

Summary
Hematopoietic stem cells (HSCs) are rare cells that generate all the various types of blood and immune cells. High-quality transcriptome data have enabled the identification of significant genes for HSCs. However, most genes are expressed in various forms by alternative splicing (AS), extending transcriptome complexity. Here, we delineate AS to determine which isoforms are expressed in mouse HSCs. Our analysis of microarray and RNA-sequencing data includes differential expression of splicing factors that may regulate AS, and a complete map of splicing isoforms. Multiple types of isoforms for known HSC genes and unannotated splicing that may alter gene function are presented. Transcriptome-wide identification of genes and their respective isoforms in mouse HSCs will open another dimension for adult stem cells.
Keywords: hematopoietic stem cell, whole-transcriptome splicing, alternative splicing, RNA-sequencing analysis, HSC, ImmGen, unannotated splicing, splicing-function, adult stem cell splicing
Graphical Abstract
Highlights
-
•
This study presents the splicing landscape in mouse hematopoietic stem cells
-
•
Many HSC genes express multiple isoforms, some with alternative isoforms
-
•
High-resolution analysis allows the discovery of unannotated splicing
-
•
HSC-expressed isoforms can have similar or divergent functions
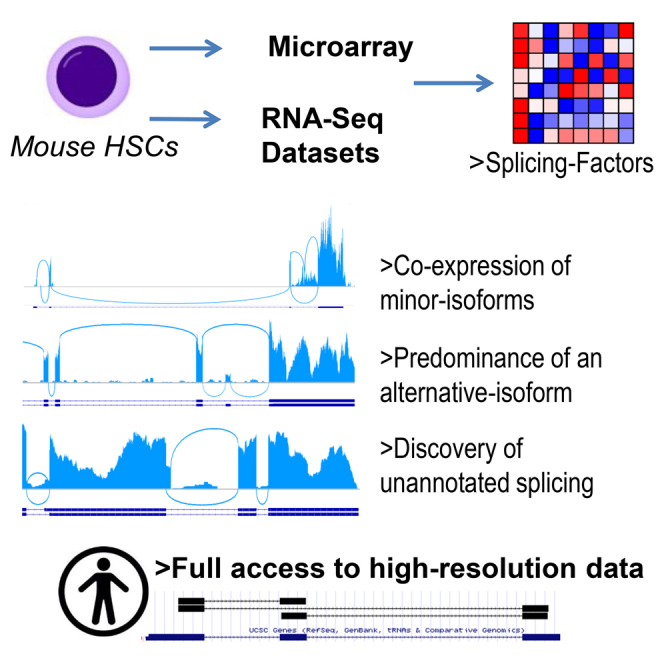
Gazit and colleagues mapped all splicing within mouse hematopoietic stem cells by analysis of microarrays and meta-analysis of RNA-sequencing datasets. They validated co-expression of minor isoforms, variant isoform predominance, multiple isoforms from one gene, and discovery of unannotated splicing. Functionally, variant isoforms may alter the activity of HSC genes, or not. Whole-transcriptome splicing data at nucleotide resolution is providing a useful tool for the community.
Introduction
Hematopoietic stem cells (HSCs) are a rare subset of cells that possess the abilities of self-renewal and multipotency (Bryder et al., 2006, Gazit et al., 2008). By virtue of these attributes, HSCs can regenerate blood and immune cells throughout life. Prospective isolation of HSCs using defined surface markers enables advanced research (Kiel et al., 2005, Osawa et al., 1996, Spangrude et al., 1988). However, our understanding of adult stem cells is incomplete, as is our ability to utilize them clinically. HSCs enable bone marrow transplantation, as they can naturally reconstitute the blood and immune systems of recipients, thus, saving tens of thousands of patients every year (Copelan, 2006, Thomas, 2005). Nevertheless, their transplantation is still limited because of the need to have a matched donor, without manipulation of the cells, further attesting to our incomplete understanding. Better knowledge of HSCs is essential for the field of adult stem cell research and to extend the application of HSCs in regenerative medicine beyond current practice.
The scientific understanding of stem cell functions advanced with molecular biology (Orkin and Zon, 2008, Rossi et al., 2012). Genetic studies have made seminal discoveries, finding the mutations in genes that are associated with hematopoietic diseases (Rosenbauer and Tenen, 2007). Such mutated genes revealed developmental, immune, and cellular processes. The development of microarray techniques has enabled the quantification of whole transcriptomes, revealing the expression of virtually all genes in their respective tissues. Transcriptome data obtained from highly purified HSCs made it possible to identify HSC-specific genes and map networks of genes (Gazit et al., 2013). We used transcriptome data to identify HSC genes that are capable of direct reprogramming blood cells into an induced HSC state (Riddell et al., 2014). However, most of the genes are transcribed into several mature mRNA by alternative splicing (AS), whereas microarray analysis is typically performed at the coarse level describing genes as discrete units. We bypassed this problem by cloning HSC factors for reprogramming directly from the cDNA of HSC (Riddell et al., 2014) but noted that the data lacked the resolution of isoform expression of each gene. High-resolution mapping of the expression of isoforms throughout the HSC transcriptome is needed for better understanding of the specific genes of interest. Fortunately, the emerging technique of RNA-sequencing (RNA-seq) can potentially identify and quantify all of the expressed isoforms simultaneously. Nevertheless, delineating the data into a complete AS map remains a challenge.
The splicing process increases transcriptome complexity: instead of having only one product per gene, there can be multiple distinguishable isoforms. Indeed, of 25,000 protein-coding genes, there are at least 10-fold more isoforms (Nilsen and Graveley, 2010). Isoforms include alternative splice-donor and/or splice-acceptor sites that change the length of each exon, or the inclusion/exclusion of an exon. If such an exon encodes for an activation domain, the isoform might invert the gene's function as the activation-deficient protein keeps its interaction and becomes an inhibitor. Knowing which isoforms are expressed is of paramount importance, since different tissues may express different isoforms of the same gene. Splicing had been recently mapped in human HSCs (Sun et al., 2014), suggesting significant roles for the maintenance and differentiation of stem cells. Hence, a map of the whole-transcriptome splicing of mouse HSCs is necessary to provide high-resolution data for future studies of these cells and advance the field of adult stem cell research.
Following the identification of embryonic stem cell (ESC)-specific AS (Gabut et al., 2011), their regulation had been studied, leading to the identification of muscleblind-like proteins as general negative regulators of stem cell-specific splicing in human cells (Han et al., 2013). Recently, unique AS in pluripotent stem cells has been discovered to be conserved in worms just as in humans (Solana et al., 2016), highlighting its fundamental role. These pioneering studies increased the interest in studying AS and regulation of ESCs, and reprogramming (Aaronson and Meshorer, 2013). Importantly, AS is implicated in many diseases after specific mutations that do not affect the coding sequence were discovered to cause aberrant splicing that can frequently disrupt gene function (Xiong et al., 2015). Clinical interest in aberrant splicing aided the discovery of mutations in splicing factors that drive hematopoietic malignancies (Steensma, 2012). This unstudied dimension of transcriptome complexity is yet to be studied in somatic stem cells.
In this study, we sought to capture the whole AS landscape of mouse HSCs. Obtaining data from extensive microarrays that cover much of the immune system was initially preferred, mainly because of the potential to undertake comparisons between cell types. RNA-seq data of highly purified HSCs were analyzed as they enable identification of both known and unknown transcripts. Microarray data were analyzed using exon ratios and RNA-seq data were analyzed by the direct observation of splice junctions. Independent validation using qRT-PCR encountered a striking preference for the RNA-seq data. Nevertheless, microarray data can still provide valuable information regarding the expression of splice factors across the hematopoietic system. RNA-seq revealed AS in all types of annotated genes, with some preference for genes involved in transcription; genes that are preferentially expressed in HSCs show extensive AS, with most having two or more isoforms. We refer to “canonical” and “variant” isoforms according to RefSeq. Some HSC genes mostly express the canonical isoform, others predominantly express variant isoforms with a potential different gene function in HSCs. Unannotated splicing was observed, resulting in isoforms not reported previously. By uploading our analyzed data on a custom track of the University of California Santa Cruz (UCSC) genome browser, we provide many scientists with the actual splicing of their genes of interest in HSCs. This study presents the comprehensive mapping of whole-transcriptome splicing in adult mouse HSCs.
Results
Microarrays and RNA-Seq Data for Splicing Analysis
Whole-transcriptome expression data of HSCs are available from extensive microarrays and from RNA-seq; the former provides well-established coverage of known genes in many cell types, while the latter can reveal both known and unknown genes. We analyzed Immunological Genome Project (ImmGen) consortium microarrays (Shay and Kang, 2013) and RNA-seq datasets that were obtained from several independent publications (Cabezas-Wallscheid et al., 2014, Qian et al., 2016, Sun et al., 2014, Venkatraman et al., 2013). The raw data output parameters (Figure 1A) suggest that the main advantage of using microarray data is that they cover many cell types (about 249): HSCs through progenitors and down to effector cells (Shay and Kang, 2013). On the other hand, RNA-seq data offer detection of all transcripts, including knowns and unknowns.
Figure 1.

Microarrays and RNA-Seq Data for Splicing in Mouse HSCs
(A) A comparison between the raw data gathered from ImmGen's microarrays and the combined results of ten RNA-seq samples (see Table S1 for details). n/a, not applicable.
(B) Gene expression correlation between a representative RNA-seq (sample A0) and the corresponding ImmGen microarray data in HSCs. A correlation for the log2 expression values of RNA-seq and ImmGen's microarrays for HSCs, both determined by their respective cell markers. Blue and red dots represent expression values having a fold-change of <4 or >4, respectively.
(C) Heatmap of splicing factor expression across hematopoietic cell types (detailed in Table S3). Relative expression value is shown on a scale of low (blue) to high (red).
To confirm the comparability of gene expression levels between microarray and RNA-seq, we calculated the correlation, which is high (r = 0.742 ± 0.019), suggesting a fairly good agreement between the two methods. Surprisingly, however, plotting the expression values, as shown in Figure 1B, indicates that there are numerous genes that “fall off” the diagonal and are substantially different between the RNA-seq and the microarray data. The ImmGen data were generated from male mice only (in order to minimize variability in this consortium dataset), while the RNA-seq data were generated from both males and females (Cabezas-Wallscheid et al., 2014, Qian et al., 2016, Sun et al., 2014, Venkatraman et al., 2013). Hence, at least some of the differences between the two datasets can be attributed to the animals' gender. Nevertheless, we find good correlative expression data across the transcriptome when analyzing genes as discrete units. Given that different cell types mostly vary in their transcriptomes, it is expected that their AS pattern will also differ. In Figure 1C we show a heatmap for the varying expression intensities of 216 splicing factors (Table S3) across the hematopoietic system and, indeed, a clear difference between cell types/groups is evident.
AS Analysis of Microarray Data
Following the good correlation between the datasets with respect to gene expression, we first analyzed the data for splicing using the microarray dataset, which provided broad coverage of many cell types. ImmGen's data were produced using Affymetrix ST 1.0 arrays, which were not originally designed to detect AS. Nevertheless, these arrays contain multiple probe sets per gene, and a previous study (Ergun et al., 2013) succeeded in retrieving splicing data. We hypothesized that we may identify exons that are either over- or underexpressed in HSCs. In brief, levels of exon expression in HSCs were compared with either all other cells, or with the “progenitors” group (Figure S1A). The seven candidate genes identified that might have exon-skipping isoforms in HSCs (Figure S1B) were validated using qRT-PCR. However, we found lack of any correlation with these predictions (Figure S1C). Despite this setback, we realized that, while microarrays might not be ideal for AS detection in HSCs using our method, the ImmGen data can provide information about splicing factors (Figure 1C); which ones are more active, or even unique, to HSCs and their close progeny. Indeed, Msi2, which plays an essential role in HSCs and in leukemia (de Andres-Aguayo et al., 2012, Kharas et al., 2010), showed preferential expression in HSCs. Our analysis finds a clear differential expression of splicing factors across the immune system (Figure 1C), suggesting there is room for further utilization of the ImmGen data for studies of splicing in immune cells.
AS Analysis of RNA-Seq
RNA-seq analysis for gene expression is established, but the analysis for splicing is not trivial yet (Hooper, 2014). We obtained the raw data for the RNA-seq of HSCs from four published studies (Cabezas-Wallscheid et al., 2014, Qian et al., 2016, Sun et al., 2014, Venkatraman et al., 2013); the file names and links are given in Table S1. The analysis included a bioinformatic pipeline using TopHat for read alignment, Cufflinks for read assembly into transcripts to determine the availability, reliability, and ability to identify previously unseen splicing (see Experimental Procedures for details). This process yielded a plethora of output data for further analysis and interpretation. Unlike microarrays, sequencing also contains reads that span exon-exon junctions (junction-spanning reads, or junction reads). Such junction reads provide direct evidence for the presence of spliced RNA. Visualization using the Integrative Genomics Viewer (IGV) (Robinson et al., 2011) highlights junction reads, enabling easy manual identification of splicing events. Importantly, our analysis is not limited to known transcripts but rather allows the discovery of unannotated genes and splicing events. We also generated tracks for the UCSC browser to enable online accessibility to any researcher to study their gene(s) of interest (see below). Some isoforms appear only in one or a few samples, which is to be expected since there are slight differences in sorting schemes and sequencing depth between the samples.
Splicing Is Common in Various Types of Genes
Equipped with global splicing data, we sought to focus on specific types of genes for further analysis. We used major gene ontology (GO) categories to dissect the transcriptome into subsets of biological relevance. As shown in Figure 2, the various types of genes all presented abundant splicing, with the average number of variants ranging between 4 and 5.7 isoforms per gene. Although some categories had fewer variants, such as “oxidoreductase” with only 4.06 variants per gene, and other categories had more variations, such as “transcription” with 5.72, it is evident that splicing is abundant. Averages may blur specific differences, so we plotted the actual distribution of splice variants for each gene category against the distribution in all genes, finding prominent deviations (Figures 2B–2D and S2), which are statistically significant (p values all bellow 0.001 by chi-square test). Clearly, each group of genes presents some deviations, usually on the numbers of genes with three to six expressed variants. We conclude that splicing is not limited to specific types of genes, but is common to all, with a preference for the gene group involved in transcription to have more isoforms expressed.
Figure 2.

Alternative Splicing Is Prevalent within Various Groups of Annotated Genes
(A) The average number of isoforms in each gene ontology annotation subcategory.
(B) Graphs of the number of isoforms per gene for a representative gene from each of the major gene ontology annotation subcategories shown in (A).
HSC Genes Reveal Multiple Splicing Forms
We focused next on a previously defined list of genes that are preferentially expressed in HSCs (Gazit et al., 2013), including key regulators, reprogramming factors (Riddell et al., 2014), and highly specific HSC genes (Gazit et al., 2014). Surprisingly, 248 of the 322 HSC genes captured by RNA-seq were found to have more than one isoform (range 2–22 isoforms, multiple AS types), while only 32 genes had a single isoform (and 43 genes were below the threshold). This finding highlights the need to recognize AS expression in order to better understand HSCs, as each isoform may change the function of a gene. To independently validate these results we sorted HSCs (Lineage–cKit+Sca1+CD34– flk2–) and performed qRT-PCR for 12 genes, including HoxA9, Meis1, Prdm16, Pbx1, Hlf, Nadk2, and CDKn1c (p57), which are presented below. We focused on transcription factors as they had the highest average splicing (Figure 2) and are of interest as cell-identity determinants. We found that the qPCR data were in good agreement with the RNA-seq data, supporting the validity of our analysis for AS in HSCs.
HoxA9 Expresses a Minor Retained-Intron Isoform in HSCs
Homeobox A9 (HoxA9) encodes for a DNA-binding protein that regulates morphogenesis, differentiation, and normal and malignant hematopoiesis (Collins and Hess, 2015, Fujimoto et al., 1998). We have previously identified it as having preferential expression in normal mouse HSCs (Gazit et al., 2013), but referred to HoxA9 as a discrete gene without consideration of AS (Riddell et al., 2014). This is a striking paradox, especially considering published data about HoxA9 AS in leukemia (Collins and Hess, 2015) and the possibility that such an isoform is expressed within normal HSCs. Indeed HoxA9 expresses two main transcripts in HSCs (Figure 3A): the canonical isoform (NM_010456) having two exons is the more studied, and the variant isoform of three exons, which excises an intron out of the first exon (NM_001277238). To better represent the relative expression of both isoforms, we have generated a Sashimi plot in which each splice junction is illustrated and reads are enumerated conveniently (Figure 3B). HoxA9 shows 13% ± 5% junction-spanning reads across the retained intron (Figure 3B). Independent qRT-PCR using isoform-specific primers (Figures S3A and S3B) found expression of both isoforms, with a predominance of the canonical isoform in agreement with the RNA-seq data (Figure 3C). The HoxA9 variant was discovered a while ago and named HoxA9T (Fujimoto et al., 1998). Our RNA-seq analysis and qRT-PCR validation show that the well-studied HoxA9 gene has substantial AS within HSCs, and the HoxA9T variant, which was previously related to leukemia (Collins and Hess, 2015), is expressed in normal mouse HSCs.
Figure 3.

Two Isoforms of HoxA9 Are Expressed in Mouse HSCs
(A) Raw RNA-seq data showing the known isoforms of HoxA9 (canonical, NM_010456; variant, NM_001277238), read coverage (upward pointing gray bars), and junctions (red arches that face downward because HoxA9 is transcribed from the negative strand) as shown in the IGV browser. Known exons are represented by blue rectangles, which are wide for open reading frames and narrow for UTRs; both canonical and variant forms are presented.
(B) A Sashimi plot of HoxA9 from four samples (A0, B0, B1, and A3, in descending order, respectively). Raw reads are visualized by bar height and splice junctions, and their respective read numbers are shown by the connecting arcs.
(C) Histogram chart showing the expression of each isoform relative to Hprt. Data of technical triplicate average ± SD from one representative experiment out of four are shown.
Meis1 Exon-Skipping Variant Is Minor
Meis-homeobox 1 (Meis1) is a major transcription factor in HSCs and leukemia. Using IGV visualization, we noted the possible presence of multiple AS events. Since Meis1 has 13 exons and stretches over 138.5 kb, we focused on exon 8 to better visualize its splicing (Figure S3C). The Sashimi plots show clear exon skipping (Figure S3D). This suggests that, although exon 8 can be present or absent, there is a significant preference for its expression in HSCs, resulting in a junction reads ratio of about 1:10. Indeed, qRT-PCR validation found the same preference for the expression of exon 8 as part of Meis1 in HSCs (Figures S3E and S3F). Importantly, the agreement between the RNA-seq analysis and the qRT-PCR assay again supports our findings and the validity of junction-indication as a quantification proxy for AS measure in HSCs.
Prdm16 Variant Is More Common than “Canonical” Isoform in HSCs
PR-domain-containing 16 (Prdm16) is a transcription regulator in normal HSCs and an oncogene in leukemia (Aguilo et al., 2011, Matsuo et al., 2015). Analysis of Prdm16 splicing using IGV visualization surprisingly indicated that it seems to express more of the variant isoform that lacks the second-to-last exon (Figure 4A right-end). This is better visualized using Sashimi plots, which show that although “canonical” reads connect all exons sequentially, there are more “variant” junction reads, suggesting that the alternative is more abundant than the canonical form (Figure 4B). This caught our attention, as we previously found Prdm16 to be preferentially expressed in HSCs (Gazit et al., 2013). qRT-PCR validation of Prdm16 isoforms indeed found significantly more of the isoform “variant” in comparison with the “canonical” isoform (Figures S4A, S4B, and 4). The rarity of HSCs suggest that if they preferentially express an isoform that is a minor in other cells then reference data would likely annotate it as “variant.”
Figure 4.

Prdm16 Predominantly Expresses a Variant Isoform in Mouse HSCs
(A) Raw RNA-seq data showing two isoforms of Prdm16 that are transcribed in HSCs (canonical, NM_027504; variant, NM_001177995), along with splice junctions from the IGV browser.
(B) A Sashimi plot of Prdm16, zooming in on the splice junction that skips exon 16. Data for the plot were taken from four samples (A0, B0, B1, and A3, in descending order, respectively).
(C) Histogram of the expression of each isoform relative to Hprt. Data of technical triplicate average ± SD from one representative experiment out of four are shown.
Nadk2 Expresses Multiple Isoforms
We next wondered whether some genes express more than two major isoforms in HSCs. We zoomed in on the nicotinamide adenine dinucleotide (NAD) kinase 2 mitochondrial gene (Nadk2), which codes for a metabolic enzyme that phosphorylates NAD+ into NADP+. Nadk2 is a rather large gene, with 13 exons stretching over almost 40 kb (Figure S4C). Our IGV analysis had indicated that several of its exons might be spliced out or retained (Figures S4C–S4J), generating multiple isoforms expressed in HSCs (Figures S4G–S4I). qRT-PCR validation found good agreement with the RNA-seq data (Figures S4C–S4J). A Sashimi plot clearly shows multiple alternative transcripts in Nadk2 (Figure S4H). Intriguingly, it is still possible that multiple exons combine into various isoforms, and we attribute the identified junctions to specific transcripts, although this is not certain. Methods such as RNA-seq or qPCR, which record short fragments of the gene, cannot directly determine the presence of unconventional transcripts that assemble exons in a unique way when multiple splice junctions are present at substantial frequencies.
CDKn1c Expresses an Unannotated Isoform in Mouse HSCs
In addition to the successful identification of known isoforms, we sought to discover unknown splicing in HSC genes. HSCs are so rare that, if they do express unique splicing, it might not have been detected by previous studies. Indeed, we have identified that CDKn1c (also known as p57 or Kip2) expresses an unannotated isoform. As shown in Figure 5A, there is an alternative donor site for exon 2, while the acceptor site remains unchanged. The alternative form includes 36 additional bases at the 3′ of exon 2, not disturbing the reading frame. An examination of the two splicing forms in the Sashimi plot suggests they are expressed in similar levels (Figure 5B). qRT-PCR validated the expression of both the known and the previously unknown splicing in HSCs (Figure 5C). CDKn1c (p57) is a major regulator of the cell cycle and was recently reported to play a major role in HSC quiescence, which is known to be required for their long-term activity in vivo (Matsumoto et al., 2011). However, this unique splicing was not previously published in the mouse and was not present on either the Ensembl or the UCSC genome browsers when we searched them. We further searched evolutionary relevance for this splicing, finding that while it has not previously been identified in murine data, there is an indication for its presence in human data (human genome hg38 assembly, data not shown). Interestingly, this short addition includes a putative phosphoserine site on position S268 of the variant. This lends further support to the discovery of this unannotated splicing in the mouse and demonstrates that previously unrecognized transcripts can be directly revealed by the mapped RNA-seq data presented here. While CDKn1c T143 phosphorylation was reported to play a role in CDKi activity (Joaquin et al., 2012), this S268 putative site was not yet studied.
Figure 5.

CDKn1c Has an Unannotated Isoform in Mouse HSCs in Addition to the Canonical One
(A) Raw RNA-seq data showing two known isoforms (from Ensembl). Raw reads and junctions are from the IGV browser. Note that the expressed variant isoform is unannotated according to the Ensembl or RefSeq databases. The enlarged area demonstrates multiple reads from a part of the sequence thought to be part of an intron.
(B) A Sashimi plot of CDKn1c. The read number is represented by the height of the schematic; the connecting arcs represent splice junctions. The data were taken from four samples (A0, B0, B1, and A3, in descending order, respectively).
(C) Histogram chart of the expression of each isoform relative to Hprt. Data of technical triplicate average ± SD from one representative experiment out of four are shown.
Pbx1 Isoforms Present Similar Phenotypes while Hlf Isoforms Have a Different Function
To test whether different isoforms retain their function, we focused on Pbx1 and Hlf, which were mostly studied for their oncogenic role as part of E2A-PBX1 and E2A-HLF translocations, respectively. We have previously focused our attention on hepatic leukemia factor (Hlf), due to its very robust function ex vivo (Gazit et al., 2013), and both genes are part of the core-reprogramming factors for iHSC (Riddell et al., 2014). Our current RNA-seq analysis identified a variant of Hlf that lacks a short portion of its N terminus (Figures 6A and 6B), and a Pbx1 isoform that skips exon 8, which is suggested to cause a frameshift of the subsequent ninth exon and truncate the C terminus of the protein (Figure S6). To functionally test isoform activity, we cloned both genes from the primary HSC cDNA library into a lentiviral vector. Sequencing further validated these Hlf2 and Pbx1-short isoforms, and the fluorescent reporter of the lentiviral vector showed similar expression levels of the canonical and the variant forms (data not shown). Notably, a phenotypical difference between these variants could not be predicted using current tools (such as Ensembl and Pfam). Functional examination found that the canonical Hlf isoform endowed primary cells with a robust growth advantage in vitro, but the shorter Hlf variant did not (Figure 6C). This demonstrates that even one exon difference in a spliced transcript might abrogate functional activity. Pbx1-long overexpression in primary bone marrow cells enhances the expression of HSC markers cKit and Sca-1 (Figure 6D), and the isoform Pbx1-short had a similar impact (no significant difference between Pbx1-short and -long, Figure 6D). Thus, the isoforms of Pbx1, which are both expressed in HSCs, have similar functional impact as measured by in vitro assay. It would be interesting to further study the relative expression and the possible activities of these isoforms in vivo. Knowledge of the transcriptional information on each and every gene widens the array of opportunities for many future studies regarding known and unknown HSC factors.
Figure 6.

Functionally Divergent Activity of the Alternatively Spliced Variants of Hlf and Pbx1
(A) Schematic of the Hlf gene.
(B) Expression of Hlf variants 1 and 2 (Hlf1 and Hlf2) relative to Hprt in mouse HSCs in two types of multipotent progenitors (MPP1 and MPP2), and in a population of committed progenitors (Lineage-cKit+Sca1-, sometimes called LK cells). Data of technical triplicate average ± SD from one representative experiment out of four are shown.
(C) Hlf1 provides a growth advantage in culture compared with Hlf2. Graph showing frequencies of Hlf1 (blue), Hlf2 (red), and reporter-only controls (green) in separate cultures over time. Hlf1 increased in frequency over time, as reported by Gazit et al. (2013), whereas Hlf2 did not. Data of technical duplicate average ± SD from one representative experiment out of five are shown.
(D) Both canonical (Pbx1-long) and variant (Pbx1-short) forms of Pbx1 increase cKit and Sca1 expression in vitro. Fluorescence-activated cell sorting plots of cells overexpressing the indicated factor (top panels) or controls (bottom panels) show the frequencies of cKit+Sca1+ in primary cells after 4 weeks in vitro. Representative data are shown for one of three independent experiments. Control vectors are identical to Pbx1 vectors with the exception of the coding sequence. Plots are representative from one experiment out of three.
Transcriptome-wide Splicing Landscape Visualized at Single-Gene Resolution on the UCSC Genome Browser
Our finding of extensive splicing in HSC genes suggests that other researchers would be interested in knowing the actual splicing of their specific gene(s) of interest. Toward that end, we generated a visualization of all the RNA-seq data aligned to the mouse genome, with an emphasis on junction reads. While the IGV software has graphical advantages, it may not be as easily accessible to every researcher, whereas the UCSC browser is familiar to many scientists and available online. An example is shown for the Rbbp6 gene, with clear expression of its long transcript (Figure 7A) and of an AS event that skips an exon and results in two variant isoforms (Figure 7B); we added a manual in the Supplemental Information that explains how to use our tracks online. The UCSC browser is easily accessible and allows convenient viewing of additional resources from various organisms, with known and predicted isoforms of the gene of interest, as noted above for CDKn1c, making this aspect of our study helpful for many researchers.
Figure 7.

Visualizing Complete Alternative Splicing Data for the Mouse HSC on the UCSC Genome Browser
(A) The entire Rbbp6 gene on the UCSC browser. The reference gene sequence is in blue; splice junctions and numbers above in black.
(B) A zoomed-in view of the splice site over exon 16; the box between the junctions and the gene shows the number of junctions to which the marker points.
Discussion
Better understanding of adult stem cells will expand their use in a field that is led by HSCs, which already saves tens of thousands of patients every year if a matched donor is available. Recent advancements have been fueled by extensive transcriptome profiling. However, to date, such data were only analyzed at the crude discrete gene level, whereas most genes may express various AS. We have previously noted AS in a few HSC genes of interest and recognized the research need for an accessible splicing map of the mouse HSC transcriptome.
Extensive microarray datasets have been published previously. The ImmGen consortium offers unique data that were generated by multiple groups following a unified and strict protocol. We have previously analyzed this dataset to reveal genes that are preferentially expressed in stem and progenitor cells (Gazit et al., 2013). Splice variation might be extracted from microarrays, as was elegantly published with regard to B and T cells (Ergun et al., 2013). Emerging RNA-seq data offer a direct AS indication. We aimed to analyze data from both microarrays and RNA-seq in order to map splicing in HSCs. Comparing the independent sets of data bears a risk of combining unrelated pieces of information. Nevertheless, despite protocol, technical, and geographical differences, there is a good expression correlation between the microarray and RNA-seq datasets for mouse HSCs. This may attest to adequate identification and sorting of the HSC population by different laboratories. It is conceivable that some probe sets do not perfectly correlate with RNA-seq, and that some genes are less efficiently detected by current sequencing (e.g., if they have a high GC content).
Our analysis of AS in microarrays was not validated. This must be highlighted in contrast to the previous successful analysis of AS from microarrays (Ergun et al., 2013). Several differences may account for this conflict. First, B and T cells are more clearly distinguishable cell types allowing a pairwise comparison; Second, Ergun et al. used RNA-seq data that were specifically generated by the same group; Third, there might be a basic difference between effector cells that express lineage-specific genes and stem cells that have to sustain multilineage potential and yet keep lineage-specific loci open.
Microarray datasets still present a major advantage in terms of data breadth, as they cover many hematopoietic and immune cell types. Indeed, several genes that are annotated as having a role in splicing show clear preferential expression in HSCs, and other splicing factors are preferential to other cell types, suggesting a rationale and specific candidate regulators for different splicing across the hematopoietic system. Interestingly, Msi2, which is annotated as having a role in splicing and which was discovered to be an essential regulator of HSCs and aggressive leukemia (Kharas et al., 2010), shows clear preferential expression in HSCs in our data. Msi2 is better known for its role in regulating translation, and our analysis now adds another possible mechanism by which it may influence HSCs. Interestingly, overexpression of human MSI2 was reported to expand HSCs via attenuation of the aryl hydrocarbon receptor pathway as an RNA-binding protein (Rentas et al., 2016). MSI2 also plays a role in malignancy, thus understanding the possible relevance of splicing and RNA regulation in normal stem cells may reveal another layer of regulation (and mis-regulation) in cancer and in cancer stem cells (Barbouti et al., 2003, de Andres-Aguayo et al., 2011, Kharas et al., 2010).
Splicing is prevalent among all gene types, and transcription-regulating genes seem to have more isoforms on average. Notably, almost all of the HSC genes (Gazit et al., 2013) show expression of two or more isoforms. This indicates that, even within a highly purified population of primary cells, there is substantial diversity with regard to the way genes are expressed. We must stress that our analysis is at the population level. It is still possible that individual cells express just one isoform or another, such that the observed variability is between cells. Nevertheless, studies of other cell types reported the co-expression of different isoforms within single cells (Gurskaya et al., 2012), and there is no reason to suspect that HSCs would behave differently. Functional heterogeneity of HSCs had been correlated with levels of the CD150 surface marker (Hock, 2010), and it is intriguing to investigate the possible correlation of splicing variation with HSCs subtypes.
Mapping of AS within HSCs is of interest to many researchers, yet not all are comfortable with using professional software that requires downloading and managing big input files, as is the case for IGV and BAM files. The availability of a user-friendly online version is of interest and benefit to the community. We chose to upload our analyzed data onto the UCSC genome browser in order to facilitate easy access and gene-specific resolution for splicing events in HSCs. Many research groups focus on a specific gene of interest and open-access whole-genome coverage with resolution at the nucleotide level will benefit current understanding and future research.
Molecular understanding of HSCs has progressed thanks to extensive transcriptome data. Our study presents a comprehensive analysis of splice variants in mouse HSCs, revealing multiple splicing in HSC genes, and offering a valuable tool with easily accessible information on any gene of interest, including known and unknown transcripts. With the accelerated generation of high-quality RNA-seq data, similar analysis of HSCs through normal development, quiescence, and induced activation are expected to bring another dimension to understanding of the molecular mechanisms of adult stem cells.
Experimental Procedures
Identification of Splicing from Microarray Data
The exon/gene expression ratios of 4,321 differentially spliced exons in 172 cell populations were obtained from Ergun et al. (2013). Significantly low or high expression within the HSC population (sample SC.LTSL.BM) was calculated in comparison with either all other 171 cell types, or the Progenitors group (samples SC.LTSL.FL, SC.STSL.FL, SC.LTSL.BM, SC.STSL.BM, SC.GMP.BM, SC.MEP.BM, SC.MPP34F.BM, and SC.ST34F.BM). The p values were adjusted for multiple testing using a false discovery rate (FDR). Exons with an FDR-adjusted p < 0.01 and fold-change >1.2 (up- or downregulation) were considered as alternatively spliced.
HSC Sort and qRT-PCR
For the validation of splicing, we used 6-month-old C57BL/6 mice housed at BGU's animal facility. Experiments were approved according to the local and state ethics committee. Cells from the femur, pelvis, and tibia were obtained by crushing followed by cKit enrichment using magnetic beads (Miltenyi Biotec). Enriched cells were stained for Lineage (Ter119, CD11b, GR-1, CD3e, CD4, CD8, and B220), Sca1, cKit, CD34, and flk2 (BioLegend). Sorted cells (FACSAria; BD) were stored in TRIzol (Invitrogen). RNA was extracted according to the manufacturer's protocol, followed by reverse transcription using SuperScript (Invitrogen). qPCR reactions were run on LightCycler 480 (Roche); primers are given in Table S2.
Splicing-Factors Analysis and Comparison of the Microarray and RNA-Seq Datasets
Expression levels of 216 splicing factors in 248 cell types were retrieved from ImmGen's microarrays. Relative expression levels were visualized using Gene-E (Broad Institute). To compare gene expression we plotted the log2 expression values, using a cutoff of >1, from the ImmGen microarray and from each RNA-seq sample. Correlation was calculated pairwise for each RNA-seq sample.
Bioinformatics Analysis of Splicing from RNA-Seq Data
RNA-seq data on HSCs was obtained from four studies (Cabezas-Wallscheid et al., 2014, Qian et al., 2016, Sun et al., 2014, Venkatraman et al., 2013). The sample numbers are provided in the Supplemental Information (Table S1). Reads were aligned to the Mus musculus mm10 genome using TopHat (Trapnell et al., 2009). Cufflinks was used to assemble the reads into transcripts (Trapnell et al., 2012). The data from all samples were combined into one file indicating junctions and transcripts/genes of all datasets together, following data unification; expression values of genes and transcripts were defined per sample, and the results are presented per individual sample (best depicted in the Sashimi plots). For the analysis of splicing prevalence within annotated groups of genes (Figures 2 and S2), all datasets were combined, and numbers of isoforms were counted for all of the detected genes and for the indicated GO-annotated groups; distribution of the discrete proportion of the number of isoforms per gene was visualized as histograms of discrete proportions. Visualization of HSC genes and multiple other alternatively spliced transcripts was done using the IGV browser (Robinson et al., 2011) in order to validate bioinformatic data and predictions. All the splicing-junctions data obtained in this study were uploaded onto the UCSC genome browser; it can be accessed by copying the following link into a web browser: http://bioinfo.bgu.ac.il/genomes/RoiG/UCSC. To search for a gene, type its symbol on the searchbar or use genomic position. Modify the shown channels to fit your needs: we suggest starting with “HIDE” or “DENSE” for TopHat reads and “FULL” for Cufflink junctions. The actual number of counted junctions is given next to its indicator (left of the screen). The splice data viewed in the UCSC website were generated via TopHat and yielded slightly higher numbers than the “.bed” file and the Sashimi plots. This minute change does not alter splicing events. A graphical user manual can be found in the Supplemental Information.
Ex Vivo Functional Examination of Splice Variants
Experiments were carried out as previously reported previously (Gazit et al., 2013). In brief, specific splicing isoforms of the genes of interest were individually cloned into pHAGE2 (Mostoslavsky et al., 2005) to generate lentiviruses that enabled induced expression in primary cells. Infected cells were readily identified by a reporter ZsGreen fluorescent protein. Specific marker expression was detected with conjugated antibodies (BioLegend) on a Gallios Flow Cytometer (Beckman Coulter) and Kaluza software. The primary cells for the ex vivo experiments were obtained according to the guidelines of the Institutional Animal Care and Use Committee of the Ben-Gurion University of the Negev, Beer Sheva, Israel.
Author Contributions
O.G. performed the analysis, visualization, qRT-PCR, and writing of the manuscript; K.M. carried out Hlf functional experiments; N.B. helped with the computational analysis; Y.G. performed the cell culture and functional assays; M.F. did some of the qRT-PCR; H.N.G. and T.S. undertook part of the initial analysis of the ImmGen arrays and most of the RNA-seq analysis; R.G. supervised the study and wrote the manuscript.
Acknowledgments
We thank Ayla Ergun for initial discussions; Dan Peer for expert advice; Marianna Romzova for qRT-PCR advice; Vered Chalifa-Caspi and Inbar Plaschkes for their essential bioinformatics analysis of arrays and help with RNA-seq visualization on the UCSC browser. This study was supported by ISF grants 1690/13 and 2348/15, and CIG grant 618647.
Published: December 29, 2016
Footnotes
Supplemental Information includes a supplemental manual, six figures, and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2016.12.002.
Supplemental Information
References
- Aaronson Y., Meshorer E. Stem cells: regulation by alternative splicing. Nature. 2013;498:176–177. doi: 10.1038/nature12253. [DOI] [PubMed] [Google Scholar]
- Aguilo F., Avagyan S., Labar A., Sevilla A., Lee D.F., Kumar P., Lemischka I.R., Zhou B.Y., Snoeck H.W. Prdm16 is a physiologic regulator of hematopoietic stem cells. Blood. 2011;117:5057–5066. doi: 10.1182/blood-2010-08-300145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbouti A., Hoglund M., Johansson B., Lassen C., Nilsson P.G., Hagemeijer A., Mitelman F., Fioretos T. A novel gene, MSI2, encoding a putative RNA-binding protein is recurrently rearranged at disease progression of chronic myeloid leukemia and forms a fusion gene with HOXA9 as a result of the cryptic t(7;17)(p15;q23) Cancer Res. 2003;63:1202–1206. [PubMed] [Google Scholar]
- Bryder D., Rossi D.J., Weissman I.L. Hematopoietic stem cells: the paradigmatic tissue-specific stem cell. Am. J. Pathol. 2006;169:338–346. doi: 10.2353/ajpath.2006.060312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabezas-Wallscheid N., Klimmeck D., Hansson J., Lipka D.B., Reyes A., Wang Q., Weichenhan D., Lier A., von Paleske L., Renders S. Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell. 2014;15:507–522. doi: 10.1016/j.stem.2014.07.005. [DOI] [PubMed] [Google Scholar]
- Collins C.T., Hess J.L. Role of HOXA9 in leukemia: dysregulation, cofactors and essential targets. Oncogene. 2015;35:1080–1089. doi: 10.1038/onc.2015.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copelan E.A. Hematopoietic stem-cell transplantation. N. Engl. J. Med. 2006;354:1813–1826. doi: 10.1056/NEJMra052638. [DOI] [PubMed] [Google Scholar]
- de Andres-Aguayo L., Varas F., Kallin E.M., Infante J.F., Wurst W., Floss T., Graf T. Musashi 2 is a regulator of the HSC compartment identified by a retroviral insertion screen and knockout mice. Blood. 2011;118:554–564. doi: 10.1182/blood-2010-12-322081. [DOI] [PubMed] [Google Scholar]
- de Andres-Aguayo L., Varas F., Graf T. Musashi 2 in hematopoiesis. Curr. Opin. Hematol. 2012;19:268–272. doi: 10.1097/MOH.0b013e328353c778. [DOI] [PubMed] [Google Scholar]
- Ergun A., Doran G., Costello J.C., Paik H.H., Collins J.J., Mathis D., Benoist C., ImmGen C. Differential splicing across immune system lineages. Proc. Natl. Acad. Sci. USA. 2013;110:14324–14329. doi: 10.1073/pnas.1311839110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto S., Araki K., Chisaka O., Araki M., Takagi K., Yamamura K. Analysis of the murine Hoxa-9 cDNA: an alternatively spliced transcript encodes a truncated protein lacking the homeodomain. Gene. 1998;209:77–85. doi: 10.1016/s0378-1119(98)00014-6. [DOI] [PubMed] [Google Scholar]
- Gabut M., Samavarchi-Tehrani P., Wang X., Slobodeniuc V., O'Hanlon D., Sung H.K., Alvarez M., Talukder S., Pan Q., Mazzoni E.O. An alternative splicing switch regulates embryonic stem cell pluripotency and reprogramming. Cell. 2011;147:132–146. doi: 10.1016/j.cell.2011.08.023. [DOI] [PubMed] [Google Scholar]
- Gazit R., Weissman I.L., Rossi D.J. Hematopoietic stem cells and the aging hematopoietic system. Semin. Hematol. 2008;45:218–224. doi: 10.1053/j.seminhematol.2008.07.010. [DOI] [PubMed] [Google Scholar]
- Gazit R., Garrison B.S., Rao T.N., Shay T., Costello J., Ericson J., Kim F., Collins J.J., Regev A., Wagers A.J. Transcriptome analysis identifies regulators of hematopoietic stem and progenitor cells. Stem Cell Rep. 2013;1:266–280. doi: 10.1016/j.stemcr.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazit R., Mandal P.K., Ebina W., Ben-Zvi A., Nombela-Arrieta C., Silberstein L.E., Rossi D.J. Fgd5 identifies hematopoietic stem cells in the murine bone marrow. J. Exp. Med. 2014;211:1314–1330. doi: 10.1084/jem.20130428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurskaya N.G., Staroverov D.B., Zhang L., Fradkov A.F., Markina N.M., Pereverzev A.P., Lukyanov K.A. Analysis of alternative splicing of cassette exons at single-cell level using two fluorescent proteins. Nucleic Acids Res. 2012;40:e57. doi: 10.1093/nar/gkr1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H., Irimia M., Ross P.J., Sung H.K., Alipanahi B., David L., Golipour A., Gabut M., Michael I.P., Nachman E.N. MBNL proteins repress ES-cell-specific alternative splicing and reprogramming. Nature. 2013;498:241–245. doi: 10.1038/nature12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock H. Some hematopoietic stem cells are more equal than others. J. Exp. Med. 2010;207:1127–1130. doi: 10.1084/jem.20100950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper J.E. A survey of software for genome-wide discovery of differential splicing in RNA-Seq data. Hum. Genomics. 2014;8:3. doi: 10.1186/1479-7364-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joaquin M., Gubern A., Gonzalez-Nunez D., Josue Ruiz E., Ferreiro I., de Nadal E., Nebreda A.R., Posas F. The p57 CDKi integrates stress signals into cell-cycle progression to promote cell survival upon stress. EMBO J. 2012;31:2952–2964. doi: 10.1038/emboj.2012.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharas M.G., Lengner C.J., Al-Shahrour F., Bullinger L., Ball B., Zaidi S., Morgan K., Tam W., Paktinat M., Okabe R. Musashi-2 regulates normal hematopoiesis and promotes aggressive myeloid leukemia. Nat. Med. 2010;16:903–908. doi: 10.1038/nm.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel M.J., Yilmaz O.H., Iwashita T., Yilmaz O.H., Terhorst C., Morrison S.J. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- Matsumoto A., Takeishi S., Kanie T., Susaki E., Onoyama I., Tateishi Y., Nakayama K., Nakayama K.I. p57 is required for quiescence and maintenance of adult hematopoietic stem cells. Cell Stem Cell. 2011;9:262–271. doi: 10.1016/j.stem.2011.06.014. [DOI] [PubMed] [Google Scholar]
- Matsuo H., Goyama S., Kamikubo Y., Adachi S. The subtype-specific features of EVI1 and PRDM16 in acute myeloid leukemia. Haematologica. 2015;100:e116–e117. doi: 10.3324/haematol.2015.124396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostoslavsky G., Kotton D.N., Fabian A.J., Gray J.T., Lee J.S., Mulligan R.C. Efficiency of transduction of highly purified murine hematopoietic stem cells by lentiviral and oncoretroviral vectors under conditions of minimal in vitro manipulation. Mol. Ther. 2005;11:932–940. doi: 10.1016/j.ymthe.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Nilsen T.W., Graveley B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orkin S.H., Zon L.I. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osawa M., Hanada K., Hamada H., Nakauchi H. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science. 1996;273:242–245. doi: 10.1126/science.273.5272.242. [DOI] [PubMed] [Google Scholar]
- Qian P., He X.C., Paulson A., Li Z., Tao F., Perry J.M., Guo F., Zhao M., Zhi L., Venkatraman A. The Dlk1-Gtl2 locus preserves LT-HSC function by inhibiting the PI3K-mTOR pathway to restrict mitochondrial metabolism. Cell Stem Cell. 2016;18:214–228. doi: 10.1016/j.stem.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentas S., Holzapfel N.T., Belew M.S., Pratt G.A., Voisin V., Wilhelm B.T., Bader G.D., Yeo G.W., Hope K.J. Musashi-2 attenuates AHR signalling to expand human haematopoietic stem cells. Nature. 2016;532:508–511. doi: 10.1038/nature17665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell J., Gazit R., Garrison B.S., Guo G., Saadatpour A., Mandal P.K., Ebina W., Volchkov P., Yuan G.C., Orkin S.H. Reprogramming committed murine blood cells to induced hematopoietic stem cells with defined factors. Cell. 2014;157:549–564. doi: 10.1016/j.cell.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J.T., Thorvaldsdottir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbauer F., Tenen D.G. Transcription factors in myeloid development: balancing differentiation with transformation. Nat. Rev. Immunol. 2007;7:105–117. doi: 10.1038/nri2024. [DOI] [PubMed] [Google Scholar]
- Rossi L., Lin K.K., Boles N.C., Yang L., King K.Y., Jeong M., Mayle A., Goodell M.A. Less is more: unveiling the functional core of hematopoietic stem cells through knockout mice. Cell Stem Cell. 2012;11:302–317. doi: 10.1016/j.stem.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay T., Kang J. Immunological genome project and systems immunology. Trends Immunol. 2013;34:602–609. doi: 10.1016/j.it.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solana J., Irimia M., Ayoub S., Orejuela M.R., Zywitza V., Jens M., Tapial J., Ray D., Morris Q., Hughes T.R. Conserved functional antagonism of CELF and MBNL proteins controls stem cell-specific alternative splicing in planarians. Elife. 2016;5:e16797. doi: 10.7554/eLife.16797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangrude G.J., Heimfeld S., Weissman I.L. Purification and characterization of mouse hematopoietic stem cells. Science. 1988;241:58–62. doi: 10.1126/science.2898810. [DOI] [PubMed] [Google Scholar]
- Steensma D.P. Surprising splicing: the new most frequent class of genetic alteration in myelodysplastic syndromes. Hematologist. 2012;9:5–7. [Google Scholar]
- Sun D., Luo M., Jeong M., Rodriguez B., Xia Z., Hannah R., Wang H., Le T., Faull K.F., Chen R. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell. 2014;14:673–688. doi: 10.1016/j.stem.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas E.D. Bone marrow transplantation from the personal viewpoint. Int. J. Hematol. 2005;81:89–93. doi: 10.1532/ijh97.04197. [DOI] [PubMed] [Google Scholar]
- Trapnell C., Pachter L., Salzberg S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatraman A., He X.C., Thorvaldsen J.L., Sugimura R., Perry J.M., Tao F., Zhao M., Christenson M.K., Sanchez R., Yu J.Y. Maternal imprinting at the H19-Igf2 locus maintains adult haematopoietic stem cell quiescence. Nature. 2013;500:345–349. doi: 10.1038/nature12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong H.Y., Alipanahi B., Lee L.J., Bretschneider H., Merico D., Yuen R.K., Hua Y., Gueroussov S., Najafabadi H.S., Hughes T.R. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science. 2015;347:1254806. doi: 10.1126/science.1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.