

Abstract
Maize (Zea mays) possesses a large, highly repetitive genome, and subsequently a number of reduced-representation sequencing approaches have been used to try and enrich for gene space while eluding difficulties associated with repetitive DNA. This article documents the ability of publicly available maize expressed sequence tag and Genome Survey Sequences (GSSs; many of which were isolated through the use of reduced representation techniques) to recognize and provide coverage of 78 maize full-length cDNAs (FLCs). All 78 FLCs in the dataset were identified by at least three GSSs, indicating that the majority of maize genes have been identified by at least one currently available GSS. Both methyl-filtration and high-Cot enrichment methods provided a 7- to 8-fold increase in gene discovery rates as compared to random sequencing. The available maize GSSs aligned to 75% of the FLC nucleotides used to perform searches, while the expressed sequence tag sequences aligned to 73% of the nucleotides. Our data suggest that at least approximately 95% of maize genes have been tagged by at least one GSS. While the GSSs are very effective for gene identification, relatively few (18%) of the FLCs are completely represented by GSSs. Analysis of the overlap of coverage and bias due to position within a gene suggest that RescueMu, methyl-filtration, and high-Cot methods are at least partially nonredundant.
Complete genome sequences are a powerful tool being utilized by many biologists. For several model species, such as Saccharomyces cerevisiae, Drosophila melanogaster, Mus musculus, Homo sapiens, Caenorhabditis elegans, and Arabidopsis, a genome sequence of high standards for coverage and accuracy has been elucidated (Goffeau et al., 1996; C. elegans Sequencing Consortium 1998; Arabidopsis Genome Initiative, 2000; Myers et al., 2000; Venter et al., 2001). However, for most species, only partial genome sequences are available. Many of the economically valuable crop species, including maize (Zea mays), have relatively large, complex genomes, which contain a significant fraction of repetitive sequence. This repetitive fraction of the genome increases the cost of obtaining a complete genome sequence due to the larger genome size and the increased difficulties in assembly. The gene space of these complex genomes, which is the portion of the genome containing coding sequences, introns, and cis-acting regulatory sequences, can exist either in clusters of genes or as single gene units separated by repetitive regions (SanMiguel et al., 1996; Bennetzen et al., 1998; Walbot and Petrov, 2001). Because genes and their expression patterns underlie most economic and adaptive traits, there is considerable value in methods that allow genes to be isolated from the large stretches of repetitive DNA in which they reside. Alternative methods, including expressed sequence tag (EST) and reduced-representation techniques, for sequencing the gene space of many plant and animals species with relatively large genomes have been investigated.
Maize has an estimated genome size of 2,300 to 2,700 Mb (Arumuganathan and Earle, 1991), which is 20- and 6-fold greater in size than the genomes of Arabidopsis and rice (Oryza sativa), respectively. The major portion (60%) of maize nuclear DNA is composed of long terminal repeat (LTR)-retrotransposon families that vary in copy number, ranging from 5 to 30,000 copies per 1C genome (Bennetzen, 1996; SanMiguel et al., 1996; Meyers et al., 2001). The highly repetitive LTR retrotransposons comprise more than 60% of nuclear DNA (Bennetzen, 1996; SanMiguel et al., 1996; Meyers et al., 2001). In total, all repetitive DNA sequences account for approximately 80% of the maize genome (Meyers et al., 2001), while the genic regions in maize constitute a fraction of the remaining 20%.
Several approaches have been utilized to sequence the maize gene space (Rabinowicz et al., 1999; Yuan et al., 2002, 2003; Palmer et al., 2003; Whitelaw et al., 2003). Maize EST sequencing efforts have been undertaken (Fernandes et al., 2002) and have resulted in the deposition of 384,103 maize ESTs from diverse tissues and genotypes into GenBank. Clustering EST collections as part of The Institute for Genomic Research (TIGR) gene indices results in 29,414 contigs and 26,426 singletons (TIGR Gene Index from December 23, 2003), which represents 56,364 tentative consensus sequences (http://www.tigr.org/tdb/tgi/maize). While EST sequencing projects target expressed sequences, there are at least five shotgun approaches that have been used to sequence maize genomic DNA, three of which preferentially target low-copy genomic sequence (Table I). The sequences collected by these five methods are collectively termed Genome Survey Sequences (GSSs). Random sequencing of maize small-insert clones and bacterial artificial chromosome (BAC) end sequences represent GSSs that are not biased for or against highly repetitive regions of the genome. Another sequencing method known as RescueMu (RM) utilizes a transgenic Mutator transposon containing the sequences necessary to perform plasmid rescue to isolate sequences adjacent to transposon insertions. The RM sequences are likely to be gene rich due to the tendency of the Mutator transposon to insert within or near genic regions (Raizada et al., 2001). In addition, the RM sequence may provide the identification of a novel mutant allele if the insertion is germinal. To date, RM has been used to obtain flanking sequence for 178,125 insertions (Raizada, 2003; www.mutransposon.org). Selecting for hypomethylated DNA is another mechanism used to reduce the representation of repetitive sequences (Burr et al., 1988; Rabinowicz et al., 1999). The methyl-filtration (MF) method uses the endogenous restriction-modification system of Escherichia coli to eliminate clones containing methylated DNA inserts (Rabinowicz et al., 1999). The resulting libraries are highly enriched for fragments of hypomethylated DNA (Rabinowicz et al., 1999). High-Cot (HC) filtration, a form of Cot-based cloning and sequencing (Peterson et al., 2002), is a procedure in which DNA reassociation kinetics is used to separate repetitive and low-copy sequences based upon differences in their relative rates of reassociation (Peterson et al., 2002; Yuan et al., 2003). A pilot project to generate approximately 1 million HC and MF maize reads is currently under way (Whitelaw et al., 2003).
Table I.
Frequency of hits in GSS sequence libraries
| Description of GSS Library | Sequencesa | GSS Category | Number of Hitsb | Frequencyc | Sequencing Group |
|---|---|---|---|---|---|
| ZM_3.0_4.0_KB maize genomic clone | 50,877 | Random | 7 | 1.0 | TIGR (Whitelaw) |
| Maize random small-insert genomic library | 3,480 | Random | 1 | 2.1 | Dupont (Morgante) |
| ZMMBBc maize subsp. Mays genomic | 130,144 | BAC end | 14 | 0.8 | Rutgers (Messing) |
| ZMMBBb maize subsp. Mays genomic | 122,994 | BAC end | 18 | 1.1 | Rutgers (Messing) |
| Subtotal for all random sequences | 308,177 | Randomd | 42 | 1.0 | |
| RM maize genomic | 178,125 | RM | 70 | 2.9 | Stanford (Walbot) |
| ZM_0.6_1.0_KB maize genomic clone | 445,286 | HC | 481 | 7.9 | TIGR (Whitelaw) |
| ZM_0.7_1.5_KB maize genomic clone | 448,974 | MF | 488 | 8.0 | TIGR (Whitelaw) |
| WGS-ZmaysF (JM107 adapted MF) | 97,551 | MF | 78 | 5.9 | CSHLe (McCombie) |
| fzmb filtered library maize | 14,594 | MF | 5 | 2.5 | Orion (Bedell) |
| JM107 adapted MF library | 1,603 | MF | 2 | 9.2 | CSHL (McCombie) |
| ZM2_0.7-1.5_KB maize | 24,341 | MF | 1 | 0.3 | TIGR (Whitelaw) |
| Subtotal for all MF sequences | 587,063 | MF | 574 | 7.2 | |
| Total GSS hits | 1,518,651 | Total GSS | 1,167 | 5.6 | |
| Total EST hits | 384,103 | Total EST | 1,274 | 24.3 |
The total number of sequences from this library present at NCBI as of November 15, 2003.
The number of GSS sequences from each library derived from the 78 genes in the dataset is listed.
The frequency of GSS sequences from each library that were derived from our set of 78 full-length sequences was calculated. The frequency was normalized to allow the frequency of sequences in randomly sequenced libraries to equal 1.
The random sequences include the BAC and truly randomly sequenced clones since the frequencies were quite similar.
CSHL, Cold Spring Harbor Laboratory.
We collected 64,357 small-insert random maize sequences; 253,138 BAC end sequences; 178,125 RM insertions; 587,371 MF sequences; and 445,286 HC sequences from the National Center for Biotechnology Information (NCBI) sequence repository (Table I) and aligned these to a small set of well-characterized maize full-length cDNAs (FLCs) to evaluate the success of each of these approaches toward tagging and sequencing maize genes. We have used the full-length coding sequences of 78 maize genes to evaluate the frequency of EST and GSS hits as well as the coverage of the sequence. In addition, for a subset of these genes we have used the full-length genomic sequence to verify our results and determine the ability to sequence across introns. We also estimated the frequency of genes for which the GSS sequencing approaches provide upstream untranslated region (UTR) and promoter sequences. Our analyses confirm that MF and HC selected DNA sequences are highly enriched for gene sequences (Palmer et al., 2003; Whitelaw et al., 2003) and further suggest that the currently available GSSs should provide tags for the majority of maize genes.
RESULTS
Frequency of EST Sequences per Gene and Coding Sequence Coverage
A subset of 70 FLC sequences from our collection of 78 FLCs was used to perform BLASTN searches of the NCBI EST database. (We excluded the Hon genes from this analysis because they are quite highly expressed.) The distribution of the number of ESTs per gene is shown in Table II. The average number of EST sequences recognizing an FLC was 18. Eighty-four percent of the FLCs were represented by five or more EST sequences, while only two FLCs were unrepresented by ESTs. However, since many of the FLCs used in this study were originally identified via EST sequences, this collection may represent a biased assessment of the frequency of maize FLCs represented by ESTs.
Table II.
Frequency of hits for full-length cDNA sequences
| Gene | Accession No.a | Length | GSS Hitsb | GSS Clones Hitb | HC Clone Hitsb | MF Clone Hitsb | RC Clone Hitsb | HC GSS Hitsb | MF GSS Hitsb | RC GSS Hitsb | RM GSS Hitsb | Promoter Extensionc | EST Hitsd | Genomic Sequencee | Complete Gene Familiesf |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| brd103 | AF545811 | 4,089 | 27 | 17 | 9 | 6 | 2 | 15 | 10 | 2 | 0 | N.A. | 10 | ||
| cha101 | AF461813 | 2,643 | 20 | 10 | 3 | 7 | 0 | 5 | 12 | 0 | 3 | N.A. | 3 | ||
| crd101 | AF527609 | 1,436 | 22 | 13 | 5 | 7 | 1 | 8 | 13 | 1 | 0 | 256 | 20 | 1,902 | |
| dmt101 | AY093415 | 4,624 | 41 | 27 | 13 | 11 | 3 | 19 | 19 | 3 | 0 | 120 | 44 | 7,955 | # |
| dmt102 | AY027539 | 3,165 | 34 | 24 | 6 | 18 | 0 | 7 | 27 | 0 | 0 | 1,382 | 31 | 13,749 | # |
| dmt103 | AF242320 | 2,365 | 23 | 13 | 6 | 7 | 0 | 11 | 12 | 0 | 0 | 769 | 9 | 4,841 | # |
| dmt104 | AY029557 | 1,454 | 6 | 4 | 3 | 1 | 0 | 4 | 2 | 0 | 0 | 0 | 6 | # | |
| dmt105 | AY093416 | 2,996 | 17 | 9 | 4 | 5 | 0 | 7 | 10 | 0 | 0 | 0 | 11 | 11,009 | # |
| dmt106 | AF527610 | 2,197 | 15 | 9 | 3 | 5 | 1 | 6 | 8 | 1 | 0 | 230 | 3 | # | |
| dmt107 | CA399729.1* | 1,903 | 8 | 6 | 4 | 2 | 0 | 6 | 2 | 0 | 0 | 1,144 | 6 | 5,788 | # |
| ep101 | AF443599 | 1,897 | 12 | 10 | 6 | 4 | 0 | 7 | 5 | 0 | 0 | 0 | 3 | ||
| fie101 | AY061964 | 1,763 | 17 | 10 | 5 | 3 | 2 | 10 | 5 | 2 | 0 | N.A. | 2 | 4,709 | # |
| fie102 | AY061965. | 1,686 | 21 | 14 | 4 | 9 | 1 | 6 | 13 | 1 | 1 | N.A. | 8 | 3,437 | # |
| gtc102 | AF545812 | 3,505 | 10 | 5 | 2 | 3 | 0 | 4 | 5 | 1 | 0 | N.A. | 24 | ||
| gte102 | AY232822 | 2,865 | 16 | 5 | 1 | 4 | 0 | 1 | 8 | 0 | 7 | N.A. | 10 | ||
| hag101 | AF440227 | 1,863 | 23 | 11 | 6 | 5 | 0 | 9 | 10 | 0 | 4 | N.A. | 6 | ||
| hag102 | AY093417 | 1,515 | 19 | 13 | 9 | 3 | 1 | 11 | 3 | 2 | 3 | N.A. | 7 | ||
| hag104 | AY122274 | 1,757 | 21 | 16 | 11 | 5 | 0 | 14 | 7 | 0 | 0 | N.A. | 10 | ||
| hda101 | AF384032 | 1,906 | 25 | 19 | 6 | 13 | 0 | 6 | 19 | 0 | 0 | N.A. | 31 | ||
| hda102 | AF440228 | 1,694 | 6 | 4 | 3 | 1 | 0 | 4 | 2 | 0 | 0 | N.A. | 13 | ||
| hda108 | AF440226 | 1,662 | 11 | 6 | 4 | 2 | 0 | 6 | 4 | 0 | 1 | N.A. | 23 | ||
| hda110 | AF527611 | 2,500 | 12 | 10 | 4 | 6 | 0 | 5 | 7 | 0 | 0 | N.A. | 14 | ||
| hdt101 | AF384033 | 1,124 | 5 | 5 | 5 | 0 | 0 | 5 | 0 | 0 | 0 | 2,006 | 6 | # | |
| hdt102 | AF254072.1 | 1,222 | 6 | 4 | 3 | 1 | 0 | 5 | 1 | 0 | 0 | 0 | 48 | # | |
| hdt103 | U82815.1 | 1,249 | 11 | 8 | 4 | 4 | 0 | 6 | 5 | 0 | 0 | 526 | 41 | # | |
| hdt104 | AR168371.1 | 1,286 | 16 | 12 | 8 | 4 | 0 | 9 | 7 | 0 | 0 | 201 | 5 | # | |
| hon101 | AF527615 | 780 | 20 | 10 | 4 | 6 | 0 | 6 | 11 | 0 | 3 | N.A. | N.A. | ||
| hon102 | AF461814 | 991 | 16 | 5 | 1 | 4 | 0 | 1 | 6 | 0 | 9 | N.A. | N.A. | ||
| hon103 | X57077.1 | 1,157 | 14 | 11 | 4 | 6 | 1 | 4 | 8 | 2 | 0 | N.A. | N.A. | ||
| hon104 | AI881474* | 1,034 | 11 | 6 | 0 | 6 | 0 | 0 | 11 | 0 | 0 | N.A. | N.A. | ||
| hon105 | AF291748.1 | 1,059 | 15 | 9 | 1 | 8 | 0 | 1 | 14 | 0 | 0 | N.A. | N.A. | ||
| hon107 | BM335827.1* | 1,212 | 17 | 11 | 3 | 6 | 2 | 5 | 9 | 2 | 1 | N.A. | N.A. | ||
| hon108 | AF461815 | 1,003 | 19 | 15 | 7 | 7 | 1 | 8 | 10 | 1 | 0 | N.A. | N.A. | ||
| hon110 | BI245355.1* | 1,149 | 6 | 3 | 0 | 3 | 0 | 0 | 4 | 0 | 2 | N.A. | N.A. | ||
| hxa102 | AJ430205.1 | 2,350 | 22 | 13 | 5 | 8 | 0 | 7 | 15 | 0 | 0 | 1,477 | 15 | # | |
| hxa103 | AY100479 | 2,169 | 27 | 17 | 9 | 8 | 0 | 15 | 12 | 0 | 0 | 703 | 10 | # | |
| mbd101 | AY029556 | 818 | 9 | 5 | 2 | 3 | 0 | 3 | 6 | 0 | 0 | 377 | 20 | # | |
| mbd105 | AY029558 | 1,767 | 7 | 3 | 0 | 2 | 1 | 0 | 2 | 1 | 4 | 0 | 33 | # | |
| mbd106 | AY029559 | 1,690 | 7 | 6 | 0 | 4 | 2 | 0 | 5 | 2 | 0 | 433 | 33 | # | |
| mbd108 | AY029560 | 1,421 | 14 | 11 | 2 | 8 | 1 | 2 | 10 | 1 | 1 | 462 | 21 | # | |
| mbd109 | AF527618 | 1,667 | 3 | 2 | 1 | 1 | 0 | 1 | 2 | 0 | 0 | 0 | 13 | ||
| mbd111 | AY029561 | 1,442 | 8 | 6 | 3 | 3 | 0 | 4 | 3 | 0 | 1 | 2,087 | 25 | # | |
| mbd113 | BG316880.1* | 1,009 | 4 | 3 | 2 | 1 | 2 | 2 | 0 | 0 | 234 | 18 | |||
| mbd115 | AI065476.1* | 1,539 | 13 | 10 | 2 | 7 | 1 | 2 | 10 | 1 | 0 | 583 | 41 | ||
| mbd120 | CC407233.1* | 637 | 10 | 5 | 1 | 4 | 0 | 2 | 7 | 0 | 1 | 77 | 0 | 2,652 | # |
| nfa101 | AY232823 | 1,327 | 12 | 8 | 3 | 4 | 1 | 5 | 6 | 1 | 0 | N.A. | 49 | ||
| nfa102 | AY100480 | 1,291 | 12 | 8 | 5 | 1 | 2 | 8 | 2 | 2 | 0 | N.A. | 24 | ||
| nfa103 | AF384035 | 958 | 11 | 8 | 4 | 4 | 0 | 6 | 5 | 0 | 0 | N.A. | 34 | ||
| nfa104 | AF384036 | 907 | 11 | 8 | 2 | 4 | 2 | 3 | 6 | 2 | 0 | N.A. | 23 | ||
| nfc101 | AA979903* | 1,768 | 8 | 7 | 1 | 6 | 0 | 2 | 6 | 0 | 0 | 585 | 29 | ||
| nfc102 | AF384037 | 1,808 | 20 | 16 | 11 | 5 | 0 | 13 | 7 | 0 | 0 | 1,576 | 28 | ||
| nfc103 | AF440219 | 1,595 | 22 | 16 | 8 | 8 | 0 | 10 | 12 | 0 | 0 | 1,307 | 19 | ||
| nfc104 | AY093418 | 1,402 | 12 | 9 | 6 | 2 | 1 | 8 | 3 | 1 | 0 | 580 | 10 | ||
| nfd101 | AF527616 | 715 | 15 | 10 | 3 | 7 | 0 | 4 | 11 | 0 | 0 | N.A. | 48 | ||
| nfd102 | AJ006708.1 | 665 | 9 | 6 | 1 | 5 | 0 | 1 | 6 | 0 | 2 | N.A. | 23 | ||
| nfd103 | AF531431 | 887 | 10 | 4 | 1 | 3 | 0 | 1 | 6 | 0 | 3 | N.A. | 50 | ||
| nfd104 | AF440222.1 | 777 | 7 | 6 | 3 | 3 | 0 | 3 | 4 | 0 | 0 | N.A. | 48 | ||
| nfd105 | Y08298.1 | 417 | 7 | 4 | 1 | 3 | 0 | 1 | 5 | 0 | 1 | N.A. | 24 | ||
| nfd106 | AF527617 | 625 | 14 | 9 | 1 | 8 | 0 | 1 | 13 | 0 | 0 | N.A. | 46 | ||
| nfd107 | AI674006* | 742 | 7 | 4 | 1 | 3 | 0 | 1 | 3 | 0 | 3 | N.A. | 12 | ||
| sdg102 | AY122273 | 1,792 | 21 | 12 | 11 | 1 | 0 | 13 | 2 | 0 | 6 | N.A. | 4 | ||
| sdg104 | AY122272 | 2,930 | 8 | 5 | 0 | 3 | 2 | 0 | 5 | 2 | 1 | N.A. | 5 | ||
| sdg105 | AY093419 | 2,487 | 17 | 12 | 8 | 4 | 0 | 11 | 6 | 0 | 0 | N.A. | 25 | ||
| sdg110 | AF545814 | 1,460 | 10 | 6 | 2 | 4 | 0 | 3 | 7 | 0 | 0 | N.A. | 5 | ||
| sdg111 | AY187718 | 1,986 | 11 | 7 | 3 | 4 | 0 | 4 | 7 | 0 | 0 | N.A. | 14 | ||
| sdg113 | AF545813 | 2,428 | 8 | 6 | 6 | 0 | 0 | 8 | 0 | 0 | 0 | N.A. | 1 | ||
| sdg117 | AY187719 | 4,666 | 10 | 8 | 5 | 1 | 2 | 6 | 2 | 2 | 0 | N.A. | 11 | ||
| sdg118 | AY122271 | 2,180 | 20 | 16 | 7 | 8 | 1 | 9 | 9 | 1 | 1 | N.A. | 1 | ||
| sdg123 | AY172976 | 1,176 | 12 | 8 | 8 | 0 | 0 | 12 | 0 | 0 | 0 | N.A. | 22 | ||
| sdg124 | AF443596 | 3,170 | 19 | 13 | 7 | 6 | 0 | 9 | 9 | 0 | 1 | N.A. | 8 | 10,271 | # |
| sdg125 | AF443597 | 3,026 | 30 | 20 | 8 | 12 | 0 | 10 | 19 | 0 | 1 | N.A. | 1 | 7,483 | # |
| sdg126 | AF443598 | 3,133 | 14 | 7 | 4 | 3 | 0 | 6 | 6 | 0 | 2 | N.A. | 3 | 11,620 | # |
| sdg130 | AF466646 | 1,233 | 40 | 34 | 21 | 11 | 2 | 26 | 12 | 2 | 0 | N.A. | 0 | 7,099 | |
| sgb101 | AF384038 | 813 | 13 | 9 | 5 | 4 | 0 | 6 | 7 | 0 | 0 | 2,226 | 15 | # | |
| sgb102 | AF384039 | 762 | 17 | 10 | 3 | 5 | 2 | 5 | 6 | 2 | 4 | 1,625 | 19 | # | |
| sgb103 | BE510463.1* | 973 | 15 | 8 | 4 | 4 | 0 | 5 | 7 | 0 | 3 | 1,387 | 22 | # | |
| srt101 | AF384034 | 1,956 | 21 | 16 | 8 | 6 | 2 | 10 | 8 | 2 | 1 | 540 | 15 | # | |
| vef101 | AY232824 | 2,195 | 19 | 14 | 8 | 4 | 2 | 12 | 5 | 2 | 0 | 416 | 13 | ||
| Average | 1,737.3 | 14.9744 | 9.858974 | 4.5 | 4.8 | 0.5 | 6.2 | 7.4 | 0.5 | 0.9 | 706.3 | 18.2 |
For all sequences marked by an asterisk, there is not an FLC available at GenBank. For these genes, the FLC is based upon an EST contig and the assembly is available at www.chromDB.org. The accession numbers for these genes are just one of the EST sequences from the contig.
The number of HC, MF, random clone (RC), and RM or total number of GSS clones and sequences corresponding to each gene is listed.
The base pair of promoter sequence (any sequence upstream of the ATG) is indicated. For any sequences that were tested, either 0 or the base-pair length is reported; for the sequences that were not tested, a value of N.A. is reported.
The number of EST sequences matching each gene is indicated.
The length, in base pairs, for each gene that was used for performing searches against the full-length genomic sequence is indicated.
The genes marked by a # were a part of the subset of genes for which there is evidence that all genes in this gene family were included in our dataset.
Of the 122,606 bp represented in the 70 FLCs, 89,402 bp were covered by EST sequence, indicating 72.9% coverage. Assembling contigs of the EST sequences that aligned to the FLCs showed that 40% of the 70 FLCs could be represented by a single contiguous sequence while the remaining 41 FLCs contained one or more regions of the coding sequence that were not represented by EST sequence. Assembly of all the ESTs that aligned to the 70 FLCs resulted in 99 nonoverlapping contigs. Therefore, on average, each FLC from our sample dataset of 70 genes is represented by 1.41 EST contigs. This statistic indicates that in many cases multiple EST contigs from assemblies such as the TIGR Gene Index actually represent a single gene. Assuming that our set of 70 FLCs is representative of all maize genes represented by ESTs in terms of size, distribution, and expression suggests that the 56,364 maize EST clusters/singletons in the current TIGR gene index may actually represent approximately 40,000 genes.
Utility of GSSs for Tagging Maize Genes
Results of alignments of the GSSs with the 78 FLCs are presented in Table II. We attempted to identify any systematic bias within the experimental data sets that would tend to over- or underrepresent the true number of GSSs per gene. To examine this, we checked that each GSS was uniquely assigned to a single gene. Five GSSs (out of 1,167) were found to be assigned to two closely related genes: BZ375290 (Nfd103-Nfd107), BZ686390 (Hdt101-Hdt104), BZ753827 (Hdt101-Hdt104), CG288034 (Nfa103-Nfa104), and CG290386 (Hxa102-Hxa103). Our data set has multiple examples of closely related genes (>90% nucleotide identity); however, the finding that 99.6% of GSSs could be unambiguously assigned suggests that incorrect assignment of a GSS to a parologous gene was an uncommon occurrence within our dataset. Furthermore, comparisons of the rate of gene tagging among members of the subset of FLCs/genes for which we had identified all gene family members, against the rate for gene tagging among all FLCs in our study, revealed no obvious difference (data not shown).
A total of 50,877 and 3,480 GSSs used in our analysis were derived from two randomly sequenced small-insert libraries (ZM_3.0_4.0_KB from TIGR and maize random small-insert library from DuPont, respectively; Meyers et al., 2001), and 252,138 GSSs were derived from two BAC end libraries (J. Messing, unpublished data; pgir.rutgers.edu). The BAC end sequences are not randomly placed throughout the genome since they represent sequences near the HindIII (ZMMBBb) and EcoRI (ZMMBBc) restriction sites used to generate the BAC inserts. However, due to the fact that the frequency of hits in the BAC end libraries were very similar to the frequency of hits in the random small-insert libraries (Table I), these sequences were all considered random sequences. Forty-two of the small-insert/BAC end GSSs (from 39 clones) were found in the 78 query sequences. Figure 1A shows the distribution of the number of randomly sequenced GSSs per FLC. Of the 42 randomly sequenced GSSs, 32 are derived from the two BAC end libraries and tag 22 of the 78 FLCs. Consequently, 22 of 78 (28%) of our FLCs have the potential to be positioned on a BAC-based maize physical map.
Figure 1.

Distribution of the number of GSSs corresponding to our 78 FLCs. This figure describes the distribution and numbers of genes hit by the different types of GSSs but, due to different numbers of reads for the different libraries, is not a measure of relative effectiveness. A, The total number of randomly sequenced GSSs and the breakdown between BAC end and small-insert random libraries is shown. The majority of FLCs had no alignments with BAC end or small-insert random library GSSs, and very few FLCs had multiple randomly sequenced GSSs. B, The number of RM GSSs aligning to the 78 FLCs in the dataset. The majority of FLCs were not identified by an RM GSS. However, there were several FLCs with a relatively high number of RM GSSs, which may reflect insertion site preferences for the Mutator transposable element. C, The number of GSSs per FLC for both the MF- and HC-selected libraries. D, The sorted values for the number of GSSs from the MF- and HC-selected libraries per kilobase of coding region used to perform the searches. There are slightly more MF hits (most likely due to the higher number of MF sequences deposited at GenBank). However, the relatively higher density of MF hits per kilobase of coding region for some FLCs may reflect a propensity for the MF to capture certain FLCs at a higher rate. E, The total number of GSSs per FLC. Every FLC had at least three GSSs with a maximum of 41 GSSs. F, The sorted values for the total number of GSSs per kilobase of coding region used to perform the searches.
A total of 178,125 RM sequences, each corresponding to the insertion site of a transgenic RM element, have been isolated during the course of the maize gene discovery project led by Virginia Walbot (www.mutransposon.org; Raizada, 2003). Twenty-seven of our 78 FLCs have at least some portion of their length represented within 70 RM sequences from 46 individual clones. Figure 1B shows the number of RM sequences per FLC. Like the number of genes tagged by small-insert or BAC end GSS, the number of genes tagged by RM GSSs was low. However, 7 of the 28 FLCs tagged by RM sequences are tagged by multiple RM sequences. This is likely to reflect either a germinal insertion event or a Mu insertion site preference.
A total of 481 HC sequences, representing 353 individual HC clones, align to 73 of the 78 FLCs. On average, each gene was represented by 6.2 HC GSSs, which amounts to an average of 3.81 GSSs per kb of coding sequence. A total of 574 MF GSSs, derived from 376 clones, align to 75 of the 78 FLCs. On average, each gene hit by an MF GSS was represented by 7.4 MF sequences, i.e. there was an average of 5.1 GSSs per kb of coding sequence. Figure 1, C and D, shows the hit distribution of the HC and MF GSSs per FLC and the range of values for the number of GSSs per kb of FLC. The frequency of tagging an FLC by either an HC or MF GSS is 0.108% and 0.098%, respectively. Therefore, assuming our test set of 78 full-length maize genes is a faithful representation of the genes in the maize genome in terms of sequence composition, length, and distribution, the proportion of maize genes that have been tagged by MF with 95% confidence is 0.94 ± 0.05 and the proportion of maize genes tagged by an HC sequence is 0.96 ± 0.04 (see “Materials and Methods”). It should be noted that the combination of MF and HC sequences tags all 78 of the sequence set.
The total collection of 1,518,959 maize GSSs analyzed contains 1,167 sequences corresponding to parts of the 78 FLCs used in this study (0.077% of the available GSSs correspond to one of the 78 FLCs). Every FLC in the dataset was tagged by at least three GSSs, and 97% of FLCs had five or more GSS hits, while 76% of the FLCs had at least 10 GSS hits (Fig. 2E). The average gene within our dataset was 1,743 bp in length and tagged by 15.0 GSSs. Table I shows the normalized frequency for gene discovery for the different types of GSS libraries. The frequency for gene discovery in randomly sequenced clones was normalized to a value of 1. RM GSSs identified our FLCs at a 2.9-fold higher rate than random sequencing, while HC selection and MF identified the FLCs at a 7- to 8-fold higher rate than random sequencing (Table I). The EST sequencing projects were 24-fold more likely to identify these FLCs than random sequencing, but since most of these genes were initially discovered within EST libraries, we are unable to relate our observed EST hit frequencies to that of an average maize gene.
Figure 2.

Coverage of the 78 FLC sequences by EST and GSS sequencing. A, The percent of nucleotides within the FLC or genomic sequences used for the BLAST searches that are represented by each subset of sequences. The GSSs represent approximately 75% of the base pairs used to perform the FLC BLAST searches. The overlap between the MF and HC coverage is indicated in B. A total of 101,987 bp of FLC sequence (out of 135,510 bp) was covered. The overlap between the MF and HC GSS coverage is 36.9%. C and D, The distribution of positions that individual GSSs cover within those FLC sequences that they align to. The RM GSSs display a significant bias toward the 5′ end of FLC sequences. Interestingly, the MF GSSs (D) also show a bias toward the 5′ end of FLC sequences.
Comparison of Results from FLC with Genomic Searches
For 12 of the 78 FLCs we had also determined the full-length genomic sequence. Consequently, the ability of different sequencing methods to capture noncoding regions of the complete genes associated with 12 of the FLC sequences could be evaluated. Comparisons of the genes and the GSSs was performed using BLASTN, and the GSSs recognizing these gene sequences were catalogued and compared to the GSSs that were found using the FLCs. All 296 GSSs found by searches using the FLCs were also found by using the full-length genomic sequences as the query. However, the full-length genomic sequences identified another 103 GSSs that were not detected by the FLCs. Inspection of 15 of these 103 GSSs revealed that these GSSs were present entirely within introns, while others spanned exon/intron junctions and simply matched too small a region of the cDNA to be considered a valid match by our original criteria.
Utility of the Maize GSSs for Providing Full Coverage of cDNA or Genomic Sequences
The BLAST searches using FLCs and genomic sequences revealed that the currently available GSSs do an excellent job of tagging maize genes. While the ability to tag a gene is quite useful to genomic studies, it is critical that the complete sequence of genes be elucidated during sequencing. The coverage of the 78 genes was tested using the alignments of the GSSs to the FLC and genomic sequences. The 12 full-length genomic sequences have a total length of 92,515 bp, of which 60,631 bp (65.5%) is covered by the GSS entries. The total length of the 78 FLC sequences is 135,510 bp, of which 102,028 bp (75.3%) is covered by the available GSSs. The extent of coverage of the FLCs and genomic sequences by each subset of GSSs is illustrated in Figure 2A. Extrapolation from this dataset would suggest that the currently available GSSs are sufficient for providing the sequence of 75% of the coding nucleotides in the maize genome.
We sought to determine the extent of coverage of the FLC sequences by GSS contigs. These contigs were determined based on the positions of alignment rather than a computational assembly and, thus, will be more permissive and longer than those assembled by an automated approach. A total of 165 GSS contigs representing the 78 FLCs were assembled. Fifty-seven of the 78 FLCs (73%) were represented by more than one, nonoverlapping GSS contig.
While the combination of MF and HC GSS data tag all 78 members of our set of genes, only 70/78 FLCs in our gene set are tagged by both MF and HC GSS reads. If this gene set is representative of the total maize gene set, then 90% of maize genes reside in the sequence space overlapped by MF and HC, while the remaining 10% are represented only by MF or HC sequences. It is unclear whether this is due to the fact that sampling of the maize genome by MF or HC is not complete or whether this reflects that MF and HC sample overlapping but nonidentical regions of the maize genome, as suggested by Whitelaw et al. (2003).
The number of nucleotides represented within the 78 FLC sequences is 135,501. Of this, 94,987 of the nucleotides are covered by MF and HC clones, with the observed proportions illustrated in Figure 2B. The distribution suggested that the portion of the genome sampled by HC and MF may be partially nonoverlapping. One possibility to examine whether or not MF and HC sample identical sequence space is to estimate what portion of the sequence space is expected to be common to both MF and HC, given a random sampling by both methods, if they sample identical sequence space. If MF and HC sample identical sequence space, then each method should be equivalent in its ability to sample a given nucleotide within that space. Therefore, one could envision that the probability distribution of the number of nucleotides selected by both HC and MF might be approximated by a hypergeometric distribution (Sincich et al., 2002).
This model predicts the proportion of nucleotides recovered by random selection by one method (HC) that would be expected to overlap with a similarly random selection by the second method (MF). One possible issue with this modeling is that, experimentally, nucleotides are not strictly independent of one another. Rather, nucleotides that are present within the same sequenced clone are dependent. To approximate the nonrandomness of nucleotides during the cloning procedure, and to simplify the reality that these sequencing reads came from filtered cloning of randomly sheared genomic DNA and thus may overlap at high density, we assumed both a read length of 720 bases and that the reads covered the sampled sequence space in a nonoverlapping fashion. The choice of 720-bp reads is consistent with the average read length of MF and HC sequences generated by the consortium for maize genomics (http://www.tigr.org/tdb/tgi/maize).
Under the aforementioned assumptions, 188 unit reads arranged end-to-end would be required to extend across the lengths of the 78 FLCs tested. The 94,987 nucleotides aligned to MF and HC sequences could likewise be represented within 132 reads, and the 61,805 and 63,842 nucleotides covered individually by MF and HC, respectively, can be represented by 86 and 89 reads, respectively. Furthermore, the number of reads corresponding to the fraction of nucleotides that are sampled by both MF and HC sequences is 43. It is currently unknown if the collection of MF- and HC-derived sequences will cover all of the nucleotides of the 78 test FLCs, or if there is some portion of these that are unavailable to either or both HC and MF cloning methods. In other words, it is not yet clear whether 94,987 nucleotides that MF + HC sample (hypothetically represented by 132 unit reads) of the possible 135,501 (hypothetically represented by 188 unit reads) represent the limits of coverage by both methods or simply reflect the current sequence depth. However, if MF and HC sample identical sequence space, then each method should be equivalent in its ability to sample a given nucleotide within that space, and any of the 132 reads sampled by both HC and MF could therefore be obtained by MF or HC alone. Under the above assumption, the probability distribution for the number of reads selected by both MF and HC is hypergeometric (Sincich et al., 2002). The nucleotides sampled by MF can be represented by 86 reads, the nucleotides sampled by HC can be represented by 89 reads, and frequency of sampling by MF is 0.65 (86/132). Assuming that HC and MF sample identical sequence space, then a random selection of 89 reads by HC from a pool of 132 should contain 57 reads (0.67 × 89) that were also identified by MF. The observed number of reads sampled by both HC and MF is 43, and the hypergeometric distribution (Sincich et al., 2002) predicts that the probability of obtaining 43 or fewer common reads, rather than the expected 58 common reads, is less than 1e−09. Thus, it is unlikely that HC and MF sample identical sequence space. It should be noted that when the above modeling is performed on single base sampling, the difference between observed and expected overlap is magnified, and the probability that the observed data is explained by sampling of the same gene space is greatly reduced. Although the above treatment is simply an approximation, the results are consistent with our experimental observations that suggest MF and HC are sampling an overlapping but nonidentical sequence space, and it is likely that MF- and HC-specific biases extend to the sequences within genes.
We also addressed the relative distribution of different types of GSSs within the gene length. The length of each FLC was normalized to a value of 100, and the position of the alignment for each EST, GSS, and contig on the normalized scale of 1 to 100 was determined (Fig. 2, C and D). The RM sequences tended to be located near the 5′ end of the gene (Fig. 2C), while the randomly sequenced GSSs displayed a more uniform distribution across the length of the gene. This is expected based on the observation that Mutator insertion sites tend to cluster near the 5′ UTRs of some genes (R. Meeley, personal communication; Dietrich et al., 2002). The apparent fluctuation of the random-clone curve is likely a product of the low hit frequencies observed for the random GSSs. The MF and HC GSSs exhibit a nonrandom distribution where coverage is greater at the gene ends. The MF sequences had a much higher level of coverage near the 5′ end of the gene. In general, the HC GSSs did not provide as high a level of coverage of the 5′ end of the genes, which may imply that within the maize genome, the 5′ portions of maize genes are in close proximity to repetitive sequence. Both HC and MF GSSs showed a slight increase in coverage for the 3′ end of the gene relative to the middle of the sequence.
Utility of the GSSs for Promoter Discovery
Many genome-wide expression studies aim to link regulatory responses in gene-expression levels to cis-acting sequence elements in gene promoters. To date, there is relatively little information about the 5′ cis-regulatory sequences of maize genes. While the EST sequences are a rich source of coding sequences, they do not provide information about the 5′ cis-regulatory sequences. The ability of the GSSs to provide genomic sequence 5′ to the translation start site was tested for a subset of 33 of the genes in our dataset. Iterative BLAST searches were performed to extend the upstream sequence for these 33 genes. For 26 of the 33 genes (78%), at least one GSS that covered the ATG start codon was available (Table II; Fig. 3). The average gene had 891 bp of sequence 5′ to the ATG start codon, while the median length of 5′ sequence was 586 bp. Seventeen of the 33 genes had at least 500 bp of upstream sequence, 10 of the 33 genes had at least 1 kb of upstream sequence, and the longest extension was 2.2 kb. This sequence is likely to contain 5′ UTRs and promoters. We attempted to determine how often a putative PolII promoter recognition sequences could be found using the Softberry TSSP package (Mount Kisco, NY; http://www.softberry.com; Shahmuradov et al., 2003), which uses characteristics of known factor binding sites to predict potential transcription start sites in plant DNA sequence. Putative promoters were predicted in 13 of the 26 upstream sequences assayed, and putative TATA boxes were identified in 9 (70%) of these. These results suggest that the currently available GSSs are sufficient to provide promoter sequences for a substantial portion of maize genes.
Figure 3.

Utility of GSSs for promoter identification. A subset of 33 FLCs (indicated in Table II) was used to perform searches to identify GSSs that could provide 5′ UTR and promoter sequences. Twenty-seven of the 33 FLCs had at least one GSS that overlapped the ATG start codon. A, The length (in base pairs) of UTR/promoter sequence for each FLC is shown. The average length was 894 bp. The sequence 5′ of the ATG start codon was analyzed for the presence of a putative promoter using Softberry TSSP software. The sequences for which a promoter was predicted are indicated by the presence of an asterisk.
DISCUSSION
In this study a set of well-characterized maize FLC sequences was used to assess the utility of the maize EST and GSS sequencing projects for tagging and sequencing maize genes. The FLCs used within this dataset display differing patterns and levels of expression, and, other than the fact that the majority of these genes were originally identified through EST sequences, there does not appear to be any evidence to suggest that these genes will not be representative of the maize genome as a whole. To confirm that these results were not over- or underestimating the number of GSSs/gene we determined the coverage for a subset of these genes with full-length genomic sequences and for a subset of genes from families in which all cross-hybridizing sequences had been identified were used to perform BLASTN searches. No evidence was found for a systematic over- or underrepresentation by querying with the full-length coding sequences. All 78 genes used as queries were tagged by multiple GSSs. The rate of gene discovery by the GSSs indicates that the HC and the MF sequences each tag about 95% of maize genes. Therefore, it is probable that the majority of genes within the maize genome have been identified by one of these two approaches. If our FLC set is representative of all maize FLCs, then approximately 50% of coding base pairs have been sequenced by either HC or MF approaches and approximately 75% of the base pairs are sequenced by the combined GSS approaches.
While the majority of maize genes have been identified by EST or GSS approaches, relatively few genes have been completely sequenced using these approaches. The average number of contigs/singletons per gene is 1.4 EST and 2.1 GSS contigs/singletons per gene. In our analysis, 41% of the FLCs were represented by multiple EST contigs, and 73% of the FLCs were represented by multiple GSS contigs. Therefore, further sequencing is necessary to finish the sequencing of the maize gene space and provide a single contiguous sequence for each gene. In addition to identifying coding sequences, the GSSs will also provide introns and promoter sequence information. The majority of genes tested had a GSS that covered the ATG start codon, and promoter sequences could be computationally predicted in half of these genes.
In our study we were also able to address the issue of whether the two major reduced representation GSS approaches used for maize, HC and MF, sequence identical or partially nonoverlapping portions of the maize genome. The finding that many genes were represented by both MF and HC sequences indicates that these two approaches often do overlap in the portion of the genome that they are sampling. Whitelaw et al. (2003) and Palmer et al. (2003) found that MF libraries often retain a significant proportion of LTR retrotransposon sequence, which is not highly represented in HC libraries. We used our 78 genes as a dataset representative of the entire maize gene space and determined the overlap between HC and MF sequences. Using two different approaches to test this distribution, we found that it is highly unlikely that HC and MF libraries sequence identical portions of the maize gene space. In addition, we also investigated the distribution of GSSs along the length of the genes in our dataset and found that MF and HC sequences have distinct distributions along the gene length, i.e. the MF sequences are more likely to provide sequence near the 5′ end of the gene than the HC sequences.
Our data suggest that the current efforts have been very successful toward sequencing the maize gene space. The majority of maize genes have been identified by EST and/or GSSs. However, most of these genes are only partially represented. Further efforts are necessary to provide more substantial coverage of the gene sequences and to begin to link the assembled GSS contigs to the physical and genetic map of maize.
MATERIALS AND METHODS
Sequences Used for BLAST Searches
Our analyses utilized three sets of nucleic acid sequences to perform BLAST searches at NCBI during the week of November 17, 2003. The first set of sequences was 78 full-length B73 cDNA sequences obtained by cDNA clone sequencing, RACE PCR to extend an EST clone, and reverse transcription (RT)-PCR of genomic sequences that cross-hybridize to a gene of interest (further sequence details, map positions, and expression profiles for many of these genes are available at the www.ChromDB.org Web site). Further analyses were performed on a subset of the 78 genes for which we had cloned and sequenced all members of the gene family. This allowed us to unambiguously attribute the GSS or EST sequences to one gene. In addition, the full-length genomic sequence for 13 of the 78 genes was available, and these sequences were used to verify that all sequences matching the cDNA also matched the genomic sequence. Table II lists the sequences, accession numbers, and relevant attributes for the genes used in this study.
BLAST Searches
All alignments were performed with BLAST version 2.2.6 available from the NCBI (Altschul et al., 1997), using the default parameters with the low-complexity filter off. BLASTN searches were performed between our collection of gene sequences and the maize (Zea mays) EST or GSSs available at NCBI. Only those alignments that extended for a minimum of 100 bp with 98% nucleotide identity were considered for the searches against the GSSs. The identity cutoff for alignments to EST sequences was set at 97% since many of the EST sequences are derived from genotypes other than B73 and maize has a relatively high polymorphism rate. These alignments were then viewed to ensure that they did not simply match a repetitive sequence (such as a MITE) and that they matched the predicted exon-intron structure. All ESTs or GSSs that passed these criteria were then catalogued to record information about the accession number, clone name, and position of the alignment. Contigs were manually assembled by analyzing the positions of each alignment and assigning each sequence to a contig based on whether it overlapped with another sequence. These contigs were defined by the gene they corresponded to and the position of the gene that they aligned to. Contigs were assembled for each type of GSS as well as for the total GSS and EST sequences. The overlap in coverage between different sets of sequences was then compared based on the positions of alignment for each gene.
Statistical Analysis of Frequency Predictions
If the proportion of maize genes sampled by HC or MF (p) is equivalent to the proportion of those genes within our sample set successfully tagged by MF or HC reads, then the 95% confidence interval for the population proportion of maize genes sampled by HC or MF is:
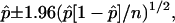 |
where p = tagged genes/total genes in sample (n; Sincich et al., 2002).
Probability Calculations Based on the Hypergeometric Distribution Model
The expected number of reads common to both MF and HC (see text) was calculated with a PERL script included in the supplemental material (available at www.plantphysiol.org). The expected number is given by the mean of the hypergeometric distribution. If the observed number is greater (or less) than the expected number, the accumulated probability associated with the departure from the expected mean was calculated.
The hypergeometric distribution probability function is f(x|A,B,n) = (combination(A,x) * combination(B,(n −x)))/combination((A + B),n), where x represents any possible integer in the interval of max{0, n− B} and min{n, A}; and combination(n1,n2) represents n1!/(n2! *(n1 − n2)!) (Sincich et al., 2002). The mean of a hypergeometric distribution is nA/(A + B). As stated (see text) the hypergeometric distribution models the number of nucleotides selected for by both MF and HC. Where the nucleotides selected by MF are represented by 86 reads, those selected by HC are represented 89 reads, the observed overlap is 43 reads, the total accessible reads are 132 reads; then, A = 89, B = 132 − 89 = 43, n = 86, x = 43. Because the actual overlap is 43, which is less than the expected observation number: nA/(A + B) = 58, the final probability was calculated as the sum of the probabilities for x = 43 to x = 1.
Supplementary Material
Acknowledgments
We thank a number of researchers who produced data that enabled this study. Peter Hermanson helped to perform and curate the BLAST searches for this study. Carolyn Napoli has performed the curation and discovery of many of the genes utilized for this study and has assembled this information at www.ChromDB.org. Shawn Kaeppler, Karen Cone, Vicki Chandler, Heidi Kaeppler, and Rich Jorgensen are principal investigators on the National Science Foundation-funded project DBI–9975930 who have been involved in the identification and studies of the chromatin genes used for this study. The authors are very appreciative of all of the projects and personnel involved in maize genome sequencing, including the V. Walbot, J. Messing, C. Shubert, Genoplante, P. Schnable, and TIGR projects. Shawn Kaeppler, Peter Tiffin, and Karen Cone read the manuscript and provided valuable suggestions and input. In addition, Robert Meeley provided valuable information about the observed distribution of Mu insertion sites. We are also very thankful for the valuable comments and suggestions provided by several anonymous reviewers.
This work was supported by the National Science Foundation (grant no. DBI–0227310 to N.M.S. and grant no. DBI–0221536 to W.B.B.).
The online version of this article contains Web-only data.
References
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796–815 [DOI] [PubMed] [Google Scholar]
- Arumuganathan K, Earle ED (1991) Nuclear DNA content of some important plant species. Plant Mol Biol 42: 251–269 [Google Scholar]
- Bennetzen JL (1996) The contributions of retroelements to plant genome organization, function and evolution. Trends Microbiol 4: 347–353 [DOI] [PubMed] [Google Scholar]
- Bennetzen JL, SanMiguel P, Chen M, Tikhonov A, Francki M, Avramova Z (1998) Grass genomes. Proc Natl Acad Sci USA 95: 1975–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burr B, Burr FA, Thompson KH, Albertson MC, Stuber CW (1988) Gene mapping with recombinant inbreds in maize. Genetics 118: 519–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- C. elegans Sequencing Consortium (1998) Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282: 2012–2018 [DOI] [PubMed] [Google Scholar]
- Dietrich CR, Cui F, Packila ML, Li J, Ashlock DA, Nikolau BJ, Schnable PS (2002) Maize Mu transposons are targeted to the 5′ untranslated region of the gl8 gene and sequences flanking Mu target-site duplications exhibit nonrandom nucleotide composition throughout the genome. Genetics 160: 697–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes J, Brendel V, Gai X, Lal S, Chandler VL, Elumalai RP, Galbraith DW, Pierson EA, Walbot V (2002) Comparison of RNA expression profiles based on maize-expressed sequence tag frequency analysis and micro-array hybridization. Plant Physiol 128: 896–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, et al (1996) Life with 6000 genes. Science 274: 546, 563–567 [DOI] [PubMed] [Google Scholar]
- Meyers BC, Tingey SV, Morgante M (2001) Abundance, distribution, and transcriptional activity of repetitive elements in the maize genome. Genome Res 11: 1660–1676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ, Kravitz SA, Mobarry CM, Reinert KH, Remington KA, et al (2000) A whole-genome assembly of Drosophila. Science 287: 2196–2204 [DOI] [PubMed] [Google Scholar]
- Palmer LE, Rabinowicz PD, O'Shaughnessy AL, Balija VS, Nascimento LU, Dike S, de la Bastide M, Martienssen RA, McCombie WR (2003) Maize genome sequencing by methylation filtration. Science 302: 2115–2117 [DOI] [PubMed] [Google Scholar]
- Peterson DG, Schulze SR, Sciara EB, Lee SA, Bowers JE, Nagel A, Jiang N, Tibbitts DC, Wessler SR, Paterson AH (2002) Integration of Cot analysis, DNA cloning, and high-throughput sequencing facilitates genome characterization and gene discovery. Genome Res 12: 795–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowicz PD, Schutz K, Dedhia N, Yordan C, Parnell LD, Stein L, McCombie WR, Martienssen RA (1999) Differential methylation of genes and retrotransposons facilitates shotgun sequencing of the maize genome. Nat Genet 23: 305–308 [DOI] [PubMed] [Google Scholar]
- Raizada MN (2003) RescueMu protocols for maize functional genomics. Methods Mol Biol 236: 37–58 [DOI] [PubMed] [Google Scholar]
- Raizada MN, Nan GL, Walbot V (2001) Somatic and germinal mobility of the RescueMu transposon in transgenic maize. Plant Cell 13: 1587–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- SanMiguel P, Tikhonov A, Jin YK, Motchoulskaia N, Zakharov D, Melake-Berhan A, Springer PS, Edwards KJ, Lee M, Avramova Z, et al (1996) Nested retrotransposons in the intergenic regions of the maize genome. Science 274: 765–768 [DOI] [PubMed] [Google Scholar]
- Shahmuradov IA, Gammerman AJ, Hancock JM, Bramley PM, Solovyev VV (2003) PlantProm: a database of plant promoter sequences. Nucleic Acids Res 31: 114–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sincich T, Levine DM, Stephan D (2002) Practical Statistics by Example, Ed 2. Prentice Hall, Upper Saddle River, NJ, pp 1–798
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al (2001) The sequence of the human genome. Science 291: 1304–1351 [DOI] [PubMed] [Google Scholar]
- Walbot V, Petrov DA (2001) Gene galaxies in the maize genome. Proc Natl Acad Sci USA 98: 8163–8164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitelaw CA, Barbazuk WB, Pertea G, Chan AP, Cheung, F, Lee Y, Zheng L, van Heeringen S, Karamycheva S, Bennetzen JL, et al (2003) Enrichment of gene-encoding sequences in maize by genome filtration. Science 302: 2118–2120 [DOI] [PubMed] [Google Scholar]
- Yuan Y, SanMiguel PJ, Bennetzen JL (2002) Methylation-spanning linker libraries link gene-rich regions and identify epigenetic boundaries in Zea mays. Genome Res 12: 1345–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, SanMiguel PJ, Bennetzen JL (2003) High-Cot sequence analysis of the maize genome. Plant J 34: 249–255 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.