

Abstract
In plants, duplication of individual genes, long chromosomal regions, and complete genomes provides a major source for evolutionary innovation. We investigated two different types of duplications, tandem and segmental duplications, in Arabidopsis for correlation, conservation, and differences of expression characteristics by making use of large genome-wide expression data as measured by the massively parallel signature sequencing method. Our analysis indicates that large fractions of duplicated gene pairs still share transcriptional characteristics. However, our results also indicate that expression divergence occurs frequently between duplicated gene pairs, a process which frequently might be employed for the retention of sequence redundant gene pairs. Preserved overall similarity between promoters of duplicated genes as well as preservation of individual cis-elements within the respective promoters indicates that the process of transcriptional neo- and subfunctionalization is restricted to only a fraction of cis-elements. We show that sequence similarities and shared regulatory properties within duplicated promoters provide a powerful means to undertake large-scale cis-regulatory element identification by applying an intragenomic phylogenetic footprinting approach. Our work lays a foundation for future comparative studies to elucidate the molecular manifestation of regulatory similarities and dissimilarities of duplicated genes.
Plant genomes are rich in duplicated genes. Various mechanisms can lead to duplication of individual genes or longer chromosomal regions. In addition to the duplication of individual chromosomal regions, polyploidization, and subsequent reorganization of the genome, tandem duplication and the generation of dispersed, duplicated genes account for the generation of sequence redundant copies. Within Arabidopsis, the evolutionary history of the modern genome structure has been exhaustively analyzed. Large fractions of the genome are derived from ancient polyploidization events, and as much as 17% of the genome reportedly is composed of tandemly repeated genes (The Arabidopsis Genome Initiative, 2000; Vision et al., 2000; Simillion et al., 2002; Bowers et al., 2003).
Gene duplications are regarded as a major source for the generation of evolutionary novelties (Ohno, 1970). Paralogous gene pairs may acquire different fates: (1) evolution of one copy to a nonfunctional pseudogene, (2) functional divergence of the two copies in which one of the two copies acquires a new function (neofunctionalization), (3) simultaneous reduction of the activity of both copies by maintaining the total capacity of the ancestral gene (subfunctionalization), and (4) the functional preservation of both copies to increase the robustness of the genetic network (Force et al., 1999; Gu et al., 2003). In principle, the first three fates can be realized by mutations both within the coding regions and within regulatory regions of the respective genes. Whereas changes within the coding region of the gene eventually result in altered biochemical properties of the protein, mutational changes within the regulatory sequences potentially alter the temporal and/or spatial expression patterns or transcriptional responses to internal and external stimuli.
Effects and consequences of mutations within the coding portion of genes have been exhaustively studied (Lynch and Conery, 2000; Parenicova et al., 2003). A comprehensive study of the evolutionary fate of duplicated genes in six eukaryotic genomes revealed a high rate of duplication, illustrating the importance of duplications for the evolution and speciation of genomes (Lynch and Conery, 2000). Although there is an ongoing debate on to what extent duplicated genes become silenced, pseudogenization likely is a significant evolutionary fate for duplicated gene pairs (Lynch and Conery, 2000; Long and Thornton, 2001; Zhang et al., 2001). In yeast (Saccharomyces cerevisiae), a genome-wide survey on the phenotypic effects of single-gene deletion mutants has been carried out. The analysis revealed that at least one-quarter of mutants with no phenotype is compensated by duplicated loci (Gu et al., 2003). This indicates that a substantial part of duplications in yeast has become at least partially redundant units and suggests that genetic redundancy increases the robustness of the genetic network.
The retention rate of duplicates within many eukaryotic genomes is significantly higher than expected from predictions by classical models (Prince and Pickett, 2002). To explain the striking discrepancies between empirical and expected retention rates for duplicated genes, the duplication-degeneration-complementation (DDC) model has been proposed (Force et al., 1999; Prince and Pickett, 2002). The model requires multifunctionality of the ancestral unit, and the function of the ancestral locus is presumed to have consisted of several discrete, independently mutable subfunctions. After a duplication event, the two copies acquire complementary amorphic or hypomorphic mutations in two separable subfunctions. Thus, both copies are required to provide the full functionality of the ancestral function and will be retained by selective pressure.
Mutable subfunctions of a duplicated pair potentially affect various sites governing the functional characteristics of the respective gene. Besides protein domains, splice sites or cis-regulatory elements can be affected. Evolution of transcriptional regulation as a crucial contributor to evolutionary changes and speciation has long been hypothesized (Britten and Davidson, 1969; Ohno, 1971; Doebley and Lukens, 1998; Carroll, 2000; Prince and Pickett, 2002). Due to their short, degenerated sequence characteristics and their combinatorial mode of action, in particular transcription factor binding sites potentially represent appropriate targets for the DDC process. Transcriptional subfunctionalization can potentially result in at least partially altered expression patterns and characteristics and might represent an important evolutionary mechanism for the retention of duplicated genes.
In contrast to exhaustive studies on the divergence of duplicated genes and their encoded proteins, little is known about how changes in gene expression affect the evolutionary fates of duplicated pairs (Wray et al., 2003). Most of our knowledge is derived from the analysis of individual genes or from well-characterized gene families, like the HOX genes in vertebrates or the MADS box genes in higher plants (e.g. Parenicova et al., 2003).
At least partial redundancy has been reported for several double or triple mutants of homologous genes, like the floral meristem identity genes AP1/CAULIFLOWER/FRUITFUL and the SHATTERPROOF genes which regulate fruit dehiscence (Ferrandiz et al., 2000; Liljegren et al., 2000; Pinyopich et al., 2003). Divergent expression patterns and putative silencing of duplicated genes were found in an expression analysis of 105 MADS box genes in Arabidopsis (Kofuji et al., 2003).
Using microarray expression data, studies in yeast have estimated the extent to which changes in gene expression contribute to the evolutionary fates of duplications (Wagner, 2000; Gu et al., 2002). Only a small fraction of duplicated gene pairs showed no or little variance, while most duplicated genes quickly diverged in their expression patterns. Expression of duplicated genes seems to be initially coupled and subsequently diverges rapidly, suggesting rapid neo- and/or subfunctionalization (Gu et al., 2002).
In higher eukaryotes (including plants), expression divergence of duplicated genes thus far has only been studied on selected examples, and genome-scale analyses haven't been reported to date. In this study, we investigate the correlation of expression of duplicated gene pairs in Arabidopsis, with an emphasis on the transcriptional characteristics of tandem and segmental duplications. We analyzed the transcriptional fate of duplicated genes by making use of genome-wide expression data derived by massively parallel signature sequencing (MPSS; Brenner et al., 2000b; http://mpss.udel.edu/at). Our results indicate that a significant portion of segmentally and tandemly duplicated genes are highly similar in their expression characteristics. However, more than two-thirds of duplicated genes exhibit divergence in their expression characteristics. By applying an intragenomic phylogenetic footprinting strategy, we demonstrate that duplicated genes still share cis-regulatory elements. These characteristics of duplicated genes can be exploited for genome-scale cis-element detection and in the future might give insights into the molecular events leading to transcriptional sub- and neofunctionalization and the mechanism by which gene duplications are stabilized.
RESULTS
Identification of Tandemly and Segmentally Duplicated Genes in Arabidopsis
To analyze similarities and dissimilarities in transcriptional characteristics of duplicated genes in Arabidopsis, we selected appropriate duplicated gene groups from the Arabidopsis genome (see “Materials and Methods”). These groups were either within segmental duplications stemming from an ancient polyploidization event that took place approximately 38 to 70 million years ago (Simillion et al., 2002; Blanc et al., 2003; Bowers et al., 2003) or genes that are organized in tandem arrays. Duplicated genes were selected using stringent similarity thresholds. For segmentally duplicated groups, we restricted our analysis to pairs that have one member in each of the respective segments (1:1 relations). This minimizes potential artifacts caused by expanded gene families not arranged in a colinear order.
We selected 2,399 groups of genes comprising 7,425 genes from previously identified segmentally duplicated regions (The Arabidopsis Genome Initiative, 2000). Most of the groups (1,680) show clear 1:1 duplicate relations, and only these were analyzed further. For the tandemly repeated duplicates, we obtained 1,564 groups comprising 4,176 genes. Most of the tandem groups (1,096) consist of two members; however, we also identified groups with up to 22 members (see supplemental material, available at www.plantphysiol.org).
To gain insight into the evolutionary relationship and the duplication age of the selected duplicated genes, the synonymous substitution rate (KS) was calculated for the corresponding gene pairs (Fig. 1). For the selected segmentally duplicated genes, the frequency distribution showed a clear peak for KS values of 0.7 to 0.8 as well as a Gaussian distribution. This confirmed that our filters successfully identified duplicated gene pairs originating from the most recent polyploidization event during the evolution of Arabidopsis (Vision et al., 2000; Simillion et al., 2002; Bowers et al., 2003). These observations are consistent with previously reported findings and date the duplication event to approximately 38 to 70 million years ago (Vision et al., 2000; Simillion et al., 2002; Blanc et al., 2003; Bowers et al., 2003). Duplicate pairs derived from earlier polyploidization events (Simillion et al., 2002; Bowers et al., 2003) were removed from the dataset.
Figure 1.

KS distributions for tandemly and segmentally duplicated genes. Divergence KS of coding sequences were determined for 1:1 segmental and tandemly repeated genes in Arabidopsis and sorted into bins of width KS = 0.1. Relative abundance of each bin is shown on the y axis.
For tandemly duplicated genes, we computed KS for all pairwise combinations of each group. For groups containing more than two members, this might impose a bias toward an overrepresentation of older pairs. To address this problem, we separately analyzed the distribution of tandem groups consisting of two genes. However, there was no striking difference between the distributions of KS between these datasets (data not shown). In both analyses, the average age of duplicates is significantly lower than that of segmentally duplicated genes. We found a pronounced peak for KS values between 0.3 and 0.4. Thus, the average age of tandem duplications is approximately one-half the age of the segmental duplications.
Expression of Duplicated Genes in Arabidopsis
For expression measurements, we made use of data generated by MPSS (Brenner et al., 2000a, 2000b; http://mpss.udel.edu/at). MPSS data from five different tissues (callus, leaf, flower, root, and silique) were used. Tag-based methods for quantifying gene expression, like MPSS, have the advantage of avoiding problems often faced in microarray analysis like cross-hybridization or low signal strength (Brenner et al., 2000a). We assigned tags to specific genes to associate them with expression abundance. Analysis was restricted to genes and tags that met high quality criteria.
The positions of all MPSS tags matching within the Arabidopsis genome were determined, and these tags were associated with annotated genes from the MAtDB (Schoof et al., 2004). To correctly map the tags to genes, cDNA information was integrated where possible (see “Materials and Methods”). Unambiguous expression data for 12,352 genes were obtained. This dataset encompasses 1,529 genes assigned to tandem groups and 2,195 grouped as segmental duplicates. Expression for 1,460 genes could not be resolved due to ambiguous tags, and 590 of these genes belonged to tandem groups. The pronounced decrease of informative tags for tandemly repeated genes largely resulted from the high similarity of their transcripts and illustrates the necessity of the strict quality criteria imposed.
For each pair of tandemly and segmentally duplicated genes with diagnostic and informative tags, the Pearson correlation r of their expression was computed. Application of our quality criteria restricted the analysis to 849 pairwise comparisons for tandemly duplicated and 777 pairwise comparisons for the segmentally duplicated genes. Negative control experiments consisted of gene pairs obtained by randomly shuffling sets of evolutionarily unrelated genes containing appropriate tags. Ten random shuffles were generated, each equal in size to the duplicated datasets. These sets represent the background level of correlated expression expected to be observed by chance. The distribution of the two duplicate classes is significantly different from the random shuffles as tested by the χ2 test (χ2 > 60, P < 10−6 for segmental pairs and χ2 > 120, P < 10−9 for tandem pairs at 19 degrees of freedom; Fig. 2). For both classes of duplicated genes, a considerable fraction showed highly similar expression characteristics (Fig. 2; Pearson correlation r > 0.8) significantly deviating from the control dataset. Approximately 26% of pairwise correlations for duplicated genes organized in tandem arrays and about 19% of segmentally duplicated gene pairs are associated with a Pearson coefficient of 0.8 or higher. Consequently, the degree of duplicated gene pairs with negatively correlated expression that had highly dissimilar expression patterns is less pronounced than expected from random associations (Fig. 2; −0.9 < r < −0.5). It is noteworthy that the distributions are similar for both classes, despite an approximately 2-fold difference in divergence time. As pairwise correlation analysis of tandem groups containing more than two members potentially bias the distribution, we additionally analyzed the expression correlations for tandem groups consisting of two genes. However, distributions between these selected and all tandem genes were highly similar (data not shown).
Figure 2.

Significant portions of tandemly and segmentally duplicated genes exhibit highly similar expression characteristics. MPSS expression data were correlated between tandemly and segmentally duplicated pairs and were compared against background distributions of random shuffled pairs. Left side of figure shows distribution for tandem, right side for segmental pairs, each in comparison to randomized sets of equal size. χ2 tests showed highly significant differences of the observed distributions against the randomized dataset. Pearson correlations r on the x axis were grouped into 20 bins of width r = 0.1, and relative abundance of each bin is indicated on the y axis.
Tandemly and Segmentally Duplicated Genes Exhibit Significant Similarities within Their Promoters
Gene duplication most likely is not restricted to the coding or transcribed portion of genes but also comprises the respective promoters. Our observations on the significant transcriptional similarities between large portions of duplicated gene pairs prompted us to undertake a systematic analysis of promoters associated to duplicated genes.
Therefore, we determined the average similarity of promoters associated with duplicated genes. This analysis was performed by aligning the promoter sequences and comparing these alignments to the expected background, obtained by the analysis of a negative control set. To avoid alignments between 5′ untranslated regions (UTRs), which frequently exhibit a high degree of similarity for duplications (G. Haberer and K.F.X. Mayer, unpublished data), we restricted our analysis to promoter pairs for which both genes have associated full-length cDNA information. Alignments of promoter sets of variable lengths were computed using DiAlign2-1 (Morgenstern, 1999). Similarity was measured as the proportion of aligned nucleotides to the total promoter length (see “Materials and Methods”). Mean similarities of tandem and segmental promoters were significantly higher than randomly selected promoters (Fig. 3). These results indicate that the similarity between promoters of duplicated genes is significantly larger than expected by chance.
Figure 3.

Tandemly and segmentally duplicated genes show significant conservation within their promoters. Means of promoter similarities for tandemly, segmentally, and randomly shuffled pairs were determined using DiAlign2. Promoter similarity was defined as length of alignable regions divided by total promoter length. Length of promoters was restricted to a maximum of 600 bp upstream of cDNA start site. Means are indicated on the y axis, and error bars represent ses.
Promoter and Protein Divergence Correlate within Tandem Duplications
To test whether promoter evolution is coupled with coding sequence divergence, we correlated the promoter similarities and the KS. To exclude potential pseudogenes and 5′ UTRs from our analysis, we restricted the dataset to pairs of genes with associated full-length cDNA information. We analyzed for potential correlations between the age of a duplicated pair measured as KS and the divergence of the respective promoters as defined by the promoter similarities (Fig. 4). As segmental duplications likely arose from an ancient polyploidization event, a time-dependent correlation can't be expected. This is confirmed by our findings of only a weak positive correlation of low significance (r = 0.095; α < 5% and no significance at the level of α ≤ 2% by Spearman rank correlation testing; Fig. 4).
Figure 4.

Promoter similarity is coupled to divergence time for tandemly repeated genes. Divergence time KS was plotted on the x axis versus promoter similarity (measured as described in Fig. 3 and “Materials and Methods”). Correlation coefficients as determined by Spearman rank correlation were highly significant for tandem genes (r = −0.45, P < 10−6) but not significant for segmentally duplicated genes at the 1% significance level (r = 0.095, P < 0.05).
In contrast to segmental duplications, divergence times of tandemly repeated genes are distributed over a larger time range. Reflective of this, we found a highly significant, strong negative correlation between protein and promoter divergence (r = −0.45, P < 10−6) for tandemly repeated gene pairs.
These findings underpin that functional selection constraints on coding sequences are more pronounced than on promoter regions and that on average more mutations within coding sequences are eliminated by negative selection than is the case for promoter regions. This is consistent with the specific structural features of cis-elements and promoters as a whole, e.g. the degenerate nature of cis-elements and the only small fraction of nucleotides constituting cis-elements within the entire promoter region.
Promoter Similarity and Expression Characteristics of Tandemly Duplicated Genes Are Correlated
As shown above, promoters associated with duplicated genes still share significant similarity. In addition, Pearson correlation of tandemly and segmentally duplicated genes revealed gene pairs that showed pronounced similarities in their expression characteristics. To test whether promoter relationships (as measured by the extent of alignable regions) correlate with expression characteristics, we plotted promoter similarity versus the Pearson coefficient r for the respective duplicated gene pairs (Fig. 5). Correlation coefficients are r = 0.17 (P < 0.01) for tandem and r = 0.12 for segmental genes (P < 0.05; Fig. 5). While the probability for segmentally duplicated genes implies only a nonsignificant correlation, we find a weak, yet significant positive correlation for tandemly duplicated pairs. This indicates that for tandemly duplicated genes, similarities in expression characteristics correlate with the similarities detected within the promoter regions, an observation which is not supported for segmentally duplicated genes. This might be attributable to the on average shorter evolutionary distance and point toward an ongoing degeneration process within promoters of tandemly duplicated genes.
Figure 5.

Promoter similarity is coupled to expression correlations for tandem genes. Expression correlation (Pearson r on the x axis) was plotted versus the promoter similarity within tandemly and segmentally duplicated genes. Maximum promoter length was 600 bp, and an alignment stringency threshold of 3 was used. Spearman rank correlation was found to be significant for tandemly repeated genes (r = 0.17, P < 0.01). Segmental duplications did not reveal a significant correlation (r = 0.12, 0.01 < P < 0.05) at the 1% significance level.
Divergence Time and Expression Characteristics Are Not Correlated in Tandemly and Segmentally Duplicated Genes
Similarity within promoters of tandemly repeated genes was found to be correlated to both the divergence time as well as to expression similarities. This suggests a continuous divergence of expression characteristics between duplicated genes. To verify this, we plotted KS against the Pearson correlation (Fig. 6). For both datasets, pronounced scattering was observed, and no significant correlation was found for both tandemly (r = −0.09, P < 0.05) as well as segmentally (r = 0.05, P < 0.2) duplicated pairs. Thus, from the divergence time of duplicated genes, conclusions on coherent expression characteristics between duplicated gene pairs cannot be derived.
Figure 6.

Expression divergence of tandemly and segmentally duplicated gene pairs is uncoupled to divergence time. Divergence time KS of tandemly and segmentally duplicated genes is plotted against the expression similarity/divergence measured as the Pearson correlation of the MPSS data. Black circles indicate value pairs derived from segmentally duplicated pairs (right); white triangles represent data from tandem genes (left). No significant correlation tested by Spearman rank correlation test was detected for both tandemly repeated (r = −0.09, 0.01 < P < 0.05) and segmentally duplicated genes (r = 0.05, P < 0.2).
cis-Regulatory Element Detection by Intragenomic Footprinting
cis-Regulatory elements are the major constituents driving gene expression. Although not restricted to these regions, these elements are predominantly located within the 5′ upstream sequence, and, consequently, analytical approaches predominantly focus on surveying these regions.
Our analysis demonstrates that duplicated promoters show both significant similarity and significant expression correlation between duplicated genes. To analyze this in more detail, we undertook several case studies, selecting duplicated pairs for which experimentally verified cis-regulatory elements have been reported. We carried out a phylogenetic footprinting analysis for the respective gene pairs. The selected promoters were subjected to an analysis by (1) detecting conserved (e.g. alignable) regions and (2) searching for statistically overrepresented sequence elements by a Gibbs sampling-based method to identify potential cis-regulatory elements (Thijs et al., 2001, 2002). We analyzed promoters of the duplicated gene pairs for histone H4 (At3g46320 and At5g59690), three copies of Rubisco (At5g38410, At5g38420, and At5g38430), and the duplicated OPR1 and OPR2 genes (At1g78680 and At1g78690; Green et al., 1988; Donald and Cashmore, 1990; Chaubet et al., 1996; Biesgen and Weiler, 1999; He and Gan, 2001). Histone H4 duplicate genes are found in segmental duplications, whereas the Rubisco and OPR1/OPR2 copies are organized in tandem arrays. The phylogenetic footprinting analysis detected the G-box and several I-boxes within the Rubisco promoters (Fig. 7B). Combination of these elements has previously been shown to be essential for the light induction of Rubisco genes in many higher plants (Green et al., 1988; Donald and Cashmore, 1990). The histone H4 gene At5g59690 has been experimentally analyzed by DNAseI footprinting, and several protected sites could be identified which previously have been reported as functional elements in maize (Zea mays; Chaubet et al., 1996). Four out of five sites were detected, indicating a conservation of these elements in duplicated promoters (Fig. 7A). The promoters of OPR1/OPR2 harbor two cis-regulatory elements involved in senescence and in response to jasmonic acid (He and Gan, 2001). Within our analysis, both elements give clear signals against the background (Fig. 7C).
Figure 7.

Detection of experimentally verified cis-regulatory elements within promoters of tandemly and segmentally duplicated genes. Promoters of the respective genes are displayed as blue horizontal bars; scale indicates bp upstream of transcriptional start site as defined by corresponding cDNA. Alignments were generated by DiAlign (threshold 3), and are shown as purple horizontal bars. Results of 100 runs of MotifSampler3 are summarized below. Height of the vertical bars indicates the number of reported motifs for the respective bp. Number of hits is also color coded (gray, < 5% of all motifs; blue, <10%; green, <20%; and red, >20%). The locations of experimentally verified elements are highlighted as red boxes. A, Results for histone H4. B, Results for three tandemly arranged Rubisco genes. C, Results for the tandemly duplicated OPR1 and 2 genes. Further details are described in the text.
We next asked the question of whether the Pearson correlation of transcriptional characteristics influences the performance of the intragenomic phylogenetic footprinting analysis. We selected duplicated gene pairs with high (KIN1 and COR6.6; r = 0.96), moderate (COR15a and COR15b; r = 0.56), and low (SCARECROW-like; r = −0.48) transcriptional correlation and subjected the respective promoters to a phylogenetic footprinting analysis (Fig. 8). Again, duplicated pairs were organized either in tandem arrays (KIN1 and COR6.6; COR15a and COR15b) or within segmental duplications (SCARECROW-like). KIN1 and COR6.6 are both up-regulated by cold treatment and abscisic acid (ABA) application. Promoters of both genes harbor sites that match consensus sequences of known cis-regulatory elements conferring ABA- and cold-responsiveness (CRT; Baker et al., 1994). The CRT-element (C-repeat; Baker et al., 1994) is found within many promoters of cold-induced genes, including COR15a/b (Thomashow, 1999). In addition to these elements, several additional elements were detected between these gene pairs (Fig. 8, A and B). Furthermore, several conserved candidate cis-regulatory elements were identified for the SCARECROW-like gene pair, yet no correlation in expression patterns was observed in the MPSS data (Fig. 8C). These results suggest that duplicate promoters harbor conserved transcription factor binding sites and that intragenomic footprinting provides a powerful tool to identify regulatory elements conserved between duplicated promoters.
Figure 8.

Intragenomic phylogenetic footprinting detects de novo candidate cis-regulatory elements in promoters of tandemly and segmentally duplicated genes. Three duplications covering a wide range of expression correlation as analyzed by the MPSS data were subjected to an intragenomic phylogenetic footprinting as described in the text. Results are presented as described in the legend of Figure 7. Locations of the ABRE (ABA responsive element, within the promoters of KIN1/COR6.6) and the cold-responsive CRT elements (within the promoters of KIN1/COR6.6 and COR15a/b) are indicated as red boxes below the MotifSampler results.
DISCUSSION
Duplication of individual genes, large genomic segments, or complete genomes provides a mechanism to introduce functional innovation and novelty into genomes. However, fixation of duplicated loci within the respective genome requires sub- or neofunctionalization of the respective loci. Besides evolution of the protein sequence and change of the biochemical properties of the protein, changes in the transcriptional regulation of duplicated genes have long been hypothesized to play an important role for the fixation of duplicated genes (Britten and Davidson, 1969; Ohno, 1971; Doebley and Lukens, 1998; Carroll, 2000). In fact, genome-wide studies in yeast revealed a rapid divergence of expression patterns of duplicated genes (Gu et al., 2002). Although expansions of gene families and both local and global duplications in plants are highly pronounced, genome-scale studies on the conservation and divergence of expression characteristics of duplicated genes have not been reported thus far. In this study, we investigated transcriptional similarities and dissimilarities among two distinct types of gene duplications, tandem and segmental duplications. The evolutionary origins of the two classes are distinct from each other. Tandemly repeated genes are located in close physical vicinity to each other and most likely are generated by illegitimate recombination. The second type of duplicated gene pairs is constituted from gene pairs located within segmental duplications, and these are most likely the remnants of an ancient polyploidization event with subsequent reorganization of the genome (The Arabidopsis Genome Initiative, 2000; Vision et al., 2000; Simillion et al., 2002; Blanc et al., 2003). We restricted our analysis to segmentally duplicated gene pairs originating from the most recent genome duplication event within Arabidopsis, approximately 38 to 70 million years ago (Vision et al., 2000; Simillion et al., 2002; Blanc et al., 2003).
Most duplicated gene pairs located within segmental duplications originated from a duplication event at a defined time point during the evolution of the Arabidopsis genome. Consistent with findings by other research groups, the age distribution within the segmentally duplicated genes shows a Gaussian distribution with a pronounced peak at KS in the range of 0.7 to 0.8 (Vision et al., 2000; Blanc et al., 2003). By contrast, genes arranged as tandem arrays exhibited a significantly lower average divergence time with a broader distribution of KS values. Surprisingly, the tandem duplications exhibited a distribution similar to the segmental duplications, with a maximum peak for KS values of 0.3 to 0.4, corresponding to approximately 20 to 40 million years ago. Assuming a continuous death and birth process for tandemly duplicated genes, an exponential decay, rather than a Gaussian distribution with a peak at a particular evolutionary time point, would be expected. As we found a similar distribution for tandem genes of groups consisting of two members, we exclude a bias potentially caused by pairwise combinations of all members of expanded tandem groups. A recent decline in the rate of generation of tandem arrays or, alternatively, an evolutionary time span (indicated by the peak at KS = 0.3–0.4) with above-average generation of tandemly duplicated genes could explain this observation. However, to our knowledge, there is no report supporting one or the other possibility to date. The causes leading to the unexpected KS distribution for tandemly duplicated genes therefore remain to be elucidated in future studies.
The tandemly repeated genes we analyzed circumvent a broad range of divergence times. We made use of MPSS data generated from five different libraries representing different plant tissues. The MPSS method measures relative expression values on a genome scale (Brenner et al., 2000a, 2000b). MPSS sequence tags were mapped to the respective loci within the Arabidopsis genome. Due to the high sequence similarity of transcripts from duplicated genes, a precise assignment of tags to matching genes is crucial for the analysis. The disproportionate loss of informative tags for the tandem and segmental duplications emphasizes the importance of this analysis step. However, one advantage of the sequence-based MPSS tags is that interference caused by sequence similarities is readily detected and, thus, noisy data can be excluded, unlike poorly characterized cross-hybridization on microarrays.
The analysis of MPSS expression data shows that a significant portion of duplicated genes retained a high degree of highly similar expression characteristics. Correlations against a negative control set are substantially different: 26.3% (20.1%) of the tandem repeats and 19.4% (12.4%) of genes located within segmental duplications revealed a Pearson correlation of above 0.8 (above 0.9). It is noteworthy that the degree of duplicated genes with highly unrelated expression, i.e. negative Pearson coefficients (close to −1.0), is significantly lower than expected from a random distribution (Fig. 2), indicating that highly dissimilar expression patterns are underrepresented between duplicated genes. These observations suggest a significant conservation of expression characteristics between duplicated pairs. However, our findings also indicate that the majority of duplicated genes already experienced a significant divergence in their expression characterization. This is consistent with expectations implied by the DDC model (Force et al., 1999).
Our study has been carried out by using MPSS data derived from particular tissues. These data describe only a few dimensions of regulatory complexity and, for example, expression differences at the cellular level or in response to internal and external stimuli are not captured and reflected within the dataset. Thus, any subfunctionalization caused by such cues would necessarily escape detection.
The DDC model proposes partitioning of ancient subfunctions as a major mechanism to retain duplicated genes in a genome. Although not limited to the subfunctionalization of regulatory regions, due to their short sequences and modular organization, cis-regulatory elements are particularly well-suited candidates for mechanisms predicted by the DDC model. Thus, expression divergence potentially is a major constituent driving a degeneration and complementation process. Repeated rounds of this process potentially generate increasingly divergent expression patterns, and these would ultimately lead to neofunctionalization on a transcriptional level. Consequently, sub-, neo-, and nonfunctionalization of duplicated gene pairs are time-dependent processes. The average divergence time or evolutionary distance among tandemly repeated genes is lower compared to duplicate gene pairs residing within segmental duplications. Supportive of a time-dependent increase in expression divergence, the proportion of highly correlated pairs is significantly larger in tandemly repeated genes in comparison to segmental duplications. However, correlation analysis between divergence time KS and expression similarity measured by the Pearson coefficient revealed no correlation for duplicated genes at a significance level α < 1%. This is in contrast to findings in yeast and human, where a significant correlation between expression and protein divergence for duplicated pairs has been found (Gu et al., 2002; Makova and Li, 2003). Several reasons might account for the differing findings in Arabidopsis. The correlation of evolutionary distance and conservation of transcriptional characteristics in yeast has been drawn for duplicated gene pairs over a range of KS = 0 to KS = 1.5, including a significant portion with KS ≤ 0.3. Most of the duplicated gene pairs used in our analysis show KS values well above 0.3 and might therefore show only weak or no correlations. An alternative hypothesis is that, in contrast to yeast, expression divergence for duplicated genes might occur more rapidly in Arabidopsis.
Several observations suggest continuous degeneration within regulatory regions and are consistent to the DDC model. Repeated cycles of degeneration and complementation should lead to an increasing divergence within regulatory regions and consequently to an increase in expression divergence. On the other hand, recent duplicate promoter pairs will still share significant similarity within their regulatory regions. Both aspects are reflected in our analysis results. We analyzed promoters from duplicated genes with respect to the degree of similarity that is still retained. We found significantly higher similarities between promoters from both tandemly and segmentally duplicated gene pairs in comparison to random expectations. This supports the hypothesis that duplicated genes share common regions within their regulatory sequences. In addition, promoter similarities between tandemly duplicated genes showed a highly significant negative correlation against the divergence time of the proteins. This suggests a continuous degeneration within regulatory regions. A very weak positive correlation has been detected between promoter similarities of tandemly duplicated genes and their expression characteristics. These findings are not unexpected as many genes within the dataset exhibit divergence times above KS ≥ 0.3. Additionally, as we selected for functional gene pairs, our dataset likely contains those duplicated gene pairs that have undergone purifying selection and were thus retained within the genome. A recent study that investigated the evolutionary fate of three very recent duplicated pairs in Arabidopsis (KS < 0.02) is supportive of these findings (Moore and Purugganan, 2003). Although the pairs still showed a highly correlated expression, different patterns were detected, indicating that positive selection can rapidly lead to changes in expression characteristics.
In Arabidopsis, partial redundancy and overlapping functionality for duplicated genes (e.g. SHATTERPROOF 1 and 2, AP1/CAULIFLOWER, PHOT1 and 2) have been reported (Ferrandiz et al., 2000; Liljegren et al., 2000; Kinoshita et al., 2001; Pinyopich et al., 2003). These reports as well as our observations indicate that for numerous gene pairs, besides at least overlapping biochemical properties of the respective proteins, a significant overlap in regulatory characteristics still exists. To analyze this in more detail, we asked whether shared cis-regulatory elements are conserved between duplicated genes.
We subjected duplicated gene pairs to a phylogenetic footprinting analysis. Both colinearity criteria as well as heuristic criteria were used for their detection (Morgenstern, 1999; Thijs et al., 2001). For the analysis, gene pairs were selected for which previously particular cis-elements have been experimentally confirmed. For all evaluated examples that we tested (histone H4, Rubisco, OPR1/OPR2), the individually confirmed cis-elements correspond to pronounced signals (Fig. 7, A–C).
To test the applicability of the approach to duplicated gene pairs with similar transcriptional characteristics (e.g. high Pearson correlation) and duplicated gene pairs with dissimilar transcriptional characteristics (low Pearson coefficient), we selected duplicated gene pairs over a range of Pearson correlation coefficients. For all examples, clear and pronounced signals that depict potential conserved cis-elements have been found. Again for these examples, at least in part, individual cis-elements have already been described. The respective elements proved to be conserved, along with numerous additional conserved cis-elements. High-scoring candidate cis-elements are not restricted to housekeeping genes like histone H4 or Rubisco but are also apparent in duplicated genes responding to specific stimuli like cold-inducible COR15a/b and KIN1/COR6.6.
Phylogenetic footprinting of orthologous genes has been proven to be a powerful tool for the detection of cis-regulatory elements (Wasserman et al., 2000; Guo and Moose, 2003). However, in the absence of a second suitable plant genome for phylogenetic footprinting and given the complications in assigning clear orthologous relationships arising from the highly duplicated nature of plant genomes, intragenomic phylogenetic footprinting represents a powerful strategy that is complementary to classical intergenomic phylogenetic footprinting. Due to potential acquisition and loss of individual cis-elements between duplicated promoters, detection of the full complement of cis-elements is not feasible by this approach. However, our analysis results demonstrate that intragenomic phylogenetic footprinting represents a powerful means to detect cis-regulatory elements on a genome scale. Thus, apart from insights into the evolution of transcriptional characteristics of duplicated genes, our study also delineates an analytical outline for the detection of plant cis-elements, which exceeds the currently available resources by far. With the availability of complete and high-quality plant genome sequences in the foreseeable future, the opportunity to undertake classical intergenomic phylogenetic footprinting is emerging. However, the inherent duplication dynamics of plant genomes often hampers clear orthology assignment. Thus, a combination of inter- and intragenomic footprinting bears the potential to circumvent this constraint and to identify regulatory sub- and/or neofunctionalization processes on a molecular level. This will enable, on one hand, analysis for cis-elements on a genome scale and, on the other, detection and definition of the birth and death of cis-elements and the molecular evolution of transcriptional characteristics.
MATERIALS AND METHODS
Computational Methods
Computations were performed on an IBM workstation with a 2.4 GHz Pentium 4 processor running on a LINUX operating system. Python scripts (http://www.python.org) were used for all analysis and can be obtained on request. Images for the motif detection were obtained with the Python Imaging Library, the residual figures with the python module PYCHART.
Computational Selection of Tandem and Segmental Duplications
Genome annotation data stored within the Munich Information Center for Protein Sequence (MIPS) Arabidopsis database (MAtDB) were used for the analysis (Schoof et al., 2004). FASTA3 as global alignment tool was used to avoid duplications reported due to a high-scoring but localized hit of a local alignment tool (Pearson and Lipman, 1988). Default parameter settings have been used. As stringent similarity threshold, the FASTA ratio (opt score divided by self score) ≥ 0.3 was used (The Arabidopsis Genome Initiative, 2000). Duplicated pairs exceeding the given threshold and separated by less than three intermediate unrelated genes were counted as tandemly repeated gene pairs. Along with the homology criteria, location within previously described segmental duplications (The Arabidopsis Genome Initiative, 2000) was decisive to detect and assign segmentally duplicated gene pairs. To minimize potential artifacts caused by expanded gene families not arranged in a colinear order, the analysis was limited to 1:1 relations, i.e. members of one pair are present as single copies within the respective segment. A list of the selected MAtDB codes is provided in the supplemental material.
The synonymous (KS) and nonsynonymous substitution (KN) rates for the selected tandem and segmental pairs were computed using the PAML program package (Yang, 1997). Proteins were aligned using DiAlign2-1 (Morgenstern, 1999) and subsequently transformed into corresponding nucleotide alignments. Due to restrictions in accuracy and reliability for rate estimation caused by the increasing number of reversions or multiple substitutions, substitution rates above 1.5 were excluded from the analysis.
MPSS Data Generation
We used MPSS data from five different untreated organs or tissues, including untreated silique, callus, leaves, roots, and inflorescence. All plant material was from Arabidopsis, ecotype Col-0. Callus was initiated from seeds grown on media containing 0.5× Murashige and Skoog salts, 3% Suc in presence of 2,4-dichlorophenoxyacetic acid (0.5 mg/L), indole-3-acetic acid (2 mg/L), and kinetin (0.1 mg/L). Floral buds (up to Stage 11/12) were harvested from plants grown under 16 h of light for 5 weeks. Developing siliques (Stage 16/17) were harvested from plants grown under conditions identical to the floral library. For the leaf and root libraries, plants were grown in 16 h of light for 21 d under sterile conditions in vermiculite and perlite. For each library, total RNA was isolated using TRIzol (Invitrogen, Carlsbad, CA). For tissues derived from whole plants, samples were taken approximately 2 h after dark.
MPSS was performed as described by Brenner et al. (2000b). For the five libraries, a total of 12,273,934 signatures were obtained in multiple sequencing runs and in two sequencing frames. The abundance for each signature was normalized to transcripts per million (TPM) to facilitate comparisons across libraries. The merging of the sequencing runs and the normalization steps are described in more detail by Meyers et al. (2004).
MPSS Tag Mapping/Quality Determination
MPSS tags were anchored to the Arabidopsis genome and mapped to the transcribed part of the genome. In addition to the coding region, 5′ and 3′ UTRs were included into the assignment. Based on full-length cDNA information for 46.6% (12,314 out of 26,444) of all annotated genes, a 3′ UTR was determined. For genes with no or only incomplete cDNA information, a default length of 200 bp has been used. For genes lacking cDNA information for the 5′ UTR, the default length used was 100 bp. Tags matching more than one gene were treated as ambiguous, while uniquely occurring tags were regarded as diagnostic. According to technical aspects of the MPSS method (Brenner et al., 2000b), tags located closest to the 3′ end of the gene were treated as informative, while residual matching tags were counted as noninformative tags. To lower the background noise, both a total minimal abundance of greater than 10 TPM over all MPSS libraries and greater than 5 TPM in at least one of the MPSS libraries was required to consider a tag as informative.
Expression Correlation
To determine the similarities and dissimilarities between expression characteristics of duplicated genes, only pairs for which both members had an informative, diagnostic tag were considered. The correlation of expression between the respective gene pairs was determined using Pearson's coefficient r:
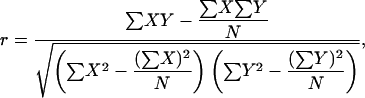 |
with X,Y representing expression data vectors of x1,x2,…xN, y1,y2,…yN respectively and N representing the number of cDNA libraries.
To compare the results against the background expectation, we analyzed negative control sets consisting of pairwise comparisons of randomly selected expression profiles from all Arabidopsis genes associated with an informative tag. Random shuffles with gene pairs overlapping with the datasets of tandemly and segmentally duplicated genes were excluded. Expression correlations were analyzed in analogous manner for 10 different random sets of equal size to the datasets of tandemly and segmentally duplicated genes.
Significance was tested using the χ2 test. χ2 values for both tandem and segmental duplications exhibited highly significant differences compared to the background distributions (χ2 > 60, P < 10−6 for segmental pairs and χ2 > 120, P < 10−9 for tandem pairs at 19 degrees of freedom; see supplemental material for details on the statistical analysis).
Promoter Alignments
Pairwise alignments of promoters from tandem and segmental duplications were computed using DiAlign2 (Morgenstern, 1999). To avoid alignments of 5′ UTRs, which generally exhibit a higher degree of similarity between duplicated pairs (G. Haberer, unpublished data), we restricted our analysis to duplicated gene pairs with associated full-length cDNA information. These criteria confined the analyses to 655 pairwise alignments for the tandemly duplicated and to 663 segmentally duplicated gene pairs. Promoter regions analyzed circumvent a 1-kb 5′ nontranscribed sequence or were delimited by the transcriptional start or stop of the previous gene. As a negative control, we used pairwise alignments of randomly shuffled promoter pairs. For each pairwise alignment, we computed the promoter similarity between two promoters as the length of aligned regions divided by their total length (see supplemental material).
As it can't be assumed that the promoter similarities follow a normal distribution, we carried out a nonparametric ranking test (Mann-Whitney-Wilcoxon test) to evaluate differences between means. Ties between ranks were resolved as midranks. For large data sizes (as analyzed in this study), the distribution of the ranks is approximately normally distributed (Bickel and Doksum, 1977). Significance levels P were highly significant (P ≪ 10−6) for all analyzed promoter lengths above a DiAlign2 threshold >1 (details on the statistical analysis are given within the supplemental material).
Statistical Analysis of Correlations
Two-dimensional samples were tested for correlations determining the Spearman rank correlation coefficient. Significance levels for each tested correlations are given in the text. Details about the tests are provided in the supplemental material.
Intragenomic Phylogenetic Footprinting of Duplicated Genes
A literature survey identified several tandemly and segmentally duplicated genes for which functional regulatory elements have been reported. Consensus sequences or the respective sites were extracted from these reports, and their positions were determined within the respective promoters. DiAlign2-1 (Morgenstern, 1999) and MotifSampler3 (Thijs et al., 2001, 2002) were used to detect candidate cis-regulatory elements and conserved promoter regions shared by duplicated promoters. Promoters were selected as described above. DiAlign2-1 was run with default settings. To exclude nonspecific alignments within promoter regions, a stringent threshold parameter of 3 has been used. MotifSampler3 is based on a heuristic algorithm which can converge to local optima. To avoid problems arising from this potential bias, results from 100 runs (50 runs with octamer and decamer motif length setting each) were sampled, and the reported motifs per site were counted. Default values were used as residual parameter settings with a maximum of five reported motifs per run. A background model of order 3, which has been determined from a large set of promoters derived from genes with full-length cDNA information, has been used for the analysis.
Supplementary Material
Acknowledgments
We thank Shin-Han Shiu and Michael Mader for comments and critical reading of the manuscript.
This work was supported by the Deutsche Forschungsgemeinschaft (grant no. MA 2522/1–1 to K.F.X.M.) and by the National Science Foundation Plant Genome Research Program (DBI–0110528 to B.C.M.).
The online version of this article contains Web-only data.
References
- The Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796–815 [DOI] [PubMed] [Google Scholar]
- Baker SS, Wilhelm KS, Thomashow MF (1994) The 5′-region of Arabidopsis thaliana cor15a has cis-acting elements that confer cold-, drought- and ABA-regulated gene expression. Plant Mol Biol 24: 701–713 [DOI] [PubMed] [Google Scholar]
- Bickel PJ, Doksum KA (1977) Mathematical Statistics. Holden-Day, Oakland, CA
- Biesgen C, Weiler EW (1999) Structure and regulation of OPR1 and OPR2, two closely related genes encoding 12-oxophytodienoic acid-10,11-reductases from Arabidopsis thaliana. Planta 208: 155–165 [DOI] [PubMed] [Google Scholar]
- Blanc G, Hokamp K, Wolfe KH (2003) A recent polyploidy superimposed on older large-scale duplications in the Arabidopsis genome. Genome Res 13: 137–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers JE, Chapman BA, Rong J, Paterson AH (2003) Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 422: 433–438 [DOI] [PubMed] [Google Scholar]
- Brenner S, Johnson M, Bridgham J, Golda G, Lloyd DH, Johnson D, Luo S, McCurdy S, Foy M, Ewan M, et al (2000. b) Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nat Biotechnol 18: 630–634 [DOI] [PubMed] [Google Scholar]
- Brenner S, Williams SR, Vermaas EH, Storck T, Moon K, McCollum C, Mao JI, Luo S, Kirchner JJ, Eletr S, et al (2000. a) In vitro cloning of complex mixtures of DNA on microbeads: physical separation of differentially expressed cDNAs. Proc Natl Acad Sci USA 97: 1665–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britten RJ, Davidson EH (1969) Gene regulation for higher cells: a theory. Science 165: 349–357 [DOI] [PubMed] [Google Scholar]
- Carroll SB (2000) Endless forms: the evolution of gene regulation and morphological diversity. Cell 101: 577–580 [DOI] [PubMed] [Google Scholar]
- Chaubet N, Flenet M, Clement B, Brignon P, Gigot C (1996) Identification of cis-elements regulating the expression of an Arabidopsis histone H4 gene. Plant J 10: 425–435 [DOI] [PubMed] [Google Scholar]
- Doebley J, Lukens L (1998) Transcriptional regulators and the evolution of plant form. Plant Cell 10: 1075–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donald RG, Cashmore AR (1990) Mutation of either G box or I box sequences profoundly affects expression from the Arabidopsis rbcS-1A promoter. EMBO J 9: 1717–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrandiz C, Gu Q, Martienssen R, Yanofsky MF (2000) Redundant regulation of meristem identity and plant architecture by FRUITFULL, APETALA1 and CAULIFLOWER. Development 127: 725–734 [DOI] [PubMed] [Google Scholar]
- Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J (1999) Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151: 1531–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green PJ, Yong MH, Cuozzo M, Kano-Murakami Y, Silverstein P, Chua NH (1988) Binding site requirements for pea nuclear protein factor GT-1 correlate with sequences required for light-dependent transcriptional activation of the rbcS-3A gene. EMBO J 7: 4035–4044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Nicolae D, Lu HH, Li WH (2002) Rapid divergence in expression between duplicate genes inferred from microarray data. Trends Genet 18: 609–613 [DOI] [PubMed] [Google Scholar]
- Gu Z, Steinmetz LM, Gu X, Scharfe C, Davis RW, Li WH (2003) Role of duplicate genes in genetic robustness against null mutations. Nature 421: 63–66 [DOI] [PubMed] [Google Scholar]
- Guo H, Moose SP (2003) Conserved noncoding sequences among cultivated cereal genomes identify candidate regulatory sequence elements and patterns of promoter evolution. Plant Cell 15: 1143–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Gan S (2001) Identical promoter elements are involved in regulation of the OPR1 gene by senescence and jasmonic acid in Arabidopsis. Plant Mol Biol 47: 595–605 [DOI] [PubMed] [Google Scholar]
- Kinoshita T, Doi M, Suetsugu N, Kagawa T, Wada M, Shimazaki K (2001) Phot1 and phot2 mediate blue light regulation of stomatal opening. Nature 414: 656–660 [DOI] [PubMed] [Google Scholar]
- Kofuji R, Sumikawa N, Yamasaki M, Kondo K, Ueda K, Ito M, Hasebe M (2003) Evolution and divergence of the MADS-box gene family based on genome-wide expression analyses. Mol Biol Evol 20: 1963–1977 [DOI] [PubMed] [Google Scholar]
- Liljegren SJ, Ditta GS, Eshed Y, Savidge B, Bowman JL, Yanofsky MF (2000) SHATTERPROOF MADS-box genes control seed dispersal in Arabidopsis. Nature 404: 766–770 [DOI] [PubMed] [Google Scholar]
- Long M, Thornton K (2001) Gene duplication and evolution. Science 293: 1551. [DOI] [PubMed] [Google Scholar]
- Lynch M, Conery JS (2000) The evolutionary fate and consequences of duplicate genes. Science 290: 1151–1155 [DOI] [PubMed] [Google Scholar]
- Makova KD, Li WH (2003) Divergence in the spatial pattern of gene expression between human duplicate genes. Genome Res 13: 1638–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers BC, Tej SS, Vu TH, Haudenschild CD, Agrawal V, Edberg SB, Ghazal H, Decola S (2004) The use of MPSS for whole-genome transcriptional analysis in Arabidopsis. Genome Res 14: 1641–1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RC, Purugganan MD (2003) The early stages of duplicate gene evolution. Proc Natl Acad Sci USA 100: 15682–15687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern B (1999) DIALIGN 2: improvement of the segment-to-segment approach to multiple sequence alignment. Bioinformatics 15: 211–218 [DOI] [PubMed] [Google Scholar]
- Ohno S (1970) Evolution by Gene Duplication. Springer-Verlag, New York
- Ohno S (1971) An argument for the genetic simplicity of man and other mammals. J Hum Evol 1: 651–662 [Google Scholar]
- Parenicova L, de Folter S, Kieffer M, Horner DS, Favalli C, Busscher J, Cook HE, Ingram RM, Kater MM, Davies B, et al (2003) Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: new openings to the MADS world. Plant Cell 15: 1538–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson WR, Lipman DJ (1988) Improved tools for biological sequence comparison. Proc Natl Acad Sci USA 85: 2444–2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinyopich A, Ditta GS, Savidge B, Liljegren SJ, Baumann E, Wisman E, Yanofsky MF (2003) Assessing the redundancy of MADS-box genes during carpel and ovule development. Nature 424: 85–88 [DOI] [PubMed] [Google Scholar]
- Prince VE, Pickett FB (2002) Splitting pairs: the diverging fates of duplicated genes. Nat Rev Genet 3: 827–837 [DOI] [PubMed] [Google Scholar]
- Schoof H, Ernst R, Nazarov V, Pfeifer L, Mewes HW, Mayer KF (2004) MIPS Arabidopsis thaliana Database (MAtDB): an integrated biological knowledge resource for plant genomics. Nucleic Acids Res 32 (Database issue): D373–D376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simillion C, Vandepoele K, Van Montagu MC, Zabeau M, Van de Peer Y (2002) The hidden duplication past of Arabidopsis thaliana. Proc Natl Acad Sci USA 99: 13627–13632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thijs G, Lescot M, Marchal K, Rombauts S, De Moor B, Rouze P, Moreau Y (2001) A higher-order background model improves the detection of promoter regulatory elements by Gibbs sampling. Bioinformatics 17: 1113–1122 [DOI] [PubMed] [Google Scholar]
- Thijs G, Marchal K, Lescot M, Rombauts S, De Moor B, Rouze P, Moreau Y (2002) A Gibbs sampling method to detect overrepresented motifs in the upstream regions of coexpressed genes. J Comput Biol 9: 447–464 [DOI] [PubMed] [Google Scholar]
- Thomashow MF (1999) Plant cold acclimation: freezing tolerance genes and regulatory mechanisms. Annu Rev Plant Physiol Plant Mol Biol 50: 571–599 [DOI] [PubMed] [Google Scholar]
- Vision TJ, Brown DG, Tanksley SD (2000) The origins of genomic duplications in Arabidopsis. Science 290: 2114–2117 [DOI] [PubMed] [Google Scholar]
- Wagner A (2000) Decoupled evolution of coding region and mRNA expression patterns after gene duplication: implications for the neutralist-selectionist debate. Proc Natl Acad Sci USA 97: 6579–6584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman WW, Palumbo M, Thompson W, Fickett JW, Lawrence CE (2000) Human-mouse genome comparisons to locate regulatory sites. Nat Genet 26: 225–228 [DOI] [PubMed] [Google Scholar]
- Wray GA, Hahn MW, Abouheif E, Balhoff JP, Pizer M, Rockman MV, Romano LA (2003) The evolution of transcriptional regulation in eukaryotes. Mol Biol Evol 20: 1377–1419 [DOI] [PubMed] [Google Scholar]
- Yang Z (1997) PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13: 555–556 [DOI] [PubMed] [Google Scholar]
- Zhang L, Gaut BS, Vision TJ (2001) Gene duplication and evolution. Science 293: 1551. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.