

Abstract
It has been known for decades that the delivery of T cell help to B cells is antigen-specific, MHC-restricted, and depends CD40L (CD154). It has been thought that when a T cell recognizes an antigen-presenting B cell, CD40L expressed on the T cell surface engages with CD40 on the surface of B cells as long as the cells remain conjugated. Addition of fluorescently labeled anti-CD40L antibody during overnight incubation of antigen-presenting B cells with antigen-specific T cells allowed us to discover that CD40L does not remain on the surface of the T cell, but is transferred to and endocytosed by B cells receiving T cell help. In the presence of anti-CD40L antibody, transferred CD40L is nearly absent on bystander B cells that are not presenting antigen, and the bystander cells do not become activated. Because transfer of CD40L to B cells correlates with B cell activation, we speculate that persistence of helper T cell-derived CD40L on or in B cells could permit sustained CD40 signaling enabling survival and proliferation of antigen-presenting B cells following brief interactions with helper T cells in vivo in germinal centers.
Keywords: CD40 ligand, CD40L, CD154, membrane transfer, T cell help, immunological synapse
Introduction
CD40L (CD154) is a type II transmembrane cytokine expressed by activated CD4+ T cells upon TCR stimulation. It engages CD40 on antigen-presenting cells (APCs) to activate macrophages, license dendritic cells for CD8+ T cell responses, and activate B cells for T cell-dependent antibody responses [1-4]. CD40L is essential for the development and maintenance of germinal centers and the processes of B cell affinity maturation, immunoglobulin class switching, and generation of long-lived plasma cells that occur there [5-7]. In the process of affinity maturation in the germinal center reaction, a limiting helper T cell signal allows selective survival and proliferation of rare B cells with somatic mutations that increase B cell receptor affinity [8]. Although CD40L is the essential, cell contact-dependent signal in T cell help for B cells, the mechanism for the antigen-specific delivery of this signal to antigen-presenting B cells is not fully understood.
Upon T cell recognition of antigen in the context of peptide-MHC, an immunological synapse is formed. Some cytokines are delivered directly to the point of T cell/APC contact, while others exit the cell surface in all directions [9]. Although directional secretion of CD40L has not been investigated, CD40L accumulates within the immunological synapse [10, 11], and T cell microtubule organizing center polarization towards the synapse is necessary for CD40L-dependent production of IL-12 by dendritic cells [10, 12, 13]. CD40L is rapidly removed from the T cell surface in vivo [14] or when T cells are cultured with CD40-expressing APC [15, 16]. Engaged CD40L has been shown both to be internalized by the T cell [15] and to be cleaved by metalloproteases to a soluble form [17].
Two photon microscopy studies have determined that interactions between antigen-specific B and T cells in the germinal center last for only a few minutes [18-22]. Because a few minutes of TCR stimulation is not long enough for de novo production of CD40L, CD40L must pre-exist in the T cell and be delivered in a rapid, antigen-specific fashion. We have shown that preformed CD40L exists in a TCR-regulated secretory compartment [23] in follicular helper T cells (Tfh) and all other memory and effector helper T cell subsets [24], and that preformed CD40L is sufficient to upregulate activation markers and induce proliferation of antigen-specific B cells after overnight incubation with T cells in the presence of IL-4 [25]. But we also know that a few minutes of CD40L stimulation is insufficient to activate resting B cells [26], and we speculate that a few minutes of CD40/CD40L engagement may also be insufficient to sustain the germinal center reaction. Recently the immunological synapse has been recognized as a site for the delivery of extracellular vesicles [27, 28], including TCR-containing microvesicles that sustain calcium signaling in antigen-presenting B cells [29]. It has been proposed without experimental evidence that CD40L is released as a vesicle onto the B cell surface and continues to deliver an activating signal to the B cell following a brief interaction with a Tfh cell in the germinal center [30]. We report here for the first time that CD40L is transferred to antigen-presenting B cells in a contact-dependent and antigen-specific manner.
Results
Antigen-specific, CD40-dependent transfer of CD40L to antigen-pulsed B cells
As stated in the introduction, CD40L is a transmembrane cytokine thought to remain on the T cell surface while it engages CD40 on an antigen presenting cell for a period long enough to induce downstream signaling in the antigen presenting cell. To determine if instead CD40L is transferred from T cells to B cells during antigen specific interactions, we used fluorescent anti-CD40L to stain permeabilized, antigen peptide-pulsed (Ag+) or mock-pulsed bystander B (Ag-) cell populations after overnight incubation at 37° with activated, Th1-polarized, TCR transgenic T cells (Fig. 1). Antigen-pulsed and bystander B cells were differentially labeled with CTV to distinguish B cell populations following overnight incubation (Fig. 1A). CD40L knockout (KO), antigen-pulsed B cells show anti-CD40L staining following overnight incubation with T cells, while bystander B cells in the same well do not (Fig 1E). Most of the transferred CD40L appears to be internalized by the B cells, because transfer was barely detectable by staining with anti-CD40L without permeabilization (data not shown). The transfer of CD40L depends on CD40 on the B cells (Figs. 1C, D). If the antigen-presenting B cells lack CD40, then CD40+ bystander B cells do obtain CD40L (Fig. 1D). CD40 KO antigen-pulsed B cells activate and induce surface expression of CD40L on the T cells but cannot downmodulate CD40L from the T cells, allowing accumulation of CD40L on the T cells [23]. We suggest that accumulation of CD40L on the T cells allows transfer to bystander B cells in a non-antigen specific manner. Together, these findings show that during antigen-specific T-B cell interactions, T cell-derived CD40L is transferred to and internalized by the B cells in antigen-specific and CD40-dependent process.
Figure 1. Antigen-specific, CD40-dependent transfer of CD40L to antigen-presenting B cells.

(A) The gating scheme for the in vitro overnight CD40L transfer assay is shown. (B, C, D, E) CD40L KO antigen-pulsed (Ag+), CD40 KO Ag+, and CD40L KO Ag- B cell populations were incubated with Th1 cells overnight, then fixed, permeabilized, and stained with anti-CD40L-PE antibody. The control histogram (solid grey) represents intracellular staining with a hamster IgG-PE antibody of CD40LKO Ag+ B cells incubated with Ag- B cells and Th1 cells. The experiment with CD40L KO Ag+ and CD40 KO Ag- B cells was performed twice. The experiment with CD40L KO Ag+ and CD40L KO Ag- B cells was performed 3 times with similar results.
Transfer of CD40L to antigen-pulsed B cells is much more apparent when fluorescent anti-CD40L antibody is added during the T/B interaction
Owing to CD40-dependent downmodulation, CD40L accumulation on the surface of activated T cells is difficult to detect following activation in the presence of CD40+ APCs unless fluorescently labeled anti-CD40L antibody is added during T cell stimulation at 37° [16, 23]; addition of blocking anti-CD40L allows accumulation of CD40L on the T cell surface to sufficient levels to allow unambiguous identification of antigen-specific T cells [31, 32]. To determine whether addition of fluorescently labeled anti-CD40L antibody during the incubation might also increase detection of transferred CD40L on antigen-presenting B cells, we set up an overnight in vitro assay with TCR transgenic T cells and target B cells in which we included fluorescently labeled anti-CD40L antibody at 1μg/ml. Under these conditions, a large percentage of antigen-pulsed B cells became strongly CD40L positive, while bystander B cells remained mostly CD40L negative (Figs. 2A, B). The labeled CD40L is T cell-derived and transferred to the antigen-pulsed B cells, because accumulation of CD40L on or in the B cells depends upon CD40L expression by T cells (Fig. 2A) and does not depend on the ability of the B cells to make CD40L (Fig. 2C). Transfer and/or accumulation of CD40L on or in the B cells is partially dependent on CD40 expression by the B cells, because antigen-pulsed CD40 KO B cells had less than half of the number of CD40L positive cells after overnight incubation with CD40L-sufficient T cells when compared to CD40-expressing antigen-pulsed B cells (Figs. 2A, B). Moreover, the fluorescence intensity of CD40L staining was higher on CD40-sufficient than CD40 KO cells, indicating that CD40-sufficient cells may obtain or retain more CD40L on a per cell basis (Fig. 2A). B cells express surface Fc receptors (FcRs) that could promote transfer and internalization of fluorescently labeled anti-CD40L/CD40L complexes. Therefore, to determine whether the transfer of CD40L to the antigen-specific B cells depends on FcRs, we blocked FcRs in vitro with an antibody. Indeed, blocking anti-FcR antibody decreased both the percent of CD40L positive B cells and fluorescence intensity of anti-CD40L on antigen-pulsed B cells (Fig. 2D), although the decrease in percent CD40L positive B cells did not reach statistical significance (Fig. 2D, right panel). However, the blocking anti-FcR antibody had a much larger effect on CD40L transfer to CD40-deficient, antigen-pulsed B (Fig. 2E), reducing transfer to near the same level as bystanders not pulsed with antigen (Fig 2E, right panel). To further verify FcR-independent transfer to antigen-specific B cells, we obtained B cells from FcR KO mice. These results recapitulate the finding with the addition of the FcR blocking antibody (Fig. 2D). Therefore, transfer or persistence of transferred CD40L complexed with fluorescent anti-CD40L is largely independent of FcRs in the case of CD40-sufficient B cells, but can be mediated by FcRs when CD40 is absent.
Figure 2. Enhanced detection of CD40L transfer in the presence of fluorescent anti-CD40L antibody.

(A, C) Each contour plot shows the percentage of B cells that have acquired CD40L following overnight incubation with Th1 cells in the presence of fluorescently labeled anti-CD40L antibody in a representative experiment. (B) The graph shows the results from seven independent experiments (mean +/- SD). p values were calculated using an unpaired, two-tailed Mann-Whitney test: Ag+ vs. Ag- p = 0.0006, Ag+ vs. CD40KO Ag+ p = 0.0061, Ag- vs. CD40KO Ag+ p = 0.0061. ** p ≤ 0.01, *** p ≤ 0.001. (D, E) Contour plots and graphs show the effects of CD40, FcR deficiency, and blocking anti-FcR antibody on detection of CD40L transfer. The graph shows the percentage of B cells bearing transferred CD40L in five (D top panels) or three individual experiments (D bottom panels, E) (mean +/- SD).
Antigen-specific transfer of CD40L in the presence of cyclosporin A (CsA) to block de novo CD40L synthesis
We have previously shown that treatment of T cells with CsA blocks de novo production of CD40L by helper T cells, thereby limiting the amount of CD40L to the small amount that is stored as protein in an intracellular secretory compartment [25]. Preformed CD40L is delivered to the T cell surface within minutes of TCR stimulation, and that small amount is sufficient to induce upregulation of co-stimulatory markers and proliferation of antigen-pulsed B cells [25]. In order to evaluate whether preformed CD40L is transferred to antigen-pulsed B cells, we treated T cells with CsA during overnight incubation to block de novo CD40L production (Fig. 3A), and then evaluated transfer following overnight incubation in the presence of fluorescently labeled anti-CD40L antibody. As shown in Fig. 3C, the small amount of preformed CD40L available in CsA-treated T cells is transferred efficiently to antigen-presenting B cells following overnight incubation. This indicates that the large amount of de novo CD40L made by T cells after antigen recognition in vitro is not required to detect CD40L transfer to B cells, although the amount of CD40L transferred to individual cells during overnight incubation in the presence of CsA was reduced as indicated by the reduction in CD40L fluorescence intensity of the B cells (geometric mean fluorescence intensity of the CsA-treated cells in Fig. 3C is 726 versus 1475 for the non-treated cells in Fig. 3B). Transfer could be detected as early as two hours of culture (Fig. 3D).
Figure 3. De novo synthesis of CD40L is not required for Ag-specific CD40L transfer.

(A) Histograms depict T cell surface CD40L after overnight culture with Ag+ B cells and anti-CD40L in the absence or presence of CsA. (B, C) Contour plots show representative transfer of CD40L to B cells in the absence of antigen (no Ag) and to bystander B cells (Ag-) and antigen-pulsed B cells (Ag+) within the same well following overnight incubation with Th1 cells and CsA. (D) Contour plots show representative transfer of CD40L to the bystander and Ag+ B cells during a two-hour incubation with Th1 cells and CsA. Results are representative of three independent experiments.
Visualization of transferred CD40L
Antigen-pulsed splenocytes were labeled with CTV and cultured overnight with unpulsed splenocytes and T helper cells in the presence of allophycocyanin-labeled anti-CD40L. B cells were then surface stained with anti-CD19-FITC labeled antibody and assessed for the presence of CD40L by laser scanning microscopy. In each image, B cells (CD19+) were classified as antigen-pulsed or bystander by CTV staining, and scored as CD40L (allophycocyanin) positive or negative (Fig. 4). Many more of the antigen-pulsed B cells stained positive for CD40L compared to the bystander cells (Fig. 4C). Antigen-pulsed B cells had both intracellular and surface CD40L as assessed by z-stacks and CD19-FITC surface staining (Figs. 4A, B, D, and Supporting Information Video 1 and Video 2). These microscopy results were consistent with samples from the same cultures analyzed by flow cytometry, which showed an average of 63% antigen-pulsed B cells positive for CD40L and only 6% of the bystanders. Cells incubated overnight with a control hamster IgG allophycocyanin-labeled antibody did not have positive allophycocyanin fluorescence after overnight incubation (data not shown). Very few CD40 KO cells were CD40L-allophycocyanin positive by microscopy as compared to flow cytometry (data not shown). The difference between the percentage of CD40 KO CD40L-allophycocyanin positive cells detected by microscopy as compared to flow cytometry may reflect a difference in detection sensitivity, because the geometric mean fluorescence intensity of the CD40L label in the CD40 KO B cells is much lower than that of the CD40-sufficient B cells (Fig. 2A).
Figure 4. Transferred CD40L is found intracellularly and on the surface of antigen-pulsed B cells following overnight incubation with Th1 cells in the presence of anti-CD40L.
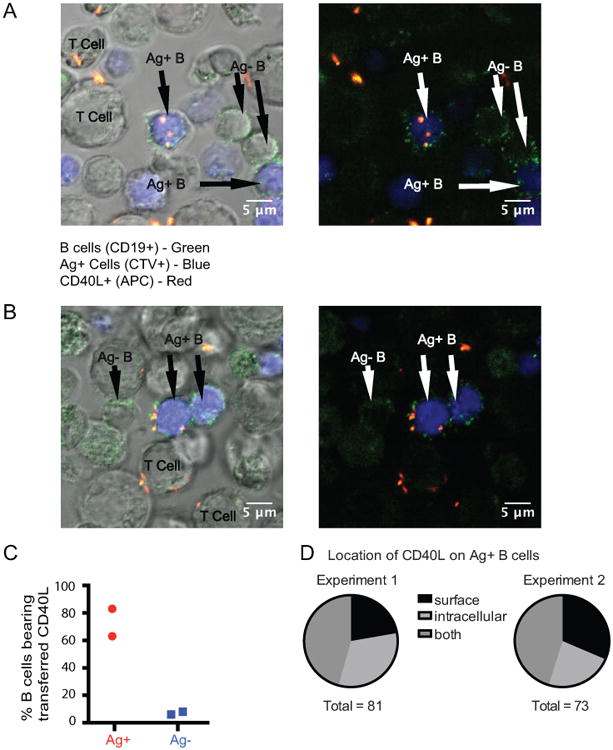
(A, B) Two representative images of Ag+ B cells (green (CD19+) and blue (CTV)) and bystander B cells (green only (CD19+)) together with T cells are shown with and without the differential interference contrast overlay following overnight incubation. The images in A and B show a frame through the middle of the cells from videos (Supporting Information Video 1 and Video 2) created from 2.5 μM z stacks. Intracellular CD40L (allophycocyanin-labeled anti-CD40L, shown in red) is detected in the Ag+ B cell in A and an example of surface CD40L is shown in B. (C) The graph shows the percentage Ag+ and Ag- B cells positive for the presence of CD40L in two individual experiments. (D) The pie charts show the relative numbers of B cells positive for surface, intracellular, or both surface and intracellular CD40L. There were 281 B cells analyzed in Experiment 1 (128 Ag+ and 153 Ag-) and 193 B cells in Experiment 2 (88 Ag+ and 105 Ag-).
Transfer of CD40L correlates with activation as measured by upregulation of ICAM-1 on B cells
Signals downstream of the CD40-CD40L interaction are necessary for T cell-dependent antibody responses. In order to determine whether transfer of CD40L could be involved in T cell-dependent B cell activation, we measured B cell activation in these cultures by CD40-dependent upregulation of ICAM-1 [25]. We chose this parameter of CD40L effector function because it was shown to be the most sensitive indicator of T cell-dependent B cell activation in our previous studies [25]. As in previous experiments, TCR transgenic Th1 cells were incubated with antigen-pulsed and mock-pulsed bystander B cells overnight at 37°. We found that the antigen-specific B cells that bore transferred CD40L were the same cells that upregulated ICAM, further supporting the idea that the transferred CD40L correlates with B cell activation (Fig. 5A). ICAM geometric mean fluorescent intensity was significantly different between those antigen-pulsed cells that had received CD40L compared to the antigen-pulsed cells that had not (Fig. 5A). When the antigen-pulsed and bystander B cells were separated from the T cells by a protein-permeable membrane in a Costar Transwell®, only the B cells in the upper chamber that were able to come into contact with T cells acquired transferred CD40L as detected by fluorescently labeled anti-CD40L antibody (Fig 5B) and received T cell help (Fig. 5D, E, F, top), indicating that upregulation of ICAM-1 on bystander B cells is dependent upon their ability to contact T cells, and is not owing to soluble CD40L or another soluble factor. Antigen-pulsed, CD40-deficient B cells did not upregulate ICAM-1 upon interaction with T cells (Figs. 5D, E), although they were able to stimulate the T cells to activate CD40-sufficient bystander B cells (Fig. 5E, a result that correlates with accumulation of CD40L on activated T cells [23] and transfer of CD40L to bystander B cells (Fig. 1D) in the presence of CD40-deficient antigen-pulsed B cells. In summary, both antigen-presenting and bystander B cells can receive contact- and CD40-dependent T cell help, and B cell activation correlates with CD40L transfer.
Figure 5. Antigen-specific CD40L transfer and help, unlike bystander help, is resistant to blocking by anti-CD40L antibody.

(A) Representative contour plot of CD40L transfer and ICAM-1 upregulation is shown for a single experiment in the antigen-pulsed B cells. Graph shows ICAM geometric mean fluorescent intensity values for CD40L positive and negative Ag-pulsed B cells after overnight incubation with Th1 cells in the presence of fluorescently labeled anti-CD40L antibody. p value was calculated using an unpaired, two-tailed Mann-Whitney test: p = 0.002 (B, C, D, E, F) Th1 and B cells were added to the top and bottom chambers of the Transwells® as depicted in the schematics in the presence (B) or absence (C, D, E, F) of fluorescently labeled anti-CD40L antibody. CD40L transfer (B) or ICAM-1 upregulation (C, D, E, F) was measured on the B cell populations in both the top and bottom chambers, and CD40L contour plots (B) or ICAM-1 histograms (C, D, E, F) are shown. The filled grey histogram on each panel is the control without antigen in the top chamber. This experiment is representative of two (B) or three (C, D, E, F) independent experiments. (G) Representative histograms of ICAM-1 upregulation on Ag+, Ag-, and no antigen control B cells after overnight incubation with 5CC7 Th1 cells. The graph shows the fold increase in ICAM-1 mean fluorescence intensity for three individual experiments (mean +/- SD) relative to the no Ag control B cell. (H, I left panel) Th1 cells were incubated with antigen-pulsed (Ag+) and bystander (Ag-) B cells in the presence or absence of neutralizing anti-CD40L antibody (10 μg/ml) and activation was assessed by ICAM upregulation. (I right panel) The graphs show the effects of different concentrations of anti-CD40L on activation of Ag+ and Ag- B cells. The left and right graphs are representative of three or two independent experiments, respectively. (J, K) Differentially labeled (CTV or CFSE) antigen-pulsed (Ag+) and CD40KO Ag+ along with Ag- B cells were incubated with Th1 cells, and proliferation was assessed after 48 hours by dilution of these dyes in the presence and absence of anti-CD40L (0.1 μg/ml). The percent of divided cells is labeled on each histogram and is representative of two independent experiments.
Antigen-specific T cell help is more resistant to inhibition by anti-CD40L blocking antibody than bystander help
In the absence of anti-CD40L blocking antibody, both bystander and antigen-pulsed B cells upregulate ICAM-1 (Fig. 5F, G). Upregulation of ICAM-1 on the antigen-pulsed B cells is greater than on bystanders, indicating a bias towards antigen-specific delivery of the CD40L/CD40 signal. Our studies show that when de novo CD40L expression is blocked by treatment with CsA as in Fig. 3A, only antigen-pulsed B cells receive T cell help as measured by upregulation of ICAM-1 [25] and transferred CD40L (Fig. 3A). Therefore, when the amount of CD40L is limiting, there is an increase in antigen specificity of both transfer of CD40L and the delivery of T cell help. As described above, we found that antigen-specific transfer of CD40L to B cells is much easier to detect when fluorescently labeled anti-CD40L antibody is added during overnight incubation with helper T cells. We wanted to determine the effects of addition of anti-CD40L antibody on T cell-dependent B cell activation of both antigen-specific and bystander B cells. Upon addition of anti-CD40L at 1μg/ml, the concentration used in the CD40L transfer experiments (Figs. 2-4, 5A, and 5B), bystander B cell activation is completely eliminated, while anti-CD40L added at 10 μg/ml attenuates but does not block activation of antigen-presenting B cells (Figs. 5H and 5I).
CD40L engagement of CD40 on B cells initiates signaling which induces B cell proliferation in the presence of soluble T cell cytokines such as IL-4 or IL-21 [23, 33, 34]. We next investigated antigen-specific delivery of T cell help by measuring proliferation of B cells following 48 hours of incubation with Th1 cells. In this assay, the antigen pulsed B cells were labeled with CFSE, while the bystanders were labeled with CTV or vice versa. After 48 hours in the presence of Th1 cells, antigen-pulsed B cells had a larger proportion of cells that had divided compared to the bystanders (Fig. 5J). Antigen-pulsed CD40-deficient B cells had almost no cells that divided following incubation with Th1 cells (Fig. 5J). When blocking antibody was added to the T/B cell cultures at the low concentration of 0.1 μg/ml, bystander proliferation is completely inhibited, while antigen-specific proliferation is largely unaffected (Fig. 5K).
Therefore, both transfer of CD40L to bystander B cells and contact- and CD40-dependent activation of bystander B cells are very sensitive to blocking with anti-CD40L, while both transfer of CD40L to and activation of antigen-presenting B cells are resistant. In the presence of anti-CD40L, only the antigen-pulsed B cells receive CD40L, upregulate ICAM, and proliferate, while bystander B cells do not accumulate CD40L and do not receive help. These results suggest that CD40L activates the antigen-presenting B cells during or after antigen-specific transfer to the B cells, and that activation of bystanders occurs by another mechanism much more sensitive to blocking by anti-CD40L. We propose that the CD40L that helps the bystander B cells needs to accumulate on the T cell surface, and thus is more readily available for neutralization by the anti-CD40L antibody, and more sensitive to inhibition of CD40L synthesis by CsA, than the CD40L delivered efficiently and directionally to the antigen-presenting B cells during antigen-specific interactions.
Discussion
The results of this study show that during antigen-specific interactions between helper T cells and B cells, CD40L is transferred to antigen-presenting B cells and remains in and on the B cells following their engagement. CD40L transfer is largely dependent on CD40, but in the absence of CD40, FcRs expressed by the B cells can mediate transfer when anti-CD40L is present. Transferred CD40L appears to accumulate in distinct dots both on the cell surface and internally in antigen-presenting B cells following overnight incubation with antigen-specific T cells. During or after transfer to B cells, CD40L induces proliferation and upregulation of ICAM-1 on antigen-presenting B cells, while bystander B cells do not acquire CD40L and do not become activated when low concentrations of anti-CD40L are present.
Antigen-specific transfer of CD40L is most readily detected when fluorescent anti-CD40L antibody is included during culture (Figs. 2-4, 5A, and 5B), but it can also be detected by intracellular staining of the B cells with anti-CD40L after overnight incubation with T cells (Fig. 1). There are several potential explanations for the enhancement of detection of transferred CD40L by co-culture with anti-CD40L. We considered the possibility that inclusion of the antibody produced immune complexes with CD40L that could enhance transfer to antigen-specific B cells by an FcR-mediated mechanism. However, addition of a blocking anti-FcR antibody or use of FcR KO B cells only slightly reduced CD40L transfer to CD40 sufficient antigen-pulsed B cells, although absence of FcR function had a much larger effect on transfer to CD40-deficient antigen-pulsed B cells (Fig 2D and 2E). Therefore, transfer of CD40L in the presence of anti-CD40L appears to depend on ligation by either CD40 or FcR. Anti-CD40L antibody may delay internalization of transferred CD40L into B cells during antigen-specific interactions to decrease the rate of degradation of CD40L and thus enhance detection. It is also possible that the fluorescent label on the antibody in complex with CD40L may be degraded more slowly than CD40L transferred in the absence of antibody, and so accumulation of the fluorescent label in the B cells in the presence of anti-CD40L may more accurately reflect the total amount of CD40L transferred during the overnight incubation.
Although it has been known for decades that the CD40L-CD40 interaction is key to T cell-dependent B cell antibody responses and is required for proliferation and differentiation of antigen-presenting B cells, the molecular details involved in the delivery of CD40L have been unclear. It is generally believed that surface CD40L engages with CD40 on the B cell long enough to initiate downstream signaling in the B cells [10, 16] and then is endocytosed back into the helper T cell [15]. However, none of these studies measured CD40L on B cells following T/B engagement. Accumulation of transferred CD40L during antigen-specific interactions may help explain how rare B cells of higher affinities can outcompete other B cells for limiting CD40L in the germinal center following short interactions with Tfh cells that last only a few minutes [18-22]. Transfer and endocytosis of CD40L by antigen-specific B cells may also explain how B cells sustain sufficient levels of CD40 signaling to induce the expression of interferon regulatory factor-4 and subsequent downregulation of B cell lymphoma-6 (Bcl-6) that are required for exit from germinal centers and terminal differentiation [35, 36]. Although CD40L transfer correlates with B cell activation in our experiments, we have not yet shown directly that transferred CD40L continues to deliver an activating signal to the B cell. CD40-mediated internalization of CD40L by antigen-specific B cells may also contribute to downregulation of CD40L from the T cell surface to prevent bystander B cell activation and autoantibody formation.
We have previously shown that CD40L is stored in a TCR-regulated secretory compartment in helper T cells [23, 24], and that this small intracellular store of CD40L is sufficient to induce upregulation of costimulatory molecules and proliferation of B cells and cytokine production and maturation of dendritic cells [25]. When we blocked de novo synthesis of CD40L by treating the T cells with CsA [23], we found that substantial amounts of CD40L are still transferred to the B cells (Fig. 3). This indicates that de novo production of CD40L is not necessary for antigen-specific transfer of CD40L, and that the small amount of preformed CD40L in the secretory compartment is efficiently transferred to antigen-presenting B cells. It seems plausible that all antigen-specific transfer of CD40L may occur through this secretory compartment. Transcriptional upregulation of CD40L through TCR signaling may be necessary to allow T cells to refill the secretory compartment when undergoing long interactions with naïve B cells [37], as in the present experiments, or multiple short interactions with B cells in germinal centers.
Delivery of extracellular vesicles by T cells during interactions with APCs has been recognized as a way in which T cell can induce changes in target cells [27, 28, 38]. TCR [29], CTLA-4 [39], and Fas ligand (FasL) [40-43] have been shown to be delivered in vesicles to target cells. CTLA-4 [44-46], CD40L [23], and FasL [47, 48] are stored in secretory lysosomes in T cells that are rapidly delivered to the T cell surface upon TCR stimulation. However, CD40L-containing vesicles were not found above background levels in exosomes in the supernatant of anti-CD3-activated human T cell cultures [49]. We have shown that CD40L is stored in the same secretory compartment as FasL, but not CTLA-4 [23], and is released to the T cell surface independently of Rab27a, an effector molecule known to be essential for the release of lytic granules [25]. The intracellular pathway and effector molecules associated with the trafficking and release of CD40L from this intracellular compartment need to be further defined.
We have shown here that helper T cells upon formation of an immunological synapse with antigen-presenting B cells deliver CD40L by a method resistant to blocking by anti-CD40L. It remains to be discovered whether transfer of CD40L involves microvesicles, like the peptide-MHC engaged TCRs delivered at the immunological synapse [29], exosomes, another type of extracellular vesicle, or another form of transfer of membrane molecules such as trogocytosis [50]. We propose that Tfh cells transfer CD40L to germinal center B cells in brief interactions, but we were unable to test that hypothesis directly because conditions to efficiently generate Tfh and germinal center B cells in vitro have not been developed. It may be possible to investigate delivery of CD40L to B cells in germinal centers in vivo via two photon microscopy using T cells with transgenic GFP-tagged CD40L.
Materials and Methods
Mice
B10.A and B10.A-Rag2tm1Fwa H2-T18a (5CC7 TCR transgenic mice on a B10A.RAG KO background), specific for a pigeon cytochrome c peptide on I-Ek and reactive against moth cytochrome c, were purchased from Taconic (Germantown, NY) [51]. B6.Cg-Tg(TcraTcrb)425Cbn/J (OT-II TCR transgenic mice), specific for chicken ovalbumin residues 323-339 on I-Ab, B6;129S-Fcgr2btm1Ttk/J (FcR KO mice), B10.BR-H2k2 H2-T18a/SgSnJ, and B6.129P2-Cd40tm1Kik/J (CD40 KO mice on a B6 background) [6] were purchased from The Jackson Laboratory (Bar Harbor, ME). B6.129P2-Cd40tm1Kik/J were bred to B10.A mice, and heterozygous mice from this breeding were crossed and selected to produce I-Ek homozygous CD40 KO mice. 5CC7 TCR transgenic male mice were crossed to female B6.129S2-Cd40lgtm1Imx/J [52] (CD40L KO mice on a B6 background, purchased from Jackson) to produce 5CC7 CD40L KO male mice. Mice were housed in specific-pathogen free conditions at Oregon Health and Science University according to institutional standards.
Antibodies and labels
The antibodies and reagents used for flow cytometry experiments were as follows: anti-CD4-PE-Cy7, anti-CD19-PerCP, anti-CD19-FITC, anti-ICAM-biotin, anti-ICAM-allophycocyanin, streptavidin-allophycocyanin, anti-CD40L-PE, anti-IL-4-PE, anti-IFNγ-allophycocyanin, and anti-CD40L-allophycocyanin (eBiosciences). Cells were differentially labeled with CFSE or CTV (Invitrogen, Carlsbad, CA) to mark antigen-pulsed or bystander cells in individual assays.
In vitro Th1 cell polarization
Spleens were harvested from 5CC7 TCR or OT-II transgenic mice. Red blood cells were removed by hypotonic lysis. Cells were suspended in complete RPMI 1640 medium (Gibco ®, Waltham, MA), supplemented as previously described [53]. For polarization to Th1, 5CC7 spleen cells were incubated with 0.1 μM moth cytochrome C peptide [ANERADLIAYLKQATK] and 10 μg/ml anti-IL-4 (11B11 [eBioscience]) for four days. OT-II spleen cells were incubated with 1 μM Ova 323-339 peptide (AnaSpec, Fremont, CA) and 10 μg/ml anti-IL-4 for four days. Th1 cells were confirmed based on production of IFNγ and absence of IL-4 by intracellular cytokine staining.
In vitro overnight CD40L transfer assay
5CC7 or OT-II Th1 cells were harvested on day four after stimulation. The OT-II cells were used only for the FcR KO and control H-2b B cells in the experiment shown in Fig. 2D. Lympholyte M (Cedarlane) was used to remove dead cells. Cells were then counted and incubated at 2 × 106 cells/ml either with or without 2 μM CsA (Sigma-Aldrich) for 3 hours to block de novo TCR-induced CD40L production. Spleens were harvested from 5CC7 CD40L KO, B10.A, B10.A CD40 KO, B6, FcR KO, or CD40 KO mice, and target splenocytes were prepared by hypotonic lysis. Splenocytes were labeled with CFSE or CTV following the manufacturer's protocol and pulsed with 2.5 μM peptide antigen for two hours at 37° and washed. Bystander B cells were labeled with lower concentrations of CTV or CFSE and mock-pulsed to allow identification of antigen-pulsed and bystander B cells following overnight incubations. The splenocytes were then washed and 4 × 105 cells were combined with 1 × 106 T cells, either treated or untreated with CsA (final concentration 1 μM). Cells were cultured overnight in the presence or absence of 1 μg/ml unlabeled (Bio × Cell) or fluorescently labeled anti-CD40L (eBiosciences) antibody, with or without addition of anti-FcR blocking antibody, anti-CD16/CD32, clone 2.4G2 (BioLegend, San Diego, CA). After overnight incubation, cells were stained with antibodies to CD4, CD19, and ICAM-1, and analyzed on the LSRII flow cytometer (Becton Dickinson). Cell doublets were excluded from analysis to eliminate T/B conjugates. Proliferation was assessed based on the dilution of CFSE or CTV. In some experiments, cells were fixed (BD Biosciences Cytofix/Cytoperm) and permeabilized (BD Biosciences Perm/Wash Buffer) before staining with PE-labeled anti-CD40L or hamster IgG-PE control (eBioscience).
Transwell® Assay
1 × 106 Th1 cells and 8 × 105 target (B10.A or B10.BR) splenocytes (4 × 105 Ag-pulsed and 4 × 105 unpulsed, differentially labeled) were placed in the top chamber of the Costar Transwell® system separated by a cell-impermeable membrane (0.4 μM pore size) from 8 × 105 splenocytes (Ag-pulsed and unpulsed). Following overnight incubation in the presence or absence of 1 μg/ml fluorescently labeled anti-CD40L-PE antibody, cells were stained with antibodies to CD4, CD19, and ICAM and analyzed by flow cytometry.
Fluorescent Cell Microscopy
Imaging of CD40L on Ag-pulsed and bystander cells after overnight incubation with T cells in the presence of allophycocyanin-labeled anti-CD40L antibody was performed on a Zeiss LSM 780 laser-scanning confocal microscope with a 32 channel gallium-arsenide-phosphide detector for capturing spectral information for linear unmixing. Overnight cultured T and B cells were stained with CD19-FITC, fixed, and placed into a single chamber of a 1 % poly-L-lysine (Sigma, St. Louis, MO #P8920)-treated 8-chambered glass slide (Lab-Tek® II, Nalgene Nunc International Corporation, Penfield, NY). Eleven z stack images were acquired for each field analyzed with the motorized stage. An image was acquired every 2.5 μm movement in the z axis, allowing for the entire cell to be visualized from the point of cell attachment to the glass chamber to the cell surface opposite the point of attachment. Images and movies were developed and analyzed using ImageJ.
B cells were selected for quantification based on positive CD19-FITC staining. Cells were excluded from analysis if they were not attached to the glass surface and were mobile at any point during acquisition of z stacks, if it was not possible to distinguish which cell in the image was positive for CD40L (cells were too close in proximity or overlapping), if cells appeared dead, or if CD19 staining did not correspond to the cell surface only. The CD19 surface stain served as a marker to allow us to distinguish between surface CD40L and intracellular CD40L on selected B cells.
Supplementary Material
Acknowledgments
We thank Drs. Yoshinobu Koguchi and Amy Moran for thoughtful review of this manuscript and Drs. Susan Murray and Yoshinobu Koguchi for experimental suggestions. This work was supported by the National Institutes of Health grants R01 AI050823 (D.C.P.) and R01 AI092080 (D.C.P.) from the National Institute of Allergy and Infectious Diseases. J.L.G. was supported by National Institute of Health Training Grants T32 AI078903, Inflammation and T Lymphocyte Immunoregulation, and T32 AI007472, Interactions at the Microbe-Host Interface.
Abbreviations used in this manuscript
- APC
antigen-presenting cell
- Ag
antigen
- Tfh
follicular helper T cell
- CTV
CellTrace® Violet
- CFSE
carboxyfluorescein succinimidyl ester
- CsA
cyclosporin A
- FcR
Fc receptor
- FasL
Fas ligand
- KO
knockout
Footnotes
Conflict of Interest Disclosure: The authors declare no commercial or financial conflict of interest.
References
- 1.Noelle RJ, Ledbetter JA, Aruffo A. Immunol Today. 1992;13:431–433. doi: 10.1016/0167-5699(92)90068-I. [DOI] [PubMed] [Google Scholar]
- 2.Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- 3.Grewal IS, Flavell RA. Annu Rev Immunol. 1998;16:111–135. doi: 10.1146/annurev.immunol.16.1.111. [DOI] [PubMed] [Google Scholar]
- 4.Parker DC. Annu Rev Immunol. 1993;11:331–360. doi: 10.1146/annurev.iy.11.040193.001555. [DOI] [PubMed] [Google Scholar]
- 5.Han S, Hathcock K, Zheng B, Kepler TB, Hodes R, Kelsoe G. J Immunol. 1995;155:556–567. [PubMed] [Google Scholar]
- 6.Kawabe T, Naka T, Yoshida K, Tanaka T, Fujiwara H, Suematsu S, Yoshida N, et al. Immunity. 1994;1:167–178. doi: 10.1016/1074-7613(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 7.Xu J, Foy TM, Laman JD, Elliott EA, Dunn JJ, Waldschmidt TJ, Elsemore J, et al. Immunity. 1994;1:423–431. doi: 10.1016/1074-7613(94)90073-6. [DOI] [PubMed] [Google Scholar]
- 8.Gitlin AD, Shulman Z, Nussenzweig MC. Nature. 2014;509:637–640. doi: 10.1038/nature13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huse M, Lillemeier BF, Kuhns MS, Chen DS, Davis MM. Nat Immunol. 2006;7:247–255. doi: 10.1038/ni1304. [DOI] [PubMed] [Google Scholar]
- 10.Boisvert J, Edmondson S, Krummel MF. J Immunol. 2004;173:3647–3652. doi: 10.4049/jimmunol.173.6.3647. [DOI] [PubMed] [Google Scholar]
- 11.Barcia C, Gomez A, de Pablos V, Fernandez-Villalba E, Liu C, Kroeger KM, Martin J, et al. J Virol. 2008;82:9978–9993. doi: 10.1128/JVI.01326-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tourret M, Guegan S, Chemin K, Dogniaux S, Miro F, Bohineust A, Hivroz C. J Immunol. 2010;185:6809–6818. doi: 10.4049/jimmunol.1001501. [DOI] [PubMed] [Google Scholar]
- 13.Bertrand F, Esquerre M, Petit AE, Rodrigues M, Duchez S, Delon J, Valitutti S. J Immunol. 2010;185:2887–2894. doi: 10.4049/jimmunol.1000739. [DOI] [PubMed] [Google Scholar]
- 14.Lesley R, Kelly LM, Xu Y, Cyster JG. Proc Natl Acad Sci U S A. 2006;103:10717–10722. doi: 10.1073/pnas.0601539103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yellin MJ, Sippel K, Inghirami G, Covey LR, Lee JJ, Sinning J, Clark EA, et al. J Immunol. 1994;152:598–608. [PubMed] [Google Scholar]
- 16.Roy M, Aruffo A, Ledbetter J, Linsley P, Kehry M, Noelle R. Eur J Immunol. 1995;25:596–603. doi: 10.1002/eji.1830250243. [DOI] [PubMed] [Google Scholar]
- 17.Yacoub D, Benslimane N, Al-Zoobi L, Hassan G, Nadiri A, Mourad W. J Biol Chem. 2013;288:36083–36093. doi: 10.1074/jbc.M113.506220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allen CD, Okada T, Tang HL, Cyster JG. Science. 2007;315:528–531. doi: 10.1126/science.1136736. [DOI] [PubMed] [Google Scholar]
- 19.Liu D, Xu H, Shih C, Wan Z, Ma X, Ma W, Luo D, Qi H. Nature. 2015;517:214–218. doi: 10.1038/nature13803. [DOI] [PubMed] [Google Scholar]
- 20.Qi H. Immunol Rev. 2012;247:24–35. doi: 10.1111/j.1600-065X.2012.01119.x. [DOI] [PubMed] [Google Scholar]
- 21.Shulman Z, Gitlin AD, Targ S, Jankovic M, Pasqual G, Nussenzweig MC, Victora GD. Science. 2013;341:673–677. doi: 10.1126/science.1241680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, Nussenzweig MC. Cell. 2010;143:592–605. doi: 10.1016/j.cell.2010.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koguchi Y, Thauland TJ, Slifka MK, Parker DC. Blood. 2007;110:2520–2527. doi: 10.1182/blood-2007-03-081299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koguchi Y, Buenafe AC, Thauland TJ, Gardell JL, Bivins-Smith ER, Jacoby DB, Slifka MK, Parker DC. PloS one. 2012;7:e31296. doi: 10.1371/journal.pone.0031296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koguchi Y, Gardell JL, Thauland TJ, Parker DC. J Immunol. 2011;187:626–634. doi: 10.4049/jimmunol.1004083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whalen BJ, Tony HP, Parker DC. J Immunol. 1988;141:2230–2239. [PubMed] [Google Scholar]
- 27.Gutierrez-Vazquez C, Villarroya-Beltri C, Mittelbrunn M, Sanchez-Madrid F. Immunol Rev. 2013;251:125–142. doi: 10.1111/imr.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mittelbrunn M, Vicente-Manzanares M, Sanchez-Madrid F. Traffic. 2015;16:327–337. doi: 10.1111/tra.12258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choudhuri K, Llodra J, Roth EW, Tsai J, Gordo S, Wucherpfennig KW, Kam LC, et al. Nature. 2014;507:118–123. doi: 10.1038/nature12951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dustin ML. Mol Cell. 2014;54:255–262. doi: 10.1016/j.molcel.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frentsch M, Arbach O, Kirchhoff D, Moewes B, Worm M, Rothe M, Scheffold A, Thiel A. Nat Med. 2005;11:1118–1124. doi: 10.1038/nm1292. [DOI] [PubMed] [Google Scholar]
- 32.Chattopadhyay PK, Yu J, Roederer M. Nat Med. 2005;11:1113–1117. doi: 10.1038/nm1293. [DOI] [PubMed] [Google Scholar]
- 33.Noelle RJ, Roy M, Shepherd DM, Stamenkovic I, Ledbetter JA, Aruffo A. Proc Natl Acad Sci U S A. 1992;89:6550–6554. doi: 10.1073/pnas.89.14.6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moens L, Tangye SG. Front Immunol. 2014;5:65. doi: 10.3389/fimmu.2014.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Polo JM, Ci W, Licht JD, Melnick A. Blood. 2008;112:644–651. doi: 10.1182/blood-2008-01-131813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saito M, Gao J, Basso K, Kitagawa Y, Smith PM, Bhagat G, Pernis A, et al. Cancer Cell. 2007;12:280–292. doi: 10.1016/j.ccr.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 37.Garside P, Ingulli E, Merica RR, Johnson JG, Noelle RJ, Jenkins MK. Science. 1998;281:96–99. doi: 10.1126/science.281.5373.96. [DOI] [PubMed] [Google Scholar]
- 38.Robbins PD, Morelli AE. Nat Rev Immunol. 2014;14:195–208. doi: 10.1038/nri3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Esposito L, Hunter KM, Clark J, Rainbow DB, Stevens H, Denesha J, Duley S, et al. J Immunol. 2014;193:889–900. doi: 10.4049/jimmunol.1303389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alonso R, Rodriguez MC, Pindado J, Merino E, Merida I, Izquierdo M. J Biol Chem. 2005;280:28439–28450. doi: 10.1074/jbc.M501112200. [DOI] [PubMed] [Google Scholar]
- 41.Martinez-Lorenzo MJ, Anel A, Gamen S, Monlen I, Lasierra P, Larrad L, Pineiro A, et al. J Immunol. 1999;163:1274–1281. [PubMed] [Google Scholar]
- 42.Mazzeo C, Calvo V, Alonso R, Merida I, Izquierdo M. Cell Death Differ. 2016;23:99–109. doi: 10.1038/cdd.2015.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Monleon I, Martinez-Lorenzo MJ, Monteagudo L, Lasierra P, Taules M, Iturralde M, Pineiro A, et al. J Immunol. 2001;167:6736–6744. doi: 10.4049/jimmunol.167.12.6736. [DOI] [PubMed] [Google Scholar]
- 44.Alegre ML, Noel PJ, Eisfelder BJ, Chuang E, Clark MR, Reiner SL, Thompson CB. J Immunol. 1996;157:4762–4770. [PubMed] [Google Scholar]
- 45.Iida T, Ohno H, Nakaseko C, Sakuma M, Takeda-Ezaki M, Arase H, Kominami E, et al. J Immunol. 2000;165:5062–5068. doi: 10.4049/jimmunol.165.9.5062. [DOI] [PubMed] [Google Scholar]
- 46.Linsley PS, Bradshaw J, Greene J, Peach R, Bennett KL, Mittler RS. Immunity. 1996;4:535–543. doi: 10.1016/s1074-7613(00)80480-x. [DOI] [PubMed] [Google Scholar]
- 47.He JS, Gong DE, Ostergaard HL. J Immunol. 2010;184:555–563. doi: 10.4049/jimmunol.0902465. [DOI] [PubMed] [Google Scholar]
- 48.Bossi G, Griffiths GM. Nat Med. 1999;5:90–96. doi: 10.1038/4779. [DOI] [PubMed] [Google Scholar]
- 49.Blanchard N, Lankar D, Faure F, Regnault A, Dumont C, Raposo G, Hivroz C. J Immunol. 2002;168:3235–3241. doi: 10.4049/jimmunol.168.7.3235. [DOI] [PubMed] [Google Scholar]
- 50.Davis DM. Nat Rev Immunol. 2007;7:238–243. doi: 10.1038/nri2020. [DOI] [PubMed] [Google Scholar]
- 51.Seder RA, Paul WE, Davis MM, Fazekas de St Groth B. J Exp Med. 1992;176:1091–1098. doi: 10.1084/jem.176.4.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Renshaw BR, Fanslow WC, Armitage RJ, Campbell KA, Liggitt D, Wright B, Davison BL, Maliszewski CR. J Exp Med. 1994;180:1889–1900. doi: 10.1084/jem.180.5.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wetzel SA, McKeithan TW, Parker DC. J Immunol. 2002;169:6092–6101. doi: 10.4049/jimmunol.169.11.6092. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.