

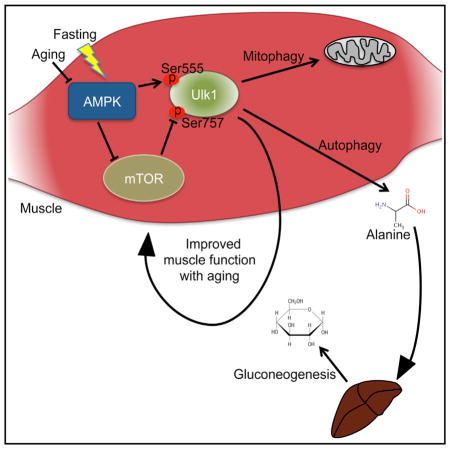
SUMMARY
The AMP-activated protein kinase (AMPK) activates autophagy, but its role in aging and fasting-induced muscle function has not been defined. Here we report that fasting mice lacking skeletal muscle AMPK (AMPK-MKO) results in hypoglycemia and hyperketosis. This is not due to defective fatty acid oxidation, but instead is related to a block in muscle proteolysis that leads to reduced circulating levels of alanine, an essential amino acid required for gluconeogenesis. Markers of muscle autophagy including phosphorylation of Ulk1 Ser555 and Ser757 and aggregation of RFP-LC3 puncta are impaired. Consistent with impaired autophagy, aged AMPK-MKO mice possess a significant myopathy characterized by reduced muscle function, mitochondrial disease, and accumulation of the autophagy/mitophagy proteins p62 and Parkin. These findings establish an essential requirement for skeletal muscle AMPK-mediated autophagy in preserving blood glucose levels during prolonged fasting as well as maintaining muscle integrity and mitochondrial function during aging.
In Brief
Bujak et al. highlight a critical role for the energy sensor AMPK in maintaining glycemia and muscle homeostasis. During fasting, muscle AMPK induces autophagy and muscle protein breakdown to prevent hypoglycemia. With old age, AMPK delays the onset of muscle myopathy and mitochondrial dysfunction.
INTRODUCTION
The healthcare costs of both age-related muscle wasting ($18 billion) and the loss of muscle strength ($10 billion) are well established, with severe consequences for rates of permanent disability and mortality in the elderly (Janssen et al., 2004). These statistics are set to rise with an expanding aging population, yet there is still no known treatment for age-related muscle dysfunction and myopathy besides exercise. Additionally, it is often observed that muscle quality control processes are impaired with aging, but the precise effectors mediating this decline are not yet clear.
Autophagy is an evolutionarily conserved recycling pathway whereby cellular materials are engulfed into an autophagosome and degraded through fusion with a lysosome (Vainshtein et al., 2014). Macroautophagy (herein referred to as autophagy) is robustly activated in response to nutrient deprivation (Mizushima et al., 2004). Skeletal muscle is required to supply gluconeogenic amino acids (such as alanine) for the maintenance of blood glucose levels during prolonged fasting, a process known as the glucose-alanine cycle (Felig et al., 1970). Autophagy is also essential for the maintenance of skeletal muscle integrity as mice with defects in this pathway exhibit muscle degeneration and dysfunctional mitochondria, phenotypes that are accentuated with aging (Masiero et al., 2009; Nemazanyy et al., 2013). Thus, skeletal muscle autophagy is an essential process required for maintaining muscle function and whole-body energy metabolism (Sandri, 2010).
The adenosine monophosphate-activated protein kinase (AMPK) is an αβγ heterotrimeric protein kinase that responds to energetic stress to maintain energy balance through phosphorylation of a diverse network of key metabolic pathways (Steinberg and Jørgensen, 2007). In skeletal muscle, exercise, hormones, nutrients, and insulin sensitizing therapeutics have been shown to increase AMPK activity. In contrast, the genetic removal of AMPK from skeletal muscle dramatically reduces exercise capacity and mitochondrial function (Lantier et al., 2014; O’Neill et al., 2011).
AMPK has recently been recognized as an activator of autophagy that occurs through a multi-pronged mechanism involving inhibition of the mammalian target of rapamycin (mTOR) complex and direct activation/phosphorylation of Forkhead box O3 (FoxO3) and the Unc-51-like kinase 1 (Ulk1). Specifically, AMPK inhibits mTOR through phosphorylation of TSC2 (Inoki et al., 2003) and raptor (Gwinn et al., 2008) to promote the formation of an Ulk1-Atg13-FIP200 complex. AMPK phosphorylates Beclin 1 to activate the pro-autophagy Vps34 complex and inhibits the non-autophagy Vps34 complex (Kim et al., 2013a). AMPK also promotes autophagy by activating FoxO3 (Sanchez et al., 2012), which upregulates the transcription of numerous muscle-specific atrogenes (Mammucari et al., 2007). Further, AMPK directly activates both autophagy and mitophagy through phosphorylation of Ulk1 at several primary sites, including Ser555 (Egan et al., 2011), Ser317, and Ser777, while mTOR inhibits this process via Ulk1 phosphorylation at Ser757 (Kim et al., 2011).
In the current study, we investigated the importance of muscle AMPK in controlling autophagy. We find that during prolonged fasting, AMPK muscle-specific null (AMPK-MKO) mice fail to induce sufficient autophagy and muscle breakdown to protect against hypoglycemia and hyperketosis, indicating that muscle AMPK is an essential component of the glucose-alanine cycle (Felig et al., 1970). We also show that aged AMPK-MKO mice have impaired muscle autophagy and develop a dramatic age-associated myopathy and mitochondrial disease.
RESULTS
AMPK-MKO Mice Have Impaired Muscle Wasting during Fasting and Develop Hypoglycemia and Hyperketosis
During prolonged fasting, blood glucose levels are maintained by switching to fatty acid oxidation and breaking down skeletal muscle for gluconeogenic substrates (Felig et al., 1970). Skeletal muscle AMPK is activated by fasting, resulting in phosphorylation and inhibition of acetyl-CoA carboxylase 2 (ACC2), causing a reduction in malonyl-CoA and an increase in fatty acid oxidation (Steinberg and Jørgensen, 2007). Therefore, we fasted mice lacking skeletal muscle AMPK (O’Neill et al., 2011) and mice with an ACC S212A knockin mutation (ACC2 KI) (O’Neill et al., 2014), which have an impaired ability to regulate fatty acid oxidation in muscle in response to pharmacological AMPK activators such as AICAR. We found that both AMPK-MKO (Figures 1A and S1A–S1C) and ACC2 KI mice (Figures S1D–S1G) were able to adapt to this energetic stress by increasing rates of fatty acid oxidation, while maintaining normal voluntary activity levels (Figures S1H and S1I). These data indicate that fasting-induced increases in fatty acid oxidation can occur independent of AMPK/ACC2 signaling.
Figure 1. During Fasting, AMPK-MKO Mice Have Hypoglycemia, Hyperketosis, and Impaired Breakdown of Muscle.

(A and B) Fatty acid oxidation (A) and blood glucose (B) during fasting (n = 6–12).
(C) Liver and gastrocnemius muscle glycogen (n = 5–6).
(D) ATT after 24 hr fast (n = 6–10).
(E and F) Serum alanine (E, n = 4–5) and serum β-hydroxybutyrate (F, n = 4–6). Data are means ± SEM.
(G) Myofiber CSA change of the TA muscle presented as median ± SEM (n = 4–6). * WT versus AMPK-MKO p < 0.05, **p < 0.01, # fed versus fasted p < 0.05, ## p < 0.01, ### p < 0.001. See also Figure S1 and Table S1.
Despite normal rates of whole-body fatty acid oxidation, blood glucose levels were significantly lower in fasted AMPK-MKO (Figure 1B) but not ACC2 KI (Figure S1J) mice compared to wild-type (WT) littermates. Hypoglycemia in AMPK-MKO mice was not due to alterations in basal levels of muscle or liver glycogen (Figure 1C). Serum lactate, glycerol, and non-esterified fatty acids (NEFAs) were also not different between genotypes (Table S1).
During prolonged fasting, alanine is a liver-specific gluconeogenic precursor that is used to maintain blood glucose (Felig et al., 1970). We therefore conducted an alanine tolerance test (ATT) following a 24 hr fast and found that while AMPK-MKO mice had lower fasting blood glucose levels, injection with alanine restored blood glucose in both WT and AMPK-MKO mice (Figure 1D). These data indicate that liver gluconeogenesis was not compromised in AMPK-MKO mice. We therefore hypothesized that the impaired maintenance of blood glucose in fasting AMPK-MKO mice might be due to an inability to supply the liver with gluconeogenic substrates. Compared to WT mice, alanine levels were dramatically reduced with fasting in AMPK-MKO mice (Figure 1E). Consistent with lower serum alanine levels and an inability to maintain gluconeogenesis, fasting resulted in a 45-fold increase in serum β-hydroxybutyrate in AMPK-MKO mice (Figure 1F).
Skeletal muscle contains the largest source of amino acids, and during prolonged fasting, the gluconeogenic demands for alanine are maintained by catabolizing muscle, resulting in a reduction in muscle size (Sandri, 2010). We found that tibialis anterior (TA) myofiber cross-sectional area (CSA) of WT mice was reduced by 18% after a 48 hr fast (from 1,812 to 1,481 μm2), but the myofiber size of AMPK-MKO mice remained unaffected (from 1,608 to 1,618 μm2) (Figure 1G). These findings indicate that skeletal muscle AMPK is indispensable for activating muscle wasting to supply the essential gluconeogenic amino acids that prevent hypoglycemia and hyperketosis.
AMPK-MKO Mice Have Impaired Induction of Autophagy with Fasting
Fasting-induced muscle wasting involves the induction of autophagy; therefore, we tested the hypothesis that a lack of skeletal muscle AMPK may have resulted in an inability to activate this pathway. Fasting increased activating phosphorylation of AMPK at Thr172 and inhibitory phosphorylation of ACC (a marker of AMPK activation) at Ser212 in WT but not AMPK-MKO mice (Figures 2A–2C and Table S2), indicating that AMPK activation is blocked in muscle of AMPK-MKO mice.
Figure 2. AMPK-MKO Mice Fail to Induce Autophagy Signaling in Response to Fasting.

(A) Representative western blots from TA muscle.
(B–I)Analysis of western blots for the following proteins: (B) AMPK Thr172 (n = 3–6), (C) ACC Ser212/ACC (n = 6–8), (D) Bnip3 (n = 5–12), (E) S6K Thr389/S6K (n = 5–12), (F) Ulk1 Total (n = 6–8), (G) Ulk1 Ser555 (n = 6–8), (H) Ulk1 Ser555/Ulk1 (n = 6–8), and (I) Ulk1 Ser757/Ulk1 (n = 5–8).
Data are means ± SEM. * WT versus AMPK-MKO p < 0.05, **p < 0.01, ***p < 0.001, # fed versus fasted p < 0.05, ### p < 0.001, † main effect of genotype p < 0.05, †† p < 0.01, ††† p < 0.001, § main effect of fasting p < 0.05. See also Figure S2 and Table S2.
FoxO3 is activated by AMPK and is an important modulator of muscle fiber size during prolonged fasting (Mammucari et al., 2007; Sanchez et al., 2012). Since the transcriptional regulation of muscle atrophy precedes muscle loss, we investigated protein and mRNA expression of the FoxO3 pathway at 12 and 24 hr of fasting in addition to the 48 hr time point. Fasting increased total FoxO3 expression, and there was a main effect of higher total and phosphorylated FoxO3 at Ser413 in the AMPK-MKO mice (Figures S2A–S2C); however, phosphorylation of FoxO3 Ser413/FoxO3 was not different between genotypes (Figures S2A and S2D). The muscle-specific atrophy protein and FoxO3 target Bnip3 was, however, significantly lower in AMPK-MKO mice in response to fasting (Figure 2D). Protein expression of another muscle-specific atrophy protein and primary FoxO3 transcriptional target, Atrogin1, was increased with fasting independent of genotype (Figures S2A and S2E). A third FoxO3 target, MuRF1, showed no changes in response to fasting in either genotype (Figures S2A and S2F). The mRNA expression of these FoxO3 family members increased with fasting as expected, but no differences were detected between WT and AMPK-MKO mice (Figure S2G). Since mitochondrial shaping dynamics have been shown to regulate muscle atrophy via the AMPK-FoxO3 axis (Romanello et al., 2010), we investigated the mRNA expression of primary mitochondrial fission and fusion proteins. Mitofusin 1 expression was significantly lower in fed AMPK-MKO mice and there was a main genotype effect of lower mitofusin 2, but other related transcripts were not altered (Figure S2H). Increased PGC1α has been shown to inhibit muscle atrophy by blocking autophagy (Cannavino et al., 2014), but this was not altered in the AMPK-MKO mice (Figure S2H). These data indicate that during fasting AMPK is required for the induction of the FoxO3 transcriptional target and autophagy protein Bnip3, but that AMPK is not essential for the control of all FoxO3 targets.
mTOR and Ulk1 form a unified feedback loop that reciprocally regulates protein balance through the control of synthetic pathways and the initial steps of autophagosome formation (Egan et al., 2011; Kim et al., 2011). To determine if mTOR was activated in AMPK-MKO mice, we measured the downstream target S6 kinase (S6K) and found a significant main effect of elevated S6K Thr389 phosphorylation/total S6K in AMPK-MKO mice (Figure 2E). Activated Ulk1 forms an isolation membrane, which is an essential first step in autophagosome formation (Vainshtein et al., 2014). Fasting increased the expression of total Ulk1, but this was independent of AMPK (Figures 2A and 2F). In contrast, Ulk1 Ser555 phosphorylation mirrored changes in AMPK activity, in that it was lower in AMPK-MKO mice under fed and fasted conditions and increased in fasted WT but not AMPK-MKO mice (Figures 2A, 2G, and 2H). Phosphorylation of Ulk1 at Ser317/total Ulk1 showed no differences between genotypes or fasting (Figures 2A and Table S2). There was also a main effect of higher phosphorylation of Ulk1 at the mTOR-dependent inhibitory site Ser757 in AMPK-MKO compared to WT mice (Figures 2A and 2I). These findings support the concept that AMPK activation of Ulk1 likely involves direct and indirect mechanisms including both the phosphorylation of Ser555 and the prevention of inhibitory mTOR phosphorylation at Ulk1 Ser757.
Lipidation of LC3 is one of the last essential steps in the formation of an activated autophagosome and is considered one of the most reliable markers of autophagy (Kabeya et al., 2000). To examine whether this parameter was altered, the TA muscle was electroporated with fluorescently tagged LC3 prior to fasting for 48 hr. The accumulation of RFP-LC3 puncta represents active autophagosomes aggregating during autophagy and was not different between genotypes under fed conditions, a finding consistent with low levels of AMPK activity and unaltered basal autophagy. However, during fasting RFP-LC3 puncta were increased in WT but not AMPK-MKO littermates (Figures 3A and 3B). This effect was not observed with an empty RFP construct (Figure S2I). To confirm that the lower amount of LC3 puncta in AMPK-MKO mice is not due to the exhaustive degradation of autophagosomes, we injected mice with colchicine (for 2 days), which prevents autophagic flux by inhibiting autophagosomelysosome fusion in skeletal muscle (Ju et al., 2010). LC3 lipidation was not altered in response to colchicine treatment during the fed condition, suggesting basal-general autophagic flux was not altered. However, WT mice did experience the expected increase in LC3 lipidation with fasting (as determined by the ratio of LC3II/LC3I) and a substantially greater increase when injected with colchicine prior to fasting (Figure 3C). Importantly, these changes were absent in AMPK-MKO mice.
Figure 3. AMPK-MKO Mice Have Impaired Autophagic Flux in Response to Fasting.

(A and B) Photomicrographs (A, scale bar, 10 μm) and quantification (B, n = 3–4) of RFP-LC3 puncta in TA muscle.
(C and D) LC3II/LC3I expression in mice treated with sterile water or colchicine (C, n = 9–19) and Parkin expression (D, n = 4–5).
(E and F) Photomicrographs of Parkin (scale bar, 10 μm), Tom20, and merged image (scale bar, 1 μm) (n = 3 per group) (E) and quantification in TA muscle (F).
Data are means ± SEM. * WT versus AMPK-MKO p < 0.05, **p < 0.01, ***p < 0.001, ### fed versus fasted p < 0.001, Φ Φ Φ fast saline versus fast colchicine p < 0.001.
Analysis of the individual LC3 isoforms showed that WT mice had the expected reduction in LC3I (Figure S2J) and increase in LC3II (Figure S2K) observed with fasting and colchicine, consistent with activation of autophagy. In contrast, AMPK-MKO mice had increased LC3I and LC3II with colchicine treatment during fasting. This finding of simultaneously higher LC3I and LC3II in AMPK-MKO mice mirrors other mouse models with reductions in autophagic flux including muscle-specific HDAC1/2 KO mice (Moresi et al., 2012) and in WT mice given colchicine for extended periods (beyond 5 days) (Ju et al., 2010). Since LC3 mRNA expression was indistinguishable between genotypes during fed, fasting, and colchicine treatments (Figure S2L), these data point to AMPK-MKO mice having increased LC3 protein stability in response to a blockade in autophagic flux.
Protein and mRNA expression of the autophagy adaptor protein p62 was increased with fasting, but was not different between genotypes or with colchicine treatment (Figures S2M and S2N). Corresponding with a reduction in autophagic flux, accumulation of the mitophagy protein Parkin, a protein that tags dysfunctional mitochondria for degradation, was over 2-fold higher in both the fed and fasted AMPK-MKO mice (Figure 3D). As the mRNA expression of Parkin was similar between genotypes (Figure S2O), this indicates that the accumulation of this protein in AMPK-MKO mice is due to the impaired removal of mitochondria tagged for degradation. This finding was further corroborated by immunostaining for Parkin and the mitochondrial protein Tom20 in TA muscle, which demonstrated an ~2.5 fold greater accumulation of Parkin in both fed and fasted AMPK-MKO mice and the co-localization of this protein with mitochondria (Figures 3E and 3F). These data indicate that skeletal muscle AMPK is required for the maintenance of basal mitophagy and for the induction of autophagy and mitophagy during fasting.
AMPK-MKO Mice Experience Severe Age-Related Myopathy and Mitochondrial Disease
Although acute metabolic stressors like nutrient deprivation strongly induce autophagy, tissues undergo a basal-level of autophagic flux even during nutrient abundance in order to recycle older proteins and maintain quality control for those newly synthesized (Mizushima et al., 2004). Aging reduces autophagic flux and mitochondrial function, effects that have been associated with lower skeletal muscle AMPK activity (Salminen and Kaarniranta, 2012). We therefore examined whether a loss of muscle AMPK may promote aging-induced muscle dysfunction and found that in the absence of AMPK, TA muscles had more fibers containing centrally located nuclei (Figure 4A), accumulated more collagen (Figure 4B), and had a greater number of large rounded fibers, small angular fibers, and necrotic fibers than aged WT littermates (Figure 4C). Consistent with the dramatic reductions in muscle quality, aged AMPK-MKO mice had lower maximal and specific force production, indicating impaired muscle function compared to aged WT littermates (Figures 4D and 4E). In contrast young-adult WT and AMPK-MKO mice had comparable maximal force production (WT = 923.8 ± 20.5 mN, AMPK-MKO = 931.4 ± 28.4 mN, n = 8). Cardiac cachexia can contribute to muscle weakness/atrophy and is characterized by cardiac hypertrophy, increased circulating inflammatory markers such as TNF-α, IL-6, and IL-1β, and loss of adipose tissue and lean mass (Anker and Sharma, 2002). We found that cardiac mass (WT = 139.8 ± 4.4 mg, n = 4, AMPK-MKO = 139.9 ± 5.0 mg, n = 6), systemic inflammation (Figure S2P), and adiposity/lean mass (Figure 4F) were all comparable between aged WT and AMPK-MKO mice. These data indicate that AMPK-MKO mice have an age-related myopathy that impairs muscle function that is unlikely the result of cardiac cachexia.
Figure 4. Aged AMPK-MKO Mice Develop Exacerbated Myopathy and Mitochondrial Disease.

(A) Percent of fibers with centrally located nuclei (n = 4–10) and (B) collagen accumulation (n = 3–10) in TA muscle.
(C–K) Aged AMPK-MKO and WT mice. (C) Photomicrograph (scale bar, 100 μm) showing large rounded fibers (closed arrow), small angular fibers (open arrow), and necrotic fibers (asterisk) in TA. (D) Maximum isometric and (E) specific force production of TA muscle (n = 4–6). (F) Percent adiposity with representative CT images (n = 4–7). Mitochondrial size in the (G) IMF and (H) SS regions of the myofiber (n = 3 per group). Number of mitochondria in the (I) IMF and (J) SS myofiber regions (n = 3 per group). (K) Representative electronmicrograph showing enlarged mitochondria (closed arrow) (scale bar, 4 μm).
(L and M) Number of mtDNA copies (L, n = 6–9) and mtDNA deletions (M, n = 5–10) in quadriceps muscle.
(N and O) p62 (N) and Parkin (O, n = 7–11) expression in extensor digitorum longus muscle in fed state.
Data are means ± SEM. * WT versus AMPK-MKO p < 0.05, **p < 0.01, ***p < 0.001, # fed versus fasted p < 0.05, ## p < 0.01, ### p < 0.001.
Alterations in mitophagy are thought to result in the decline of mitochondrial content and quality with aging (Terman et al., 2010). Electron microscopy imaging revealed that the average size of mitochondria from both the intermyofibrillar (IMF) and sub-sarcolemmal (SS) regions were twice as large in aged AMPK-MKO mice compared to WT (Figures 4G and 4H). Aged AMPK-MKO mice also had a substantially lower number of mitochondria present in the IMF and SS regions (27% and 51%, respectively) (Figures 4I–4K) and reduced mitochondrial DNA (mtDNA) copy number (Figure 4L), consistent with what is seen in human aging (Crane et al., 2010). These changes in mitochondrial size were much more dramatic than we have observed previously in young adult AMPK-MKO (O’Neill et al., 2011). Mutations in mtDNA are linked to mitochondrial disease (Park and Larsson, 2011) and we found that aged AMPK-MKO mice had significantly more mtDNA deletions than young AMPK-MKO mice (Figure 4M). Aged ad libitum-fed AMPK-MKO mice also had a 2-fold increase in p62 and Parkin accumulation, suggesting that basal autophagic clearance of mitochondria is chronically impaired in muscle of these mice (Figures 4N and 4O). These findings identify that muscle AMPK is essential to prevent aging-associated myopathy.
DISCUSSION
Autophagy is an essential mediator of cellular maintenance, particularly in tissues containing long-lived post-mitotic cells such as muscle. Skeletal muscles serve as the largest reserve of protein in the body. In periods of prolonged fasting, alanine is the primary amino acid to be released peripherally from muscle and converted into glucose within the liver during the glucose-alanine cycle (Felig et al., 1970). The deletion of muscle AMPK activity prevented mice from fasting-induced muscle wasting, resulting in lower levels of serum alanine and blood glucose and the development of hyperketosis. Since increased ACC activity caused by the loss of muscle AMPK could potentially lower fatty acid oxidation in these mice, we also determined the fasting response in ACC2 KI mice, but found that these mice did not develop hypoglycemia, suggesting the hypoglycemia observed in AMPK-MKO mice is due to lower amino acid availability and not from altered fatty acid metabolism.
In order to identify the mechanisms by which skeletal muscle AMPK prevents fasting-induced hypoglycemia, hyperketosis, and reductions in muscle size, we assessed the localization of LC3, a protein that is required for autophagosome activation (Kabeya et al., 2000). We found that fasted WT mice had a significantly higher amount of RFP-LC3 puncta compared to AMPK-MKO mice. Reduced LC3 puncta in combination with the lower state of autophagic flux observed in AMPK-MKO mice indicates that in the absence of AMPK there is impaired autophagosome formation and induction of autophagy during fasting. AMPK has been shown to regulate both autophagy and mitophagy in liver cells via phosphorylation of Ulk1 at Ser555 in addition to three other AMPK-dependent sites (Egan et al., 2011). In agreement with this study, we found that only WT mice increased phosphorylation at the Ser555 site in response to fasting and that the ratio of Ser555/total Ulk1 was substantially decreased in fed and fasted AMPK-MKO mice. mTOR has also been found to regulate autophagy via inhibitory Ulk1 phosphorylation at Ser757 (Castets et al., 2013; Kim et al., 2011). Consistent with these reports, we found AMPK-MKO mice had higher activation of mTOR and greater inhibitory phosphorylation of Ulk1 at Ser757. Additionally, we observed lower Bnip3 and prevention of fasting-induced muscle loss in AMPK-MKO mice. This supports the findings of Romanello et al. that overexpression of FoxO3 and its target protein Bnip3 activated autophagy and muscle atrophy in an AMPK-dependent manner (Romanello et al., 2010). In addition, liver-specific deletion of the autophagy protein Atg7 also lowers serum amino acids and results in hypoglycemia (Ezaki et al., 2011). Thus, it appears that both skeletal muscle and liver autophagy are required to provide amino acids to sustain hepatic gluconeogenesis during prolonged food deprivation.
Aging is commonly associated with muscle wasting (sarcopenia), inappropriate fiber repair, and mitochondrial dysfunction (Johnson et al., 2013). Aged AMPK-MKO mice have increased centrally located nuclei, increased muscle collagen, and impaired muscle function compared to WT. These features resemble the accelerated age-induced myopathy and mitochondrial dysfunction observed in mice lacking Atg7 in muscle (Carnio et al., 2014; Masiero et al., 2009). In parallel to the pronounced myopathy, the mitochondria of aged AMPK-MKO mice have an aberrant enlarged morphology and an increased amount of mtDNA deletions, indicative of accelerated mitochondrial dysfunction. This mitochondrial phenotype of aged AMPK-MKO mice resembles effects seen in TSC-1-deficient mice, which have chronically impaired autophagy due to hyper-activation of mTORC1 (Castets et al., 2013) and with muscular dystrophy (Grumati and Bonaldo, 2012).
In summary, we demonstrate the essential role of muscle AMPK in regulating autophagy and glucose homeostasis during prolonged fasting and limiting aging-induced muscle and mitochondrial dysfunction. We show that this is likely mediated via directly phosphorylating Ulk1 at Ser555 and by indirectly blocking mTOR inhibitory Ulk1 phosphorylation at Ser757. These studies provide an important molecular link between muscle breakdown/alanine release and gluconeogenesis originally described by the Cahill laboratory (Felig et al., 1970). In addition, as aging is associated with reduced skeletal muscle AMPK (Reznick et al., 2007) and reduced autophagic function (Kim et al., 2013b), our studies suggest that activation of AMPK may be beneficial in mitigating aging-induced reductions in muscle function.
EXPERIMENTAL PROCEDURES
A detailed description of all methods can be found in the Supplemental Experimental Procedures. Briefly, all animal studies were approved by McMaster’s Animal Research Ethics Board (AUP #12-12-44). AMPK-MKO mice (O’Neill et al., 2011) were studied at 2 (young) or 18 (aged) months of age. ACC2 KI mice (Fullerton et al., 2013) were studied at 5 months of age. For fasting experiments, food was removed for a total of 12, 24, or 48 hr prior to sacrifice and tissue collection. ATT was performed following a 24 hr fast by injecting 2 g/kg L-alanine (Sigma) in PBS intraperitoneally (i.p.). Autophagic flux was evaluated following i.p. injection with 100 μl of either sterile water (control) or colchicine (0.4 mg/kg/day) daily for 2 days. TA muscles were electroporated with tandem fluorescent-tagged LC3 plasmid or RFP plasmid, and tissues were collected 5 days later. Contractile properties of the TA muscle were measured in situ. Immunostaining, transmission electron microscopy, western blotting, and RT-qPCR conditions and reagents used are described in the Supplemental Experimental Procedures. Serum metabolites and cytokines were measured according to manufacturer’s instructions. Liver and muscle glycogen were determined via acid hydrolysis and colorimetric detection of glucose. mtDNA copy number and deletions were assessed using primers targeted to the Nd1, Nd4, and β-globin genes. All statistical analysis was performed using Student’s t test or two-way ANOVA as appropriate on GraphPad Prism 5. p < 0.05 was considered statistically significant. Data are presented as mean ± SEM except where stated otherwise.
Supplementary Material
Highlights.
Muscle AMPK is required for the induction of autophagy during fasting
AMPK is required for proteolysis to maintain blood glucose during fasting
A loss of AMPK accelerates aging-induced myopathy and mitochondrial dysfunction
Acknowledgments
We thank Alba Guarné for the use of her laboratory equipment. This work was supported by grants and fellowships from the Natural Sciences and Engineering Research Council of Canada (G.R.S), the Australian National Health and Medical Research Council (B.E.K. and G.R.S.), and the Canadian Institutes of Health Research (G.R.S.) and supported in part by the Victorian Government’s OIS Program (B.E.K.) and Canadian Foundation for Innovation (G.R.S. and T.J.H.). J.D.C. is supported by a MAC-Obesity postdoctoral fellowship. A.E.G. is supported by PhD scholarships from CIHR and MitoCanada. J.D.S is a Canadian Diabetes Association Scholar. G.R.S. is a Canada Research Chair in Metabolism and Obesity and the McMaster University J. Bruce Duncan Chair in Metabolic Diseases.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, two figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.cmet.2015.05.016.
References
- Anker SD, Sharma R. The syndrome of cardiac cachexia. Int J Cardiol. 2002;85:51–66. doi: 10.1016/s0167-5273(02)00233-4. [DOI] [PubMed] [Google Scholar]
- Cannavino J, Brocca L, Sandri M, Bottinelli R, Pellegrino MA. PGC1-α over-expression prevents metabolic alterations and soleus muscle atrophy in hindlimb unloaded mice. J Physiol. 2014;592:4575–4589. doi: 10.1113/jphysiol.2014.275545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnio S, LoVerso F, Baraibar MA, Longa E, Khan MM, Maffei M, Reischl M, Canepari M, Loefler S, Kern H, et al. Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep. 2014;8:1509–1521. doi: 10.1016/j.celrep.2014.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castets P, Lin S, Rion N, Di Fulvio S, Romanino K, Guridi M, Frank S, Tintignac LA, Sinnreich M, Rüegg MA. Sustained activation of mTORC1 in skeletal muscle inhibits constitutive and starvation-induced autophagy and causes a severe, late-onset myopathy. Cell Metab. 2013;17:731–744. doi: 10.1016/j.cmet.2013.03.015. [DOI] [PubMed] [Google Scholar]
- Crane JD, Devries MC, Safdar A, Hamadeh MJ, Tarnopolsky MA. The effect of aging on human skeletal muscle mitochondrial and intramyocellular lipid ultrastructure. J Gerontol A Biol Sci Med Sci. 2010;65:119–128. doi: 10.1093/gerona/glp179. [DOI] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezaki J, Matsumoto N, Takeda-Ezaki M, Komatsu M, Takahashi K, Hiraoka Y, Taka H, Fujimura T, Takehana K, Yoshida M, et al. Liver autophagy contributes to the maintenance of blood glucose and amino acid levels. Autophagy. 2011;7:727–736. doi: 10.4161/auto.7.7.15371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felig P, Pozefsky T, Marliss E, Cahill GF., Jr Alanine: key role in gluconeogenesis. Science. 1970;167:1003–1004. doi: 10.1126/science.167.3920.1003. [DOI] [PubMed] [Google Scholar]
- Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen ZP, O’Neill HM, Ford RJ, Palanivel R, O’Brien M, et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med. 2013;19:1649–1654. doi: 10.1038/nm.3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumati P, Bonaldo P. Autophagy in skeletal muscle homeostasis and in muscular dystrophies. Cells. 2012;1:325–345. doi: 10.3390/cells1030325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- Janssen I, Shepard DS, Katzmarzyk PT, Roubenoff R. The healthcare costs of sarcopenia in the United States. J Am Geriatr Soc. 2004;52:80–85. doi: 10.1111/j.1532-5415.2004.52014.x. [DOI] [PubMed] [Google Scholar]
- Johnson ML, Robinson MM, Nair KS. Skeletal muscle aging and the mitochondrion. Trends Endocrinol Metab. 2013;24:247–256. doi: 10.1016/j.tem.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju JS, Varadhachary AS, Miller SE, Weihl CC. Quantitation of “autophagic flux” in mature skeletal muscle. Autophagy. 2010;6:929–935. doi: 10.4161/auto.6.7.12785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013a;152:290–303. doi: 10.1016/j.cell.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YA, Kim YS, Oh SL, Kim HJ, Song W. Autophagic response to exercise training in skeletal muscle with age. J Physiol Biochem. 2013b;69:697–705. doi: 10.1007/s13105-013-0246-7. [DOI] [PubMed] [Google Scholar]
- Lantier L, Fentz J, Mounier R, Leclerc J, Treebak JT, Pehmoller C, Sanz N, Sakakibara I, Saint-Amand E, Rimbaud S, et al. AMPK controls exercise endurance, mitochondrial oxidative capacity, and skeletal muscle integrity. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2014;28:3211–3224. doi: 10.1096/fj.14-250449. [DOI] [PubMed] [Google Scholar]
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S, Sandri M. Autophagy is required to maintain muscle mass. Cell Metab. 2009;10:507–515. doi: 10.1016/j.cmet.2009.10.008. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moresi V, Carrer M, Grueter CE, Rifki OF, Shelton JM, Richardson JA, Bassel-Duby R, Olson EN. Histone deacetylases 1 and 2 regulate autophagy flux and skeletal muscle homeostasis in mice. Proc Natl Acad Sci USA. 2012;109:1649–1654. doi: 10.1073/pnas.1121159109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemazanyy I, Blaauw B, Paolini C, Caillaud C, Protasi F, Mueller A, Proikas-Cezanne T, Russell RC, Guan KL, Nishino I, et al. Defects of Vps15 in skeletal muscles lead to autophagic vacuolar myopathy and lysosomal disease. EMBO Mol Med. 2013;5:870–890. doi: 10.1002/emmm.201202057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill HM, Maarbjerg SJ, Crane JD, Jeppesen J, Jørgensen SB, Schertzer JD, Shyroka O, Kiens B, van Denderen BJ, Tarnopolsky MA, et al. AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci USA. 2011;108:16092–16097. doi: 10.1073/pnas.1105062108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill HM, Lally JS, Galic S, Thomas M, Azizi PD, Fullerton MD, Smith BK, Pulinilkunnil T, Chen Z, Samaan MC, et al. AMPK phosphorylation of ACC2 is required for skeletal muscle fatty acid oxidation and insulin sensitivity in mice. Diabetologia. 2014;57:1693–1702. doi: 10.1007/s00125-014-3273-1. [DOI] [PubMed] [Google Scholar]
- Park CB, Larsson NG. Mitochondrial DNA mutations in disease and aging. J Cell Biol. 2011;193:809–818. doi: 10.1083/jcb.201010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ, Liu ZX, Dong J, Mustard KJ, Hawley SA, et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007;5:151–156. doi: 10.1016/j.cmet.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanello V, Guadagnin E, Gomes L, Roder I, Sandri C, Petersen Y, Milan G, Masiero E, Del Piccolo P, Foretz M, et al. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 2010;29:1774–1785. doi: 10.1038/emboj.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A, Kaarniranta K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res Rev. 2012;11:230–241. doi: 10.1016/j.arr.2011.12.005. [DOI] [PubMed] [Google Scholar]
- Sanchez AM, Csibi A, Raibon A, Cornille K, Gay S, Bernardi H, Candau R. AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1. J Cell Biochem. 2012;113:695–710. doi: 10.1002/jcb.23399. [DOI] [PubMed] [Google Scholar]
- Sandri M. Autophagy in skeletal muscle. FEBS Lett. 2010;584:1411–1416. doi: 10.1016/j.febslet.2010.01.056. [DOI] [PubMed] [Google Scholar]
- Steinberg GR, Jørgensen SB. The AMP-activated protein kinase: role in regulation of skeletal muscle metabolism and insulin sensitivity. Mini Rev Med Chem. 2007;7:519–526. doi: 10.2174/138955707780619662. [DOI] [PubMed] [Google Scholar]
- Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT. Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid Redox Signal. 2010;12:503–535. doi: 10.1089/ars.2009.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainshtein A, Grumati P, Sandri M, Bonaldo P. Skeletal muscle, autophagy, and physical activity: the ménage à trois of metabolic regulation in health and disease. J Mol Med. 2014;92:127–137. doi: 10.1007/s00109-013-1096-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.