

Abstract
Carney complex (CNC) is a rare dominantly inherited multiorgan tumoral disorder that includes Cushing syndrome (CS). To establish the Mayo Clinic experience with the CS component, including its clinical, laboratory, and pathological findings, we performed a retrospective search of the patient and pathological databases of Mayo Clinic in Rochester, Minnesota, for patients with CNC and clinical or laboratory findings of CS. Thirty-seven patients with CNC were identified. Twenty-nine had clinical, pathological, or laboratory evidence of an adrenocortical disorder. Seventeen had classic CS; 15 underwent bilateral, subtotal, or partial unilateral adrenalectomy, and 2 had no treatment. Pathologically, the glands were normal sized or slightly enlarged with multiple small (1–4 mm), brown, black, and yellow micronodules (primary pigmented nodular adrenocortical disease) (PPNAD). Three glands each had a mass: a 2-cm adenoma, a 1.5-cm macronodule, and an unencapsulated 1.8-cm myelolipoma. Fourteen of the patients were alive at follow-up and 3 were deceased; 2 of the latter had PPNAD at autopsy, and the third had PPNAD at surgery. Twelve patients without clinical features of classic CS had abnormal adrenocortical testing results; none developed classic CS during follow-up (mean, 10 years). Autopsy findings in 1 showed bilateral vacuolated cell cortical hyperplasia.
Keywords: adrenal, Carney complex, Cushing syndrome, genetics, pathology
Introduction
In 1932, in a report based on clinical and pathological observations in 12 patients, Harvey Cushing (1) enunciated the findings known today as Cushing syndrome (CS). The clinical findings were adiposity, kyphosis, amenorrhea in females and impotence in males, hypertrichosis, purple lineae atrophicae, osteoporosis, and skin abscesses and ulcers. The pathological findings included pituitary tumors and unilateral and bilateral adrenal enlargement.
In 1939, 17-year-old fraternal twins were evaluated at Mayo Clinic in Rochester, Minnesota. They had the clinical features described by Cushing and also intense facial and body pigmented spots. Adrenal exploration in 1 of the twins showed “small adrenal glands with multiple small brown nodules.” These findings were not mentioned by Cushing, and the twins were thought to have an “adrenocortical syndrome” different from that described by Cushing.
In 1984, Shenoy et al (2) reported on 4 young Mayo Clinic patients who had CS and a bilateral adrenocortical pathology that they termed primary pigmented nodular adrenocortical disease (PPNAD). The adrenal glands were small and studded with brown and black cortical micronodules.
In 1985, Carney et al (3,4) recognized that PPNAD was a component of a familial multiorgan tumoral syndrome that included pigmented skin lesions (lentiginosis and blue nevi), myxomas (cardiac, cutaneous, and mammary), and endocrine disorders (CS, acromegaly, and sexual precocity) and was subsequently referred to as Carney complex (CNC) (5). A provisional diagnosis of CNC was considered to be warranted when 2 of its components occurred in characteristic fashion (3)—when the lentiginosis affected the face and vermilion border of the lips, the cardiac tumor was multifocal and occurred in any or all cardiac chambers, and the endocrine neoplasms were multifocal and bilateral (the pituitary involvement was multifocal).
In 2000, Kirschner et al (6) discovered that CNC was caused by heterogeneous inactivating mutations of PRKAR1A in more than 60% of affected patients. PRKAR1A, an apparent tumor suppressor gene, is located at chromosome 17q22–24, encodes protein kinase A regulatory subunit 1α, and is a central component of the cyclic adenosine monophosphate signaling pathway.
CS has occurred in about 25% of patients with CNC (7,8). The syndrome requires treatment because of its serious long-term consequences. Herein, we describe the adrenocortical clinical, pathological, and molecular genetic findings in the Mayo Clinic patients with CNC, focusing on the CS component.
Patients and Methods
This study was approved by the Mayo Clinic Institutional Review Board (protocols 979-01 and 14–3722). We searched the clinical, pathological, and autopsy records at Mayo Clinic in Rochester, Minnesota, for patients who had 2 or more components of CNC. Information was recorded on the patients and their families, including clinical findings, adrenal imaging and laboratory results, PRKAR1A mutational status, treatment, pathological findings, and follow-up. Histological slides and paraffin blocks of surgical and necropsy adrenal glands were retrieved from the Mayo Clinic Tissue Registry Archives. Similar information and materials were obtained for relatives of the Mayo Clinic patients who had adrenal surgery at other institutions and were subsequently evaluated at Mayo Clinic. The study group included the grandchildren of 1 of the Mayo Clinic patients; they were evaluated at the National Institutes of Health (Bethesda, Maryland).
Categories of CS
Patients were categorized as having 1) classic CS, 2) subclinical CS, 3) possible CS, or 4) no CS. Patients with classic CS had 1) physical findings and symptoms, including weight gain (primarily abdominal); thick, purple-red abdominal striae; proximal muscle weakness; facial rounding and redness; hirsutism; and hypertension and 2) abnormal laboratory findings, including 24-hour urinary free cortisol excretion that exceeded the upper limit of the reference range, absence of diurnal variation in serum cortisol level, nonsuppressible blood cortisol levels after overnight dexamethasone administration, and suppression of baseline morning plasma corticotropin (ACTH) levels.
Patients with subclinical CS had some or none of the signs and symptoms of classic CS. Their cortisol levels were not suppressed with dexamethasone administration. Other findings included loss of diurnal serum cortisol variation and suppressed serum ACTH levels.
Patients with possible CS had 1) some of the clinical findings listed above and borderline results of 1 or more tests that define classic CS or 2) adrenal pathological findings without symptoms or signs of CS or abnormal adrenal testing results. A patient included in this group had no symptoms or signs of CS and normal circulating levels of plasma cortisol; this patient’s ectopic (spermatic cord) adrenal cortex was available for pathological study.
Patients with no CS had no clinical or other evidence of CS and, thus, underwent no laboratory adrenal testing for it. Mild biochemical abnormalities could have been undetected in these patients.
Diagnostic Studies
Patients underwent adrenal and pituitary imaging when available. Adrenocortical laboratory testing included measurement of 24-hour urinary cortisol excretion, assessment of diurnal serum cortisol rhythm, measurement of percentage change in 24-hour urinary cortisol with dexamethasone administration, and measurement of plasma ACTH. Urinary ketosteroid and ketogenic steroid excretion was measured in some patients who presented before 1975.
For histological and immunocytochemical study, the adrenal glands from surgical resection and necropsy and the adrenal biopsy specimens were immersed in 10% neutral buffered formalin and embedded in paraffin. Sections 4-μm thick were cut for hematoxylin-eosin and Masson trichrome staining and for immunostaining with antibodies to synaptophysin, inhibin-A, melan A, and CD56.
DNA for PRKAR1A mutation testing was extracted from peripheral blood leukocytes of 13 patients.
Treatment
Treatment of classic CS was surgical: bilateral adrenalectomy (12 patients—3 after initial unilateral adrenalectomy); subtotal adrenalectomy (2 patients—excision of 1 adrenal gland and partial removal of the contralateral gland); and subtotal unilateral adrenalectomy (1 patient—partial removal of 1 adrenal gland). Two patients with classic CS and 12 with subclinical or possible CS received no treatment.
Results
Clinical Findings
Thirty-seven patients with 2 or more components of CNC were identified. The group included the CNC index patient, who was recognized in 1984 from findings in a 1959 autopsy (9) and the dizygotic twins mentioned above, who were identified in 1989 from clinical photographs obtained 50 years earlier (10). Of the 37 patients, 26 (70%) had classic CS, subclinical CS, or possible CS (Table 1 and Figures 1–3), and 3 others (patients 27–29) had adrenal or ectopic adrenal pathological findings typical of CNC but no CS. These 29 patients formed the study group. The other 8 patients, who were excluded from the study because they lacked clinical features of CS and had no laboratory testing for it, were followed for 2 to 49 years (mean, 19.5 years), but CS did not develop in any of them.
Table 1.
Clinical and Selected Family Findings for 29 Patients With Carney Complex (CNC)
| Patient/Reference | Sex/Age at First Manifestation of CNC/Age at Diagnosis of CNC | Cushing Syndrome (Age, y) | Components of CNC/Other Conditions | Family Identification/No. of Generations Affected | Germline PRKAR1A Mutation |
|---|---|---|---|---|---|
| 1/10 | F/4 y/65 y | Classic (17) Remitted spontaneously (24) Subclinical (65) |
Lentiginosis/pilonidal cyst, pancreatic carcinoma | A/2 | Not tested |
| 2/10 | M/4 y/44 y (at autopsy) | Classic (17) Persistent (44) |
Lentiginosis/bilateral simple mastectomy (gynecomastia), pilonidal cyst, inguinal hernia (prolapsed omental fat) | A | Not tested |
| 3/11 | F/12 y/37 y | Classic (12) | Lentiginosis, nasal schwannoma/depression, Graves disease, cerebral ischemic event, bilateral ovarian carcinoma | A | None |
| 4 | F/2 y/48 y | Classic (3) | Multiple left atrial myxomas,a blue nevus/osteopenia, 1-cm cystic pancreatic mass, 1.7-cm hypodense splenic mass | B/4 | c496C>T/p.Gln166X |
| 5 | M/Birth/4 y | Classic (4) | Facial and labial lentiginosis, bilateral punctate testicular calcifications, cutaneous myxomas, osteopenia with multiple fractures | B | c496C>T/p.Gln166X |
| 6 | F/3 y/14 y | Classic (4, left adrenalectomy) Recurrent classic (14, right adrenalectomy) |
Facial and mucosal lentiginosis, blue nevus | B | c496C>T/p.Gln166X |
| 7 | F/12 y/12 y | Classic (14) | Lentiginosis, multichamber cardiac myxoma, external ear canal myxoma | B | c496C>T/p.Gln166X |
| 8 | M/2 y/2 y (family screening for CNC) | Classic (10) | Lentiginosis, bilateral testicular calcifications, pituitary tumor | B | c496C>T/p.Gln166X |
| 9 | F/3 y/42 y | Classic (cyclic) (2) | Lentiginosis, left atrial myxoma/Ebstein anomaly | C/1 | Not tested |
| 10/12 | F/Birth/32 y | Classic (34) | Lentiginosis, multichamber cardiac myxoma, cutaneous and mammary myxomas, pituitary adenoma/thyroid adenoma | D/3 | c.340delG/p.Val113fsX15 |
| 11 | F/<6 y/43 y | Classic (6) | Lentiginosis, blue nevus, multichamber cardiac myxoma, cutaneous myxomas/bilateral multifocal follicular thyroid carcinoma with lymph node metastasis, hepatic cirrhosis, nephrolithiasis (left nephrectomy), type 2 diabetes mellitus | E/2 | c.712insAA/p.SER238fsX3 |
| 12 | F/Birth/18 y | Classic (15) | Lentiginosis, right atrial myxoma, bilateral mammary myxomas, pituitary adenoma | F/3 | Declined testing |
| 13/13 | F/12 y/28 y | Classic (14, left adrenalectomy; 17, right adrenalectomy) | Multichamber cardiac myxoma, psammomatous melanotic schwannoma | G/2 | C.286C>T/p.Arg96X |
| 14/13 | F/Birth/32 y | Classic (8) | Lentiginosis, multichamber cardiac myxoma (hemiplegia due to myxoma embolus) | G | C.286C>T/p.Arg96X |
| 15 | F/Childhood/46 y | Classic (46) | Lentiginosis, right atrial myxoma, cutaneous and bilateral mammary myxomas | H/2 | IVS4+1G>C |
| 16 | F/Birth/11 y | Classic (11) | Lentiginosis, mitral valve myxoma, bilateral mammary myxomas | I/1 | p.arg228term |
| 17 | F/2 y/29 y | Nonfunctioning adrenal mass (2, right adrenalectomy) Classic (27, left adrenalectomy) |
Short stature (142 cm), bilateral mammary myxoid fibroadenomas, lentiginosis, elevated insulinlike growth factor 1/developmental delay, epilepsy, mesial temporal sclerosis, osteopenia, stress fractures, necrotizing pneumonia (right upper lobectomy) | J/1 | Gene deleted Partial duplication of chromosome 2 |
| 18 | M/Birth/37 y | Subclinical (45) | Lentiginosis, unilateral LCCSCT, psammomatous melanotic schwannoma | K/1 | c.348+1g>c |
| 19 | F/6 y/26 y | Subclinical (57) | Lentiginosis, mammary myxoma, blue nevus/depression | D | c.340delG/p.Val113fsX15 |
| 20/14 | M/6 mo/35 y | Subclinical (46) | Lentiginosis, multichamber cardiac myxoma (hemiplegia due to myxoma embolus), bilateral testicular calcifications, cutaneous myxoma, pituitary microadenoma | L/1 | Declined testing |
| 21 | M/Childhood/15 y | Subclinical (22) | Lentiginosis, multiple left atrial myxomas (cerebral embolus) bilateral testicular calcifications | H | Not tested Mother (patient 15) had mutation |
| 22 | F/8 y/19 y | Subclinical (19) | Lentiginosis, multichamber cardiac myxoma with residual hemiplegia cutaneous and mammary myxomas | M/1 | Not tested |
| 23 | M/Birth/29 y | Subclinical (29) | Lentiginosis, LCCSCT/splenic hemangiomas | N/1 | Not tested Cytogenic analysis showed 2 abnormal metaphases at chromosome 17q25 |
| 24 | M/12 y/40 y | Possible (41) | Lentiginosis, multichamber cardiac myxoma, cutaneous myxoma, bilateral testicular calcifications/1.5-cm liver mass, nephrolithiasis | O/2 | c.891G>A |
| 25 | F/34 y/49 y | Possible (34) | Pituitary adenoma, recurrent left atrial myxoma, mammary myxoma/multinodular goiter, pilonidal cyst, left superior vena cava entry into coronary sinus | Not tested | Not tested |
| 26 | F/4 y/18 y | Possible (22) | Lentiginosis, café au lait spot, cutaneous “dermatofibromas” | Q/1 | C992–993delGA 2-bp deletion in exon 10 |
| 27 | F/3 y/34 y (autopsy) | Possible (34) | Lentiginosis, multichamber cardiac myxoma, cutaneous myxomas, blue nevus (surgical), right atrial myxoma, bilateral adrenal vacuolated cell hyperplasia with micronodules and a unilateral eosinophilic cell macronodule with lipochrome (at autopsy) | R/2 | Not tested |
| 28 | F/Unknown/33 y (autopsy) | Possible (33) | Lentiginosis and mammary myxomas (surgical), left atrial myxoma, PPNAD, pituitary Crooke cells, osteopenia (at autopsy) | S/1 | Not tested |
| 29 | M/Birth/37 y | Possible (42) | Lentiginosis, bilateral LCCSCT, PPNAD in rete testis, spermatic cord ectopic adrenal rest with PPNAD-type features, left atrial myxoma, pituitary adenoma/submandibular pleomorphic adenoma, inflammatory bowel disease (colectomy), depression | T/1 | Not tested |
Abbreviations: LCCSCT, large-cell calcifying Sertoli cell tumor; PPNAD, primary pigmented nodular adrenocortical disease.
Cardiac myxoma is usually a single left atrial tumor affecting middle-aged patients; 75% of patients are women.
Figure 1.
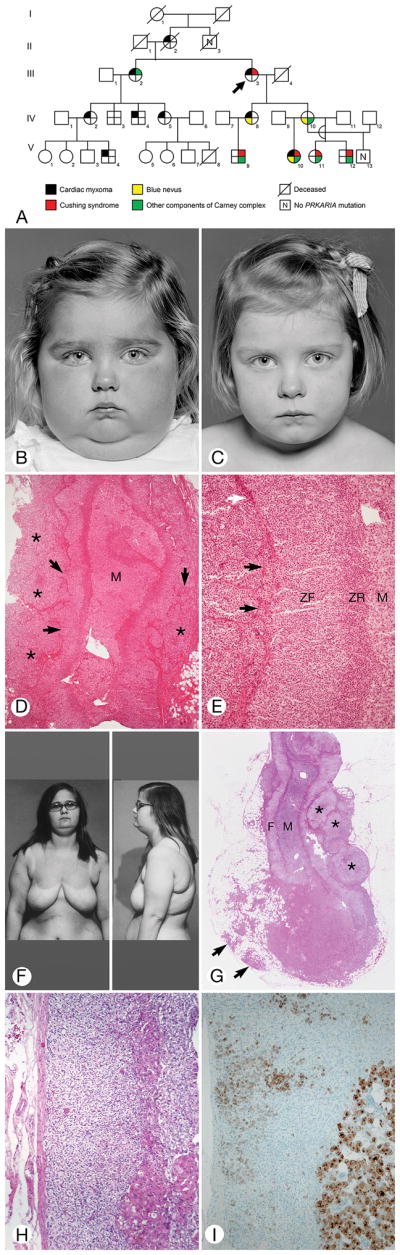
Classic Cushing Syndrome. A, Pedigree of family B. Arrow indicates index patient (III.3); circles, females; squares, males; diagonal line through symbol, deceased. B through E, Patient 4. F through I, Patient 13. B, Two-year-old child with florid facial fullness and double chin. The skin was clear. C, The face appeared normal 4 months after unilateral subtotal left adrenalectomy. D, Transverse section of the left adrenal gland showed extension of the cortex outside the capsule (asterisks) into the periadrenal fat (lower right) (hematoxylin-eosin, original magnification ×40). The cortical cells had almost completely replaced the adrenal capsule (arrows). M indicates medulla. E, Transverse section of the adrenal gland showed zona fasciculata (ZF), zona reticularis (ZR), and medulla (M). Clusters of cortical cells had separated the fibroblasts of the capsule (arrows) and formed a mantle on the external surface of the gland. A portion of intact capsule was present (white arrow) (hematoxylin-eosin, original magnification ×100). F, Obese 15-year-old patient had pendulous breasts and abdomen (patient 13). G, Left adrenal gland showed zona fasciculata (F), zona reticularis (narrow band of pink cells deep to the zona fasciculata), and medulla (M). Several aggregates of cells (asterisks) were applied to the external surface of the capsule. A large circumscribed aggregate of dark cells extended from the adrenal gland into the periadrenal fat (arrows) (hematoxylin-eosin, original magnification ×20). H, Intact adrenal capsule, zona fascicula with vacuolated cells, and aggregates of enlarged eosinophilic cells were seen deep in the cortex (hematoxylin-eosin, original magnification ×100). I, Large aggregate of cells strongly positive for synaptophysin (bottom right) and cells with weaker positivity were scattered in the cortex. The adrenal capsule is at left (original magnification ×100).
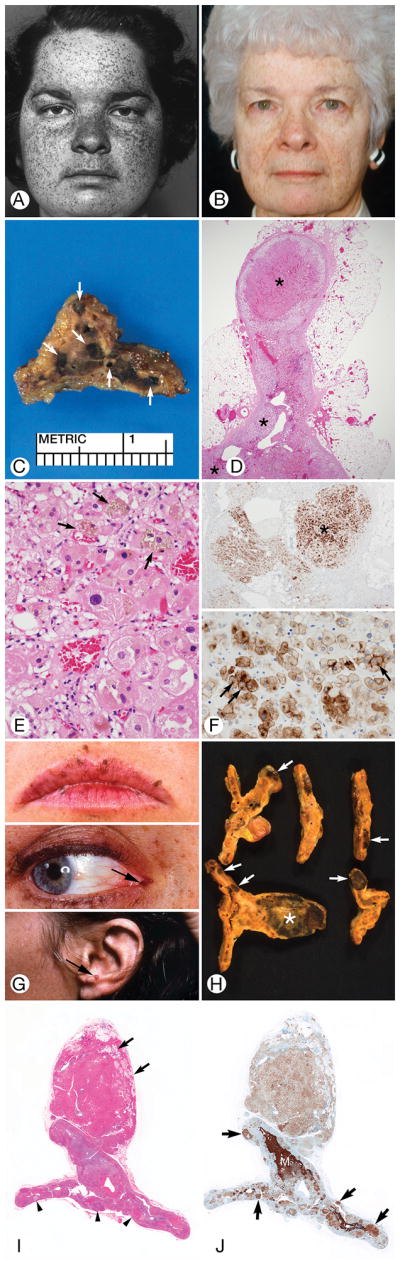
Figure 3.

Suspicious for Cushing Syndrome. A through D, Patient 28 (the index patient for Carney complex), E through H, Patient 27. A, The patient had spotty facial pigmentation (arrows). B, Cut surface of formalin-fixed adrenal gland obtained at autopsy showed brown micronodules (arrows) at junction of cortex and medulla (M). The normal shape of the gland was preserved. C, Sharply circumscribed mass of cortical cells mushroomed from the gland through a break in the capsule (stained blue with Masson trichrome technique). Multiple cortical micronodules were present (arrows) (original magnification ×40). D, A cortical micronodule was composed of round eosinophilic cells heavily stained with lipochrome. A few very large cells had little or no pigment (arrows) (hematoxylin-eosin, original magnification ×400). E, Spotty facial (upper) and forearm (lower) pigmentation and diffuse palmar pigmentation (lower). F, The cortex showed separation into zona fasciculata (vacuolated cells) and zona reticularis (eosinophilic cells). Multiple extra-adrenal mixed clear and eosinophilic ovoid cortical micronodules (arrows) indented the capsule. One intra-adrenal cortical micronodule was present (asterisk) (hematoxylin-eosin, original magnification ×1). G, Vacuolated cortical cells (right) extruded from the cortex though a break in the capsule (between arrows) to the exterior (hematoxylin-eosin, original magnification ×100). H, An aggregate of huge eosinophilic cortical cells surrounded by clusters of zona fasciculata-type vacuolated cells (hematoxylin-eosin, original magnification ×400).
The sex and age of the study patients, their CNC components and other findings, family relationships, and PRKAR1A mutational status are shown in Table 1. Seventeen patients (59%) had classic CS (1 was African American): 7 were 10 years or younger, 7 were aged 11 through 20 years, and 3 were older than 20. CS was the presenting finding for 2 of them (Figure 1B and 1F). The youngest (Figure 1B–1E) and oldest patients at diagnosis of CS were 2 and 45 years, respectively. Seven patients had adrenal surgery at Mayo Clinic; 8 were operated on at other institutions. Two patients with classic CS were not treated (Figure 2A and 2B). Six patients had subclinical CS, and 6 had possible CS. Eighteen patients (62%) were members of 6 families in which CNC was transmitted as a dominant trait (Figure 1A). Some findings in 10 of the patients were reported previously (9–16).
Figure 2.
Classic Cushing Syndrome. A through F, Patient 1; G through J, Patient 10. A, At age 17 years, the patient’s plump face was covered with small pigmented spots. B, At age 65 years, the face had lost its fullness and the facial pigmentation was less obvious. C, Cut surface of left adrenal gland at autopsy showed several dark micronodules (arrows). D, The left adrenal gland featured several micronodules (asterisks) and loss of normal cortical zonation. The capsule was intact. There was no extracapsular extension (hematoxylin-eosin, original magnification ×20). E, A cortical nodule had large and huge eosinophilic cells, many with lipochrome (arrows), and there was an exceptional cell with elongated crystal type spaces (center) (hematoxylin-eosin, original magnification ×400). F, Synaptophysin staining. Upper, Two positive areas were seen; 1 was ovoid (asterisk) (original magnification ×100). Lower, Heavily stained round paranuclear “bodies” (arrows) were seen along with cell membrane and cytoplasmic staining (original magnification ×400). G, Carney complex mucosal and skin lesions (patient 10). Upper, Mucocutaneous pigmentation. Middle, Lacrimal caruncular pigmentation. Lower, Cutaneous auricular nodule (myxoma). H, Cut surfaces of the left adrenal gland showed variably-sized pigmented micronodules and oval elongated zones (arrows) and a 1.7-cm oval darkly pigmented lesion (myelolipoma) (asterisk) attached to the gland. I, Bottom left slice of adrenal in H showed an epicapsular mass of cortical cells with some fat (arrows) (hematoxylin-eosin, original magnification ×1). J, The gland had multiple micronodules staining strongly for synaptophysin (arrows). The myelolipoma was weakly stained for synaptophysin (original magnification ×1).
Adrenal and Pituitary Imaging and Laboratory Testing
Computed tomographic (CT) imaging (Supplementary Table) showed unilateral or bilateral adrenal micronodularity, with or without thickening of the gland(s) in 7 patients; unilateral thickening or unilateral nodule or unilateral mass with contralateral micronodularity in 1 patient each; and normal adrenals in 3 patients. Four patients had indeterminate results or did not have an imaging study. Nine patients had a pituitary tumor (3 had a growth hormone–producing adenoma resected). Results of the laboratory testing are shown in the Supplementary Table.
Molecular Genetics
Six different PRKAR1A mutations were found among 12 of 13 patients (Table 1). The gene was deleted in patient 17.
Treatment and Follow-up
Nine of the 17 patients with classic CS underwent primary bilateral adrenalectomy and were cured of all manifestations of the syndrome (Table 2). Three others had recurrence of the syndrome after unilateral adrenalectomy and were cured with contralateral adrenalectomy. Two patients underwent subtotal adrenalectomy: Patient 11 (Table 2) was cured of clinical manifestations of the syndrome without need for postoperative adrenal steroid replacement (mild asymptomatic adrenal autonomy remained) (follow-up, 54 years), and patient 14 (Table 2) required postoperative adrenal support. One patient (patient 4; Table 2) underwent subtotal unilateral adrenalectomy and was cured of CS without need for postoperative adrenal support (follow-up, 66 years). Classic CS did not develop in any of the 12 patients with subclinical or possible CS during follow-up that ranged from 1 to 36 years (mean, 10 years).
Table 2.
Treatment, Adrenal Pathological Diagnosis, Duration of Follow-up, and Status of 17 Patients with Cushing Syndrome and Carney Complex
| Patient | Treatment | Pathological Diagnosis
|
Duration of Follow-up, y | Status | |
|---|---|---|---|---|---|
| Initial (Year) | Study | ||||
| 1 | None | Cortical hyperplasia (1937) | PPNAD with cortical atrophy (autopsy) | 60 | Dead at 77 y No CS Adrenal sufficient Autopsy: metastatic pancreatic carcinoma, bilateral PPNAD, pituitary normal |
| 2 | None | No adrenal tissue obtained at presentation (1937) | PPNAD with cortical atrophy (autopsy) | 27 | Dead at 44 y (sudden) Chronic Cushing syndrome Autopsy: severe coronary atherosclerosis, bilateral PPNAD, pituitary normal, testes grossly normal |
| 3 | Bilateral adrenalectomy | PPNAD | PPNAD with cortical atrophy | 27 | Alive at 67 y Well Adrenal insufficient |
| 4 | Subtotal (<90%) left adrenalectomy Biopsy right adrenal |
Left adrenal: diffuse cortical hyperplasia Right adrenal: normal cortex |
PPNAD variant | 66 | Alive at 68 y Depression Adrenal sufficient Osteopenia Type 2 diabetes mellitus 4×4×7-mm thyroid nodule |
| 5 | Bilateral adrenalectomy | Right adrenal: single cortical nodule Left adrenal: nodular hyperplasia | PPNAD variant | 4 | Well at 11 y Adrenal insufficient |
| 6 | Bilateral adrenalectomy, asynchronous (ages 4 and 13 y) | Consistent with PPNAD | PPNAD variant | 6 | Well at 19 y Adrenal insufficient |
| 7 | Bilateral adrenalectomy | Consistent with PPNAD | PPNAD variant PPNAD in adrenal ectopia |
5 | Well at 21 y Adrenal insufficient |
| 8 | Bilateral adrenalectomy | Micronodular hyperplasia | PPNAD variant | 5 | Well at 17 y Adrenal insufficient |
| 9 | Bilateral adrenalectomy | PPNAD | PPNAD | 23 | Alive at 65 y Adrenal insufficient Severe tricuspid valve regurgitation due to Ebstein anomaly |
| 10 | Bilateral adrenalectomy | PPNAD | PPNAD with 1.8-cm epicapsular myelolipoma | 29 | Well at 63 y Adrenal insufficient |
| 11 | Left: total adrenalectomy Right: subtotal (67%–75%) adrenalectomy |
Cortical hyperplasia | Suggestive of PPNAD | 54 | Alive at 61 y Adrenal sufficient Cervical nodal and pulmonary metastatic follicular thyroid carcinoma |
| 12 | Bilateral adrenalectomy | PPNAD | PPNAD | 14 | Well at 33 y Adrenal insufficient |
| 13 | Bilateral adrenalectomy, asynchronous (ages 9 and 18 y) | Left adrenal (1969): pigmented carcinoma Right adrenal (1973): adenomatous cortical hyperplasia |
PPNAD with vacuolated cell hyperplasia and massive intracapsular and epicapsular cortical hyperplasia | 42 | Alive at 59 y Adrenal insufficient Mitral and tricuspid regurgitation after 5 cardiac operations for cardiac myxoma |
| 14 | Left total and right subtotal (67%) adrenalectomy (age 10 y) Completion of right adrenalectomy (age 18 y) |
Cortical hyperplasia | PPNAD with vacuolated cell hyperplasia and massive epicapsular cortical hyperplasia | 44 | Alive at 63 y Adrenal insufficient Hemiplegia due to cardiac myxoma embolus |
| 15 | Bilateral adrenalectomy (age, 46 y) | Consistent with PPNAD | PPNAD | 2 | Well at 48 y Adrenal insufficient |
| 16 | Bilateral adrenalectomy (age, 12 y) | PPNAD | PPNAD with 1.5-cm macronodule | 1 | Well at 17 y Adrenal insufficient |
| 17 | Bilateral adrenalectomy, asynchronous (right at age 2 y and left at 29 y) | Right adrenal (1985): cortical adenoma Left adrenal (2012): consistent with PPNAD |
Right: cortical adenoma (2 cm); rare PPNAD-type micronodules; outer cortex synaptophysin positive Left: PPNAD |
27 (after right adrenalectomy) 3 (after left adrenalectomy) |
Dead at 33 y (sudden) Adrenal insufficient Autopsy: no anatomical cause of death found; brain, mesial temporal sclerosis; pituitary, normal; vitreous body electrolytes, normal; kidneys, nephrosclerosis; vertebral bone, nonspecific osteopenia |
Abbreviation: PPNAD, primary pigmented nodular adrenocortical disease.
Pathology
The initial and study adrenal pathological diagnoses are presented in Table 2. The glands were generally normal size or slightly enlarged and retained their normal shape in the patients with classic CS. Total adrenal weight ranged from 6 to 13.7 g (mean, 9 g; normal range for adult, 8–9 g). The sectioned glands showed small (1–4 mm), brown, black, yellow, and red micronodules (Figure 2). The glands were enlarged, together weighing 13.5 g, and microscopically abnormal in patient 27, who did not have classic CS. Three patients each had a cortical mass: an encapsulated 2-cm adenoma (patient 17), an unencapsulated 1.5-cm macronodule (patient 27), and an unencapsulated 1.8-cm epicapsular myelolipoma (patient 10).
Hematoxylin-eosin–stained sections of the adrenals showed 1) enlarged eosinophilic cortical cells, some with lipochrome arranged in micronodules; 2) extranodular cortical atrophy or vacuolated cell hyperplasia (patients 4–8); 3) epicapsular and intracapsular extension of cortical hyperplasia, with the former sometimes connected with the cortex proper through breaks in the gland capsule (there was no infiltration of the medulla); and 4) benign cortical tumors (mentioned in the previous paragraph). According to combinations of findings, 12 patients had PPNAD with many micronodules, extranodular cortical atrophy, and slight intracapsular and epicapsular cortical extension, and 7 patients had PPNAD variant with scattered cortical micronodules, vacuolated cell cortical hyperplasia, and prominent intracapsular and epicapsular cortical hyperplasia. The ectopic adrenal cortex found incidentally in patients 7 and 29 had findings suggestive of PPNAD variant and PPNAD, respectively. The cortical hyperplasia and micronodules in patients 13 and 14 (sisters) and patient 27 were vacuolated cell type.
The adrenal glands of 2 patients with CS (patients 1 and 2; Table 2) who were not treated were obtained at autopsy. The syndrome regressed spontaneously in patient 1. When re-examined at age 65 years, the patient had no clinical evidence of CS, but there was mild ACTH-independent adrenal autonomy. The patient’s twin brother (patient 2; Table 2) died suddenly at age 44 years with acute hypertension (blood pressure, 230/130 mm Hg) after an inguinal hernia operation. Hospital notes included the following comments: “Needs an endocrinological work-up,” “Historically, people called him ‘red-face’,” and “Has anterior vertebral body flattening, suggesting decalcification.” Both patients had PPNAD at autopsy. Patient 17 who had asynchronous bilateral adrenalectomy died suddenly at age 33 years. No cause was found at autopsy. The patient had vomiting and diarrhea for several days before death and reportedly had been taking steroid replacement.
The adrenal glands of 2 patients with possible CS were studied. Patient 27 (Table 1; Figure 3E–3H) died at age 44 years of cardiac failure due to ischemic heart disease and sequelae of multiple operations for cardiac myxoma. Necropsy showed bilateral adrenal enlargement due to vacuolated cell hyperplasia, vacuolated cell micronodules, and a 1.5-cm eosinophilic pigmented macronodule. Patient 28 (Figure 3A–3D) (the index patient for CNC) died suddenly at age 33 years of mitral valve occlusion caused by a prolapsed left atrial myxoma. PPNAD and pituitary Crooke cells (an indicator of hypercortisolemia) were found at autopsy.
Immunoperoxidase staining results were available for 14 patients (the sections in 2 patients were not immunoreactive). Synaptophysin, inhibin-A, melan A, and CD56 (cell membrane) stained the micronodules in 13, 13, 8, and 5 patients, respectively; the extranodular cortex was positive for the stains in 10, 10, 10, and 7 patients, respectively; and the epicapsular cortex was positive in 12, 11, 8, and 8 patients, respectively. The adenoma in patient 17 stained for synaptophysin (patchy), inhibin-A and melan A, and CD56 (weakly). The associated extratumoral cortex was strongly positive for synaptophysin. The ectopic adrenal cortex in patients 7 and 29 was synaptophysin positive. The micronodules in the glands obtained at the autopsy of patient 1 were synaptophysin positive. The paraffin sections in the other 2 autopsied patients (patients 27 and 28) were not immunoreactive.
Discussion
CS is usually caused by an ACTH-secreting pituitary tumor (about 80% of the cases). Infrequently, it results from a primary adrenal neoplasm, an adenoma, or (rarely) a carcinoma, and, exceptionally, it is caused by primary bilateral micronodular and macronodular adrenocortical hyperplasia resulting from genetic mutations: germline mutations of PRKAR1A (6), PDE11A (17), PDE8B (18), PRKACA (19), and ARMC5 (20) and somatic mutations of both GNAS1 and PRKACA (19,21).
Herein, we report the findings from the 29 Mayo Clinic patients with CNC and abnormal adrenocortical findings, including 1) different types of CS (persistent, cyclic, and ameliorative); 2) syndrome cure with different surgical interventions (total adrenalectomy, subtotal adrenalectomy, and partial unilateral adrenalectomy); 3) rare spontaneous resolution of the syndrome (patient 1); 4) several pathological findings, PPNAD, PPNAD variant, and benign cortical neoplasms; 5) typical CNC-type adrenal pathology (PPNAD) but no CS (silent PPNAD) (patients 27 and 28); and 6) PPNAD in ectopic adrenal cortex (patients 7 and 29).
Radiologic and laboratory testing results were incomplete because of 1) unavailability of CT and measurement of serum cortisol and cortisol suppression testing when some of the patients were examined and 2) nonsystematic performance of the testing by multiple examiners. The CT imaging results (Supplementary Table) were not diagnostic. None of the patients had the “string of beads” appearance characteristic of PPNAD (22). Half of those with classic CS had small nodules or thickening of the glands. Biochemically, the laboratory results showed that patients with classic CS had ACTH-independent cortisol synthesis and secretion (Supplementary Table).
PPNAD is cured with bilateral adrenalectomy (23,24). In the past, some patients who were treated with unilateral adrenalectomy had variable results, including unexpected “cure” of the CS for up to 9 years after unilateral adrenalectomy (25). Three different surgical treatments were used in 15 of the 17 Mayo Clinic patients with classic CS. The other 2 patients (seen in 1939) were not treated (Table 2). Twelve patients had bilateral total adrenalectomy and were cured of all manifestations of CS. Two patients underwent subtotal adrenalectomy: 1 (patient 11) was cured of the clinical manifestations of CS without need for corticosteroid replacement (mild adrenal autonomy remained); the other (patient 14) was cured of the clinical manifestations of the syndrome but had postoperative adrenal insufficiency. Patient 4 had subtotal unilateral adrenalectomy, was cured of CS, and did not need adrenal support. At follow-up of 1 of the untreated patients (patient 1) (Figure 2A–2D), the CS (present at age 17) had regressed spontaneously. At age 65, she had no clinical evidence of CS (mild adrenal autonomy remained). At age 77, she died of pancreatic carcinoma: PPNAD was found at autopsy. The syndrome persisted in the other untreated patient (patient 2) for 27 years. He died suddenly at age 44. PPNAD was found at autopsy. The mild postoperative adrenal autonomy in patients 4 and 11 apparently did not cause adverse effects. The results of less than bilateral total adrenalectomy warrant reconsideration of the surgical treatment of CS in CNC.
The majority of the patients (12 of 13) with classic CS had a PRKAR1A mutation or deletion (Table 1). The mutations caused different clinical phenotypes, and different clinical phenotypes resulted from the same mutation (Table 1 and Figures 1–3).
On the basis of the relative prominence of the observations, the findings were classified as PPNAD (12 patients) or PPNAD variant (including the 5 members of family B [patients 4–8] and the sisters [patients 13 and 14]). PPNAD histologic changes were found in accessory adrenal cortex in patients 7 and 29: Patient 7 had classic CS and histologically proven PPNAD variant, and patient 29 did not have CS, but it seems possible, likely perhaps, that the patient’s orthotopic adrenal glands had functionally silent PPNAD.
The pathologic findings described in the previous 2 paragraphs are not peculiar to CNC—they have appeared in patients with mutations of PDE11A, PDE8B, and PRKACA, thereby indicating that they are common in germline-induced adrenal cortical hyperplasia and are perhaps characteristic of it. Nongenetic (ACTH)-induced cortical hyperplasia is different histologically, being confined within the gland capsule and thus raising the following question: Why does genetically driven hyperplasia extrude outside the capsule and ACTH-driven hyperplasia not extrude? The explanation may lie in the level of the cortex or a particular subset of cortical cells affected by the abnormal stimulus.
Although all the adrenal lesions encountered in the study were benign, adrenocortical carcinoma has been reported in 2 patients with CS in CNC (25,26). It is possible that these neoplasms evolved through progression of a micronodule to a macronodule (found in patient 26) to an adenoma (found in patient 17) and eventually to carcinoma. β-Catenin mutations have been found in adenomas in PPNAD and may be involved (27).
The synaptophysin-positive cells in the micronodules and the cortex were evidently a site of cortisol overproduction and secretion. Although the molecular events leading to this are unknown, apparently a malfunction in the normal pathway of cortisol synthesis or in an upstream mechanism regulated this. The large eosinophilic cells in the micronodules were variously 1) overactive with excessive secretion of cortisol, causing classic CS (patients 2–17); 2) overactive to a lesser degree and insufficient to cause classic CS (patients 18–23); 3) overactive (causing CS) with return to “normal” function (patient 1); and 4) normoactive (patient 27) with PPNAD in adrenocortical heterotopia.
Among the other conditions listed in Table 1, pilonidal cyst, which was present in patients 1 and 2 (brother and sister), has been reported in 4 members of a family with CNC (28), making the condition almost certainly a rare manifestation of the syndrome. Also, in patient 11, the follicular thyroid carcinoma, which was bilateral with cervical lymph node metastasis, is a very unusual presentation of the tumor, and this lesion may be another rare constituent of CNC.
CS is a serious component of a rare familial multiorgan tumoral disorder, CNC. Among the 37 Mayo Clinic patients with CNC, 17 had classic CS caused by primary bilateral adrenocortical pathology. The CS was cured with bilateral adrenalectomy, with subtotal adrenalectomy, and with partial unilateral adrenalectomy; it persisted untreated for almost 3 decades in 1 patient and regressed spontaneously in another. Two patients had PPNAD but no CS, indicating that the adrenal pathology was clinically silent.
Supplementary Material
Acknowledgments
Source of Funding: The study was supported in part with funds from the National Institutes of Health under Z01HD008920.
Abbreviations
- ACTH
corticotropin
- CNC
Carney complex
- CS
Cushing syndrome
- CT
computed tomographic
- PPNAD
primary pigmented nodular adrenocortical disease
Footnotes
Conflicts of Interest: The authors have no conflicts of interest.
References
- 1.Cushing H. The basophil adenomas of the pituitary body and their clinical manifestations (pituitary basophilism) 1932. Obes Res. 1994 Sep;2(5):486–508. doi: 10.1002/j.1550-8528.1994.tb00097.x. [DOI] [PubMed] [Google Scholar]
- 2.Shenoy BV, Carpenter PC, Carney JA. Bilateral primary pigmented nodular adrenocortical disease: rare cause of the Cushing syndrome. Am J Surg Pathol. 1984 May;8(5):335–44. doi: 10.1097/00000478-198405000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Carney JA, Gordon H, Carpenter PC, Shenoy BV, Go VL. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine (Baltimore) 1985 Jul;64(4):270–83. doi: 10.1097/00005792-198507000-00007. [DOI] [PubMed] [Google Scholar]
- 4.Carney JA, Hruska LS, Beauchamp GD, Gordon H. Dominant inheritance of the complex of myxomas, spotty pigmentation, and endocrine overactivity. Mayo Clin Proc. 1986 Mar;61(3):165–72. doi: 10.1016/s0025-6196(12)61843-6. [DOI] [PubMed] [Google Scholar]
- 5.Johns Hopkins University. Online Mendelian Inheritance In Man: Carney complex Type 1 [Internet] 2012 Sep 14; [cited 2016 Jul 27]. Available from: http://omim.org/entry/160980.
- 6.Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, et al. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet. 2000 Sep;26(1):89–92. doi: 10.1038/79238. [DOI] [PubMed] [Google Scholar]
- 7.Stratakis CA, Carney JA, Lin JP, Papanicolaou DA, Karl M, Kastner DL, et al. Carney complex, a familial multiple neoplasia and lentiginosis syndrome. Analysis of 11 kindreds and linkage to the short arm of chromosome 2. J Clin Invest. 1996 Feb 1;97(3):699–705. doi: 10.1172/JCI118467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stratakis CA, Kirschner LS, Carney JA. Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab. 2001 Sep;86(9):4041–6. doi: 10.1210/jcem.86.9.7903. [DOI] [PubMed] [Google Scholar]
- 9.Discovery of the Carney Complex, a Familial Lentiginosis–Multiple Endocrine Neoplasia Syndrome: A Medical Odyssey [Internet] [cited 2016 Jul 27]. Available from: http://journals.lww.com/theendocrinologist/Abstract/2003/01000/Discovery_of_the_Carney_Complex,_a_Familial.7.aspx?trendmd-shared=0.
- 10.Young WF, Jr, Carney JA, Musa BU, Wulffraat NM, Lens JW, Drexhage HA. Familial Cushing’s syndrome due to primary pigmented nodular adrenocortical disease: reinvestigation 50 years later. N Engl J Med. 1989 Dec 14;321(24):1659–64. doi: 10.1056/NEJM198912143212407. [DOI] [PubMed] [Google Scholar]
- 11.Dao LN, Scheithauer BW, Erlandson RA, Young WF, Jr, Carney JA. Divergent myoid, neuroendocrine, and perineural differentiation in a nasal tumor of a patient with Carney complex. Am J Surg Pathol. 2008 Jan;32(1):167–71. doi: 10.1097/PAS.0b013e31813c0e11. [DOI] [PubMed] [Google Scholar]
- 12.Morgan DL, Palazola J, Reed W, Bell HH, Kindred LH, Beauchamp GD. Left heart myxomas. Am J Cardiol. 1977 Oct;40(4):611–4. doi: 10.1016/0002-9149(77)90078-9. [DOI] [PubMed] [Google Scholar]
- 13.Barlow JF, Abu-Gazeleh S, Tam GE, Wirtz PS, Ofstein LC, O’Brien CP, et al. Myxoid tumor of the uterus and right atrial myxomas. S D J Med. 1983 Jul;36(7):9–13. [PubMed] [Google Scholar]
- 14.Konecny T, Reeder G, Noseworthy PA, Konecny D, Carney JA, Asirvatham SJ. Percutaneous ablation and retrieval of a right atrial myxoma. Heart Lung Circ. 2014 Nov;23(11):e244–7. doi: 10.1016/j.hlc.2014.07.059. Epub 2014 Jul 24. [DOI] [PubMed] [Google Scholar]
- 15.Carney JA, Boccon-Gibod L, Jarka DE, Tanaka Y, Swee RG, Unni KK, et al. Osteochondromyxoma of bone: a congenital tumor associated with lentigines and other unusual disorders. Am J Surg Pathol. 2001 Feb;25(2):164–76. doi: 10.1097/00000478-200102000-00004. [DOI] [PubMed] [Google Scholar]
- 16.Fligiel Z, Kaneko M, Leiter E. Bilateral Sertoli cell tumor of testes with feminizing and masculinizing activity occurring in a child. Cancer. 1976 Oct;38(4):1853–8. doi: 10.1002/1097-0142(197610)38:4<1853::aid-cncr2820380465>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 17.Carney JA, Gaillard RC, Bertherat J, Stratakis CA. Familial micronodular adrenocortical disease, Cushing syndrome, and mutations of the gene encoding phosphodiesterase 11A4 (PDE11A) Am J Surg Pathol. 2010 Apr;34(4):547–55. doi: 10.1097/PAS.0b013e3181d31f49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horvath A, Mericq V, Stratakis CA. Mutation in PDE8B, a cyclic AMP-specific phosphodiesterase in adrenal hyperplasia. N Engl J Med. 2008 Feb 14;358(7):750–2. doi: 10.1056/NEJMc0706182. [DOI] [PubMed] [Google Scholar]
- 19.Carney JA, Lyssikatos C, Lodish MB, Stratakis CA. Germline PRKACA amplification leads to Cushing syndrome caused by 3 adrenocortical pathologic phenotypes. Hum Pathol. 2015 Jan;46(1):40–9. doi: 10.1016/j.humpath.2014.09.005. Epub 2014 Oct 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Assie G, Libe R, Espiard S, Rizk-Rabin M, Guimier A, Luscap W, et al. ARMC5 mutations in macronodular adrenal hyperplasia with Cushing’s syndrome. N Engl J Med. 2013 Nov 28;369(22):2105–14. doi: 10.1056/NEJMoa1304603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med. 1991 Dec 12;325(24):1688–95. doi: 10.1056/NEJM199112123252403. [DOI] [PubMed] [Google Scholar]
- 22.Doppman JL. Problems in endocrinologic imaging. Endocrinol Metab Clin North Am. 1997 Dec;26(4):973–91. doi: 10.1016/s0889-8529(05)70290-5. [DOI] [PubMed] [Google Scholar]
- 23.Powell AC, Stratakis CA, Patronas NJ, Steinberg SM, Batista D, Alexander HR, et al. Operative management of Cushing syndrome secondary to micronodular adrenal hyperplasia. Surgery. 2008 Jun;143(6):750–8. doi: 10.1016/j.surg.2008.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nieman LK, Biller BM, Findling JW, Murad MH, Newell-Price J, Savage MO, et al. Endocrine Society. Treatment of Cushing’s syndrome: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2015 Aug;100(8):2807–31. doi: 10.1210/jc.2015-1818. Epub 2015 Jul 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anselmo J, Medeiros S, Carneiro V, Greene E, Levy I, Nesterova M, et al. A large family with Carney complex caused by the S147G PRKAR1A mutation shows a unique spectrum of disease including adrenocortical cancer. J Clin Endocrinol Metab. 2012 Feb;97(2):351–9. doi: 10.1210/jc.2011-2244. Epub 2011 Nov 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morin E, Mete O, Wasserman JD, Joshua AM, Asa SL, Ezzat S. Carney complex with adrenal cortical carcinoma. J Clin Endocrinol Metab. 2012 Feb;97(2):E202–6. doi: 10.1210/jc.2011-2321. Epub 2011 Nov 23. [DOI] [PubMed] [Google Scholar]
- 27.Tadjine M, Lampron A, Ouadi L, Horvath A, Stratakis CA, Bourdeau I. Detection of somatic β-catenin mutations in primary pigmented nodular adrenocortical disease (PPNAD) Clin Endocrinol (Oxf) 2008 Sep;69(3):367–73. doi: 10.1111/j.1365-2265.2008.03273.x. Epub 2008 Apr 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Irvine AD, Armstrong DK, Bingham EA, Hadden DR, Nevin NC, Hughes AE. Evidence for a second genetic locus in Carney complex. Br J Dermatol. 1998 Oct;139(4):572–6. doi: 10.1046/j.1365-2133.1998.02450.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.