment of 3b or 3c to mutant 3 on the other hand did not affect the enzyme activity significantly, and to our satisfaction, covalent bond formation with the modified resins 4b and 4c did not cause significant decrease in activity either. To assess the efficiency of CYP3A4 immobilization, the enzyme activity present in the supernatant was assayed using BFC as the substrate. The activity detected in the supernatants was <5% compared to 100% for a control containing 4a, indicating an immobilization yield of ≥95%. Moreover, once immobilized, the activity of CYP3A4 mutant 3 was retained (Figure 5) and no penalty was incurred on the thermodynamic stability of the mutant over the 16 h observation period. These results were very promising with respect to applications that require oriented immobilization of enzymes onto solid surfaces such as single-molecule spectroscopy and the production of protein microarrays.
Immobilization of CYP3A4 mutant 3 onto maleimide-functionalized silica microspheres
Finally, we sought to assess the activity mutant 3 upon immobilization to a surface that would be more appropriate for applications such as those mentioned above. Thus, 0.1 μm colloidal silica microspheres were aminosilanized and treated with crosslinker 15c to produce a PEG-maleimide surface analogous to that used to immobilize biomolecules on glass slides for single-molecule experiments38 or the production of microarrays. In fact, biomolecules-modified microspheres are often used directly in single molecule experiments39,40 and in the production of liquid bead arrays. CYP3A4 mutant 3 was then immobilized onto these beads as described above for the CarboxyLink agarose resin. Under the conditions used here (see experimental section for details), the immobilization yield was ~70%. Once immobilized, mutant 3 retained 60 ± 10% of its activity compared to the activity of mutant 3 in solution when assayed with testosterone as the substrate, and 90 ± 10% activity retained versus the wild-type enzyme (Figures 5 and 10). Activity of the microsphere-immobilized mutant 3 was also observed with BFC as the substrate (60 ± 10% versus wild-type and 40 ± 10% versus mutant 3).
Figure 10.

Immobilization of CYP3A4 mutant 3 on silica microspheres. A) Control: representative HPLC UV-chromatogram of 6β-hydroxytestosterone (P, 7.1 min) formation by CYP3A4 mutant 3 in solution. B) Representative HPLC UV-chromatogram of 6β-hydroxytestosterone (P, 7.1 min) formation by microsphere-immobilized CYP3A4 mutant 3. The peaks at 10.1 min and 15.8 min correspond to the internal standard cortexolone (IS) and the substrate testosterone (T) respectively. Note: a very large excess of testosterone was used in these assays.
CONCLUSION
In summary, we have generated a triple mutant of CYP3A4 (C98S/C239S/C468G) that is singly-modified at C64 by a variety of soluble and solid-supported maleimides. While the actual chemical modification at C64 incurs a minor cost to activity, this is made up for in many cases by the fact that this mutant is more active (150 ± 30%) than the wild-type enzyme. Bioconjugation of mutant 3 with some dyes (1, 2 and 8) had a detrimental effect on the enzyme activity. We were able to rule out the implication of steric effects in this observed loss in activity and demonstrated instead that enzyme inhibition by some fluorophore moieties on the maleimide labels played a predominant role. Finally, our bioconjugation strategy was successfully applied toward the immobilization of this CYP3A4 mutant on maleimide-functionalized CarboxyLink resin and silica microspheres. The fact that the enzyme, attached via C64, remained active on these solid matrices is promising with respect to applications that favour oriented immobilization and suggests that this mutant of CYP3A4 would be an appropriate choice in the development of protein microarrays, biosensors or bioreactors.
Supplementary Material
Acknowledgments
This work was funded by research grants from the National Science and Engineering Research Council of Canada (NSERC) to KA and also to GC. AM was supported by scholarships from NSERC and the Center for Green Chemistry and Catalysis. YH’s stipend was partly covered by an NSERC undergraduate scholarship. PK was supported by the McGill Chemical Biology Fellowship Program (Canadian Institutes of Health Research) for postgraduate scholarships.
Footnotes
SUPPORTING INFORMATION AVAILABLE: Preparation, characterization, and spectra of non-commercial maleimides. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Claridge SA, Schwartz JJ, Weiss PS. Electrons, photons, and force: quantitative single-molecule measurements from physics to biology. ACS Nano. 2011;5:693–729. doi: 10.1021/nn103298x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wong LS, Khan F, Micklefield J. Selective Covalent Protein Immobilization: Strategies and Applications. Chem Rev. 2009;109:4025–4053. doi: 10.1021/cr8004668. [DOI] [PubMed] [Google Scholar]
- 3.Kalia J, Raines RT. Advances in bioconjugation. Curr Org Chem. 2010;14:138–147. doi: 10.2174/138527210790069839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chefson A, Zhao J, Auclair K. Replacement of natural cofactors by selected hydrogen peroxide donors or organic peroxides results in improved activity for CYP3A4 and CYP2D6. ChemBioChem. 2006;7:916–919. doi: 10.1002/cbic.200600006. [DOI] [PubMed] [Google Scholar]
- 5.Chefson A, Zhao J, Auclair K. Sugar-mediated lyoprotection of purified human CYP3A4 and CYP2D6. J Biotechnol. 2007;130:436–440. doi: 10.1016/j.jbiotec.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 6.Chefson A, Auclair K. CYP3A4 activity in the presence of organic cosolvents, ionic liquids, or water-immiscible organic solvents. ChemBioChem. 2007;8:1189–1197. doi: 10.1002/cbic.200700128. [DOI] [PubMed] [Google Scholar]
- 7.Larsen A, May E, Auclair K. Predictable stereoselective and chemoselective hydroxylations and epoxidations with P450 3A4. J Am Chem Soc. 2011;133:7853–7858. doi: 10.1021/ja200551y. [DOI] [PubMed] [Google Scholar]
- 8.Sletten EM, Bertozzi CR. Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew Chem Int Ed. 2009;48:2–27. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang L, Schultz PG. Expanding the genetic code. Chem Commun. 2002;1:1–11. doi: 10.1039/b108185n. [DOI] [PubMed] [Google Scholar]
- 10.Kiick KL, Tirrell DA. Protein engineering by in vivo incorporation of non- natural amino acids: control of incorporation of methionine analogues by methionyl-tRNA synthetase. Tetrahedron. 2000;56:9487–9493. [Google Scholar]
- 11.Muralidharan V, Muir TW. Protein ligation: and enabling technology for the biophysical analysis of proteins. Nat Methods. 2006;3:429–438. doi: 10.1038/nmeth886. [DOI] [PubMed] [Google Scholar]
- 12.Besanceney-Webler C, Jiang H, Zheng T, Feng L, Soriano del Amo D, Wang W, Klivanski LM, Marlow FL, Liu Y, Wu P. Increasing the efficacy of bioorthogonal click reactions for bioconjugation: a comparative study. Angew Chem Int Ed. 2011;50:8051–8056. doi: 10.1002/anie.201101817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blanden AR, Mukherjee K, Dilek O, Loew M, Bane S. 4-Aminophenylalanine as a biocompatible nucleophilic catalyst for hydrazone-ligations at low temperature and neutral pH. Bioconjugate Chem. 2011;22:1954–1961. doi: 10.1021/bc2001566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Behrens CR, Hooker JM, Obermeyer AC, Romanini DW, Katz EM, Francis MB. Rapid chemoselective bioconjugation through the oxidative coupling of anilines and aminophenols. J Am Chem Soc. 2011;133:16398–16401. doi: 10.1021/ja2033298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsalkova TN, Davydova NY, Halpert JR, Davydov DR. Mechanism of interactions of R-naphthoflavone with cytochrome P450 3A4 explored with an engineered enzyme bearing a fluorescent probe. Biochemistry. 2007;46:106–119. doi: 10.1021/bi061944p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mie Y, Suzuki M, Komatsu Y. Electrochemically driven drug metabolism by membranes containing human cytochrome P450. J Am Chem Soc. 2009;131:6646–6647. doi: 10.1021/ja809364r. [DOI] [PubMed] [Google Scholar]
- 17.Nicoli R, Bartolini M, Rudaz S, Andrisano V, Veuthey JL. Development of immobilized enzyme reactors based on human recombinant cytochrome P450 enzymes for phase I drug metabolism studies. J Chromatogr A. 2008;1206:2–10. doi: 10.1016/j.chroma.2008.05.080. [DOI] [PubMed] [Google Scholar]
- 18.Nath A, Koo PK, Rhoades E, Atkins WM. Allosteric effects on substrate dissociation from cytochrome P450 3A4 in Nanodiscs observed by ensemble and single-molecule fluorescence spectroscopy. J Am Chem Soc. 2008;130:15746–15747. doi: 10.1021/ja805772r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Das A, Zhao J, Schatz GC, Sligar SG, Van Duyne RP. Screening of Type I and II drug binding to human cytochrome P450-3A4 in nanodiscs by localized surface plasmon resonance spectroscopy. Anal Chem. 2009;81:3754–3759. doi: 10.1021/ac802612z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shumyantseva VV, Bulko TV, Kuznetsova GP, Samenkova NF, Archakov AI. Electrochemistry of cytochromes P450: analysis of current-voltage characteristics of electrodes with immobilized cytochromes p450 for the screening of substrates and inhibitors. Biochemistry (Mosc) 2009;74:438–444. doi: 10.1134/s0006297909040129. [DOI] [PubMed] [Google Scholar]
- 21.Shumiantseva VV, Shikh EV, Makhova AA, Bylko TV, Kukes VG, Sizova OS, Ramenskaia GV, Usanov SA, Archakov AI. The influence of vitamin B group on monooxygenase activity of cytochrome P450 3A4: pharmacokinetics and electro analysis of catalytic properties. Biomed Khim. 2011;57:343–54. doi: 10.18097/pbmc20115703343. [DOI] [PubMed] [Google Scholar]
- 22.Ignaszak A, Hendricks N, Waryo T, Songa E, Jahed N, Ngece R, Al-Ahmed A, Kgarebe B, Baker P, Iwuoha EI. Novel therapeutic biosensor for indinavir-a protease inhibitor antiretroviral drug. J Pharm Biomed Anal. 2009;49:498–501. doi: 10.1016/j.jpba.2008.10.025. [DOI] [PubMed] [Google Scholar]
- 23.Chang G, Morigaki K, Tatsu Y, Hikawa T, Goto T, Imaishi H. Vertically integrated human P450 and oxygen sensing film for the assays of P450 metabolic activities. Anal Chem. 2011;83:2956–2963. doi: 10.1021/ac103059k. [DOI] [PubMed] [Google Scholar]
- 24.Nath A, Grinkova YV, Sligar SG, Atkins WM. Ligand binding to cytochrome P450 3A4 in phospholipid bilayer nanodiscs. J Biol Chem. 2007;282:28309–28320. doi: 10.1074/jbc.M703568200. [DOI] [PubMed] [Google Scholar]
- 25.Domanski TL, Liu J, Harlow GR, Halpert JR. Analysis of four residues within substrate recognition site 4 of human cytochrome P450 3A4: role in steroid hydroxylase activity and a-naphthoflavone stimulation. Arch Biochem Biophys. 1990;350:223–232. doi: 10.1006/abbi.1997.0525. [DOI] [PubMed] [Google Scholar]
- 26.Omura T, Sato R. The carbon monoxide-binding pigment of liver microsomes. J Biol Chem. 1964;239:2370–2378. [PubMed] [Google Scholar]
- 27.Oprian DD, Coon MJ. Oxidation-reduction states of FMN and FAD in NADPH-cytochrome P-450 reductase during reduction by NADPH. J Biol Chem. 1982;257:8935–8944. [PubMed] [Google Scholar]
- 28.Grassetti DR, Murray JF., JR Determination of sulfhydryl groups with 2,2′- or 4,4′-dithiodipyridine. Arch Biochem Biophys. 1967;119:41–49. doi: 10.1016/0003-9861(67)90426-2. [DOI] [PubMed] [Google Scholar]
- 29.Scheer JM, Ryan CA. A method for the quantitative recovery of proteins from polyacrylamide gels. Anal Biochem. 2001;298:130–132. doi: 10.1006/abio.2001.5384. [DOI] [PubMed] [Google Scholar]
- 30.Karam P, Ngo AT, Rouiller I, Cosa G. Unraveling electronic energy transfer in single conjugated polyelectrolytes encapsulated in lipid vesicles. Proc Natl Acad Sci USA. 2010;107:17480–17485. doi: 10.1073/pnas.1008068107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ngo AT, Karam P, Fuller E, Burger M, Cosa G. Liposome encapsulation of conjugated polyelectrolytes: toward a liposome beacon. J Am Chem Soc. 2008;130:457–459. doi: 10.1021/ja076217b. [DOI] [PubMed] [Google Scholar]
- 32.Ha T. Single-molecule fluorescence energy transfer. Methods. 2001;25:78–86. doi: 10.1006/meth.2001.1217. [DOI] [PubMed] [Google Scholar]
- 33.Harada Y, Sakurada K, Aoki T, Thomas DD, Yanagida T. Mechanochemical coupling in actomyosin energy transduction studied by in vitro movement assay. J Mol Biol. 1990;216:49–68. doi: 10.1016/S0022-2836(05)80060-9. [DOI] [PubMed] [Google Scholar]
- 34.Cordoba OL, Linskens SB, Dacci E, Santome JA. ‘In gel’ cleavage with cyanogen bromide for protein internal sequencing. J Biochem Biophys Methods. 1997;35:1–10. doi: 10.1016/s0165-022x(97)00018-3. [DOI] [PubMed] [Google Scholar]
- 35.Schagger H. Tricine-SDS-PAGE. Nat Protoc. 2006;1:16–22. doi: 10.1038/nprot.2006.4. [DOI] [PubMed] [Google Scholar]
- 36.Moss JA. Current Protocols in Protein Science. Unit 18.7.8. John Wiley & Sons Inc; 2005. Guide for resin and linker selection in solid-phase peptide synthesis. [DOI] [PubMed] [Google Scholar]
- 37.Wen B, Lampe JN, Arthur G, Roberts AG, Atkins WM, Rodrigues AD, Nelson SD. Cysteine 98 in CYP3A4 contributes to conformational integrity required for P450 interaction with CYP reductase. Arch of Biochem Biophys. 2006;454:42–54. doi: 10.1016/j.abb.2006.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zimmermann JL, Nicolaus T, Neuret G, Blank K. Thiol-based, site-specific and covalent immobilization of biomolecules for single molecule experiments. Nat Protoc. 2010;5:975–985. doi: 10.1038/nprot.2010.49. [DOI] [PubMed] [Google Scholar]
- 39.English BP, Min W, van Oijen AM, Lee KT, Luo G, Sun H, Cherayol BJ, Kou SC, Xie XS. Ever-fluctuating single molecules: Michaelis-Menten equation revisited. Nat Chem Biol. 2006;2:87–94. doi: 10.1038/nchembio759. [DOI] [PubMed] [Google Scholar]
- 40.Neuman KC, Nagy A. Single-molecule force spectroscopy: optical tweezers, magnetic tweezers and atomic force microscopy. Nat Methods. 2008;5:491–505. doi: 10.1038/nmeth.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.