

Abstract
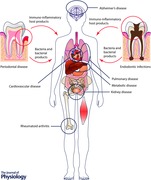
The oral microbiome is established within a few minutes after birth and consists of stable multi‐species communities that engage in a dynamic equilibrium with the host immune system. Dental caries, endodontic infections and periodontal diseases are bacterially driven diseases that are caused by dysbiotic microbiomes. Over a century ago, the focal infection theory implicated these infections in the aetiology of several systemic diseases, ranging from arthritis to neurodegenerative diseases. However, a lack of concrete evidence, combined with the urgency with which clinicians embraced this approach without regard for appropriate case selection, led to its demise within 30 years. In the last decade of the 20th century, the concept of periodontal medicine was introduced to explain the correlations that were being observed between periodontitis and cardiovascular disease, rheumatoid arthritis, Alzheimer's disease, pulmonary disease, pre‐term delivery of low birth weight infants and metabolic disease. It was proposed that periodontal pathobionts played a causal role in the initiating or exacerbating certain diseases either by direct invasion or by stimulating a florid immune‐inflammatory response that extended into the systemic circulation. This review will examine the strength of current evidence in establishing a causal link between oral pathobionts and systemic disease.
Keywords: focal infection, periodontal medicine, oral microbiome, oral pathobiont, periodontitis, systemic disease
The oral microbial ecosystem – real estate and habitats
In the early 1930s, Arthur Roy Clapham coined the word ‘ecosystem’ to describe a community that consisted of living organisms that interact with each other as a system and are linked through energy flow and nutritional and metabolic support (Blew, 1996). The oral cavity is home to arguably one of the most well studied ecosystems in the human body. This environment presents several habitats for both aerobic and anaerobic bacterial colonization: abiotic surfaces such as the tooth, dental implants and dental restorations, and biotic environments such as the subgingival crevice (space between the tooth and the gumline), keratinized mucosal surfaces on the dorsum of the tongue, hard palate and attached gingiva, and non‐keratinized epithelial surfaces on the buccal mucosa, tonsils and alveolar mucosa. The subgingival crevice provides 12 cm2 of surface area for bacterial colonization (Hartzell & Henrici, 1916), while both keratinized and non‐keratinized surfaces of the oral mucosa constitute a real estate of more than 200 cm2 (Collins & Dawes, 1987). Together with the tooth surfaces, there is 500 cm2 of space available for bacterial colonization. Bacteria colonize these niches within a few minutes of birth and co‐evolve with shifts in the host through two dentition states, concomitant changes in food habits, oral hygiene practices and lifestyle shifts.
Dysbiosis in this system underlies the aetiologies of some of the most common diseases to affect humans: caries, periodontal disease and endodontic infections. Over the past three decades, a robust body of evidence attesting to the systemic effects of these dysbiotic communities has burgeoned. However, as we examine the historical evolution of oral microbiology, it is seen that this idea had been in vogue during the turn of the 20th century as well. This review will summarize the historical as well as currently available evidence regarding the role played by the oral microbiome in causing disease in the rest of the human body.
The impact of the focal infection theory on medicine and dentistry
The focal infection theory (FIT) posits that bacteria and/or bacterial toxins and metabolic byproducts can enter the systemic circulation from a clinically asymptomatic localized lesion that contains pathogenic bacteria and translocate to distant parts, initiating disease in these organ systems. The resulting metastatic disease is chronic, but not infectious. Some examples of diseases that have been attributed to focal sepsis are gonococcal arthritis following gonorrhoeal infection, neuritis, myalgia, nephritis, osteomyelitis, emphysema, endocarditis, pneumonia, asthma, gastritis, pancreatitis, colitis, diabetes, goitre, thyroiditis and Hodgkin's disease (Roberts, 1921). The central belief of the focal infection theory is that injury occurs at a site distant from the site of infection; the focus of infection is usually unrecognizable or clinically unremarkable, as in infections of the tonsil, sinus, prostate, appendix, bladder, gall bladder and kidney; and the secondary disease occurs only in sites that
are susceptible to the bacterial species or toxins. This distinguishes it from true ‘infectious diseases’ such as cholera and typhoid, where the organ damage occurs secondary to the primary systemic infection.
While this theory has been in vogue since Hippocrates reported curing arthritis by extracting a tooth (Francke, 1973), the modern form experienced a period of popularity during the early 20th century, beginning with a lecture at McGill University by Dr William Hunter, a British physician who denounced the preservation of a carious tooth by building what he called ‘a veritable mausoleum of gold fillings, crowns and bridges over a mass of sepsis’ as the cause of a multitude of systemic diseases (Hunter, 1900). Although some investigators believe that his words were misquoted, and that he was referring to ill‐fitting crowns and dentures, influential physicians like Russell Cecil and Charles Mayo, who were significant thought leaders of the time, recommended that all teeth be extracted either to prevent or to treat any number of conditions ranging from allergy to schizophrenia, and thus earned the sobriquet ‘one hundred percenters’ (Dussault & Sheiham, 1982; Murray & Saunders, 2000). Together with tonsillectomy, full mouth extractions became routine treatment options for diverse conditions ranging from arthritis deformans to blindness (Billings, 1930; Bocca et al. 1989). Two events may have played a significant role in creating this generation of edentulous individuals. The first was an influential book by W. D. Miller in 1880 The Micro‐Organisms of the Human Mouth: The Local and General Diseases Which Are Caused by Them, in which he introduced the term ‘oral focal sepsis’ and recommended dental fillings or root canal therapy to treat tooth decay, which he said was a bacterial disease (Miller, 1880). The second was the development of the dental X‐ray, which revealed the presence of peri‐apical radiolucencies in asymptomatic teeth and periodontal bone loss. A novel research strategy called ‘reverse investigation’, where the research is instigated by a conclusion and is focused on gathering evidence to support this conclusion, was used to generate evidence to support this theory (Ingle et al. 2008).
Soon, the infected periodontal pocket drew attention as yet another, larger nidus of infection. It was held that teeth with ‘pyorrhoea’ (periodontitis) ‘shower bacteria into the blood stream’ even during everyday activities such as chewing or tooth brushing, and that these bacteria were identifiable in the circulation close to the source (peri‐apical veins) and in distal blood vessels (median basilic veins) following tooth extraction or chewing on hard candy (Fish, 1940). Rosenow proposed that oral bacteria or their toxins preferentially segregate to areas predominantly composed of mesenchymal tissues, notably joints, muscles and neuronal sheaths (Rosenow, 1930). He believed that their ‘unique functions of repair, regeneration and scavenging of waste products’ increased their susceptibility to bacteria and bacterial toxins. Rosenow proposed that certain pathogens demonstrated a predilection for specific target tissues (the theory of ‘elective localization or dissemination’) and that bacteria were capable of spontaneously changing to another species (transmutation). Transmutation was held to be the reason why results could not be replicated between researchers and labs. Thus, through case reports of diseases being identified in individuals with infected root canals or teeth with ‘pyorrhoea’ (periodontitis), animal experiments that demonstrated induction of lesions of the ‘heart muscle and endocardium, lesions of the kidney, focal and diffuse, lesions of the adventitia of the blood vessels, and iritis’ by ‘organisms taken from the dental path’ (Hartzell & Henrici, 1916), and selective or complete edentulation of subjects with arthritis and vascular disease, several researchers demonstrated the aetiological role of oral bacteria in systemic diseases (Klotz, 1913; Hartzell & Henrici, 1916).
However, it was soon apparent that the routine removal of teeth could not predictably cure circulatory, neurodegenerative, or kidney diseases. Patients demonstrated a worsening of arthritic symptoms and, not surprisingly, developed digestive complications following therapeutic edentulation (Cecil & Angevine, 1938; Vaizey & Clark‐Kennedy, 1939), and in many cases, were cured of their psychiatric ailments even in the absence of edentulation (Wessely, 2009; Shorter, 2011). As medical experimentation and animal model research grew more sophisticated, significant flaws were identified in Rosenow's, Hartzell and Henrici's, and Price's studies, notably the lack of controls and the massive doses of bacterial inoculum used. More importantly, a concerted effort by the clinical endodontic community to establish that root‐canal treatment resulted in resolution of dental and peri‐apical infection led to the demise of the focal infection theory. Thus, focal infection was disregarded as a scientific theory for several decades.
Periodontal medicine: resurrecting the focal infection theory?
The endodontic community has remained steadfast in its rejection of the infected root canal as a cause of distant, non‐infectious disease (Ingle et al. 2008). The position of the American Association of Endodontists (AAE) has been that (i) bacteraemia occurs as part of normal daily activity such as chewing and tooth brushing; (ii) there is no evidence on the inoculum size needed to generate a metastatic disease – the only consistency between the turn of the century animal studies (Rosenow, Price, Henrici and Hartzell) was that the inoculum sizes were unrealistically large; and (iii) dental extractions produce a larger circulatory bacterial load than endodontic therapy. The AAE does, however, recognize that untreated peri‐apical infections may cause distant disease by releasing bacteria and bacterial products into the circulation.
On the other hand, untreated periodontal disease has continued to be examined as a source of circulatory bacteria. This became especially important when the American Heart Association released a position paper on the role of oral streptococci in bacterial endocoarditis (Rammelkamp et al. 1957). The last decades of the 20th century saw the emergence of new techniques for bacterial identification and classification, especially oral microorganisms. Non‐targeted molecular assays such as 16S sequencing revealed the presence of novel and hitherto unsuspected organisms in the oral cavity (Paster et al. 2001; Kumar et al. 2003), while innovations in culturing and microscopical approaches allowed the identification of uncommon phenotypes in known species (Beighton et al. 1991; Kell et al. 2015). Several systemic pathogens, ranging from respiratory pathobionts such as Hemophilus influenzae, Pseudomonas aeruginosa and Acinetobacter baumanii to gut pathogens such as Trophyrema whipplei (Liljemark et al. 1984; Zinkernagel et al. 2003; Persson et al. 2008; Kumar et al. 2011; Mason et al. 2014), have been identified in significant numbers in the periodontal pocket. Explorations of atheromatous plaques, knee implants, placenta, amniotic sac, the tracheobronchial tree, joint cavities and the pancreas have revealed the presence of periodontal pathogens, for example Porphyromonas gingivalis, Treponema denticola, Fusobacterium nucleatum and Campylobacter rectus, in these areas, especially in regions that were previously considered sterile (Bearfield et al. 2002; Leon et al. 2007; DiGiulio et al. 2008; Aagaard et al. 2014). These advances in microbiological methodologies and clinical techniques produced data that suggested that the oral cavity could indeed act as a reservoir of bacteria that might metastasize to distant sites in the body and cause disease in susceptible individuals. The World Workshop in Periodontics introduced the term ‘periodontal medicine’ in 1996 to describe the role played by periodontitis in exacerbating or initiating systemic diseases (Offenbacher, 1996). Thus, the last two decades have seen what may be considered a resurrection of the focal infection theory; however, investigators are using an abundance of caution in advocating therapy based on these links.
While several lines of evidence are emerging to suggest that periodontitis may be linked to osteoporosis, diabetes, atherosclerotic circulatory disease, rheumatoid arthritis, pregnancy‐related complications, pulmonary disorders, pancreatic cancer, chronic renal disease, obesity and Alzheimer's disease, there is little evidence at this point in time that oral bacteria or bacterially driven pathways play a role in all of these linkages. Therefore, in this review, we will focus only on studies that have examined the contributions of oral bacteria to periodontal‐systemic disease.
The microbial ecosystem in periodontal disease
The oral cavity is an open microbial ecosystem in that, at any given time, it is home to several allochthonous species (transient visitors) in addition to autochthonous members (stable colonizers). Together, over 20 billion organisms can be found in this environment (Loesche, 1982), representing nearly 700 different species (Aas et al. 2005). These organisms live in a state of dynamic equilibrium with the host immune system, a situation that is reflected as clinical health. When the micro‐environment changes, as a function of systemic antibiotics that negate the protective influence of commensals, reduced oxygen tension due to increase in biofilm thickness, altered host defences, or nutritional, metabolic and structural stresses within the ecosystem, a dysbiosis occurs in the indigenous microbiome, reducing the abundance of the commensal population and creating a pathogen‐rich ecosystem (Socransky & Haffajee, 2005). The florid immune‐inflammatory response to this pathogenic colonization leads to destruction of the attachment between the tooth and the gingiva, and loss of structures that anchor the tooth to the jawbone. Together, these two events result in a deepening of the gingival sulcus, which is the space between the tooth and the gingiva (Listgarten, 1986). This inflamed sulcus, now called a periodontal pocket, provides an anaerobic, protein‐ and haem‐rich, oxidant‐rich niche that promotes colonization by anaerobes, many of which are pathogenic to humans. At a conservative estimate, there are over 10 billion bacteria in 1 mg of dental plaque (Gibbons et al. 1964). Since these bacteria are packed into the space between the tooth and the sulcular epithelium, the breakdown of epithelial integrity caused by inflammation results in seeding of the systemic circulation with these pathogens when the biofilm is disrupted. The diseased periodontal pocket also contains significant levels of inflammatory mediators, especially those that mediate chronic inflammation. Tumour necrosis factor α, interleukins 1, 2 and 8, and prostaglandins can be released into the circulation from the diseased periodontium (Offenbacher et al. 1993; Hernichel‐Gorbach et al. 1994), and may contribute to systemic inflammation.
As was demonstrated during the turn of the 20th century, newer studies have confirmed that simple oral hygiene procedures can translocate bacteria, bacterial products, toxins and inflammatory products to other sites in the body (Carroll & Sebor, 1980; Baltch et al. 1988), especially in individuals with oral infections. For example, in children with extensive dental decay, the frequency of bacteraemia following tooth brushing has been reported to range from 17 to 40% (Roberts et al. 1997), 100% following dental extraction (Heimdahl et al. 1990), 70% after professional dental cleaning (Lofthus et al. 1991), 97% following injection of dental anaesthetics and 20% following root canal treatment (Debelian et al. 1995). In immunocompetent individuals, the transient bacteraemia is eliminated from the circulation. However, individuals with a compromised immune system, e.g. diabetics and people with upper respiratory disease, may not exhibit similar abilities to clear the systemic bacteraemia, rendering them more susceptible to disease. The disrupted immune‐inflammatory axes in these individuals may also result in profuse amounts of bacterial products (e.g. lipopolysaccharide and endotoxin) as well as host response mediators to be released into the circulation, triggering inflammatory responses in the target organs. Thus, the underlying pathophysiology of systemic diseases caused by periodontal infections may be metastatic infection, metastatic injury or metastatic inflammation.
Periodontal disease and pulmonary diseases
While pneumonia can be caused by infection with a bacterium, virus, fungus or parasite, the most common type is bacterial pneumonia. Typically, the lower respiratory tract is protected from microorganisms by the cough reflex, ciliary movement of the lining cells, and innate immune mediators (Levison, 1994), which are capable of dispersing salivary bacteria aspirated during sleep or from accidental swallowing. However, impairment of these defences (as in long‐term smoking, diabetes, chronic obstructive pulmonary disease or immunosuppression, and during intubation or prolonged post‐operative hospital stay) can result in nosocomial pneumonia (Toews, 1986; Sinclair & Evans, 1994). Cross‐sectional studies have demonstrated that in dentate patients, poor oral hygiene and non‐compliance with dental hygiene visits increase the risk for developing pneumonia, indicating that oral pathobionts may be a potential link between oral and lung diseases (Terpenning et al. 1993). Hospitalized subjects suffering from pneumonia have been shown to harbour the respiratory pathogens Klebsiella pneumoniae, Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus, Haemophilus influenzae and H. parainfluenzae (Scannapieco et al. 1992; Russell et al. 1999; Scannapieco, 2006), while periodontal pathogens, for example, P. gingivalis, F. nucleatum, Prevotella oralis, Campylobacter gracilis, Fusobacterium necrophorum and Aggregatibacter actinomycetemcomitans, have been identified in lung aspirates of subjects with pneumonia (Yuan et al. 1992; Zijlstra et al. 1992; Lorenz & Weiss, 1994; Shinzato & Saito, 1995). Furthermore, periodontal treatment and improved oral hygiene decreased the incidence of pneumonia in children and hospitalized adults (Yoneyama et al. 1996; Scannapieco & Binkley, 2012).
The studies reviewed above and several others have suggested that oral bacteria may cause respiratory diseases when (i) oral bacteria or respiratory pathogens from oral reservoirs are aspirated into the lower respiratory tract, (ii) salivary enzymes released during chronic periodontal disease or smoking modify the oral mucosa and lead to increased adhesion by respiratory pathogens, and/or (iii) circulating pro‐inflammatory cytokines released as a consequence of periodontal inflammation modify the respiratory mucosa (Li et al. 2000; Paju & Scannapieco, 2007).
The consensus report of the Joint European Federation of Periodontology/American Academy of Periodontology Workshop on Periodontitis and Systemic Diseases states that while there is insufficient evidence to date to infer causal relationships in most systemic diseases due to a paucity of prospective studies, it is highly likely that organisms originating in the oral microbiome can cause lung infections (Linden et al. 2013). A recent finding from the Women's Health Initiative Observational Study was that while periodontal disease was not independently associated with lung cancer in non‐smoking postmenopausal women, in smokers this risk was increased beyond what could be expected from the sum of the each effect separately (Mai et al. 2014).
Periodontal disease and cardiovascular diseases
Cardiovascular disease is an umbrella term that encompasses a range of conditions, from high blood pressure to acute myocardial infarction, angina and stroke. The central pathophysiology of these diverse diseases is the atheromatous plaque. The infection hypothesis of atherosclerosis sprang up in the early 19th century, when Gilbert and Lion identified that systemic inoculation of the bacterium Bacillus typhosus could induce fatty sclerosis in the aortal wall in rabbits (Nieto, 1998). Osler, who is credited with the infection hypothesis of atherosclerosis, listed major wear and tear, acute infections, ‘intoxications’ (smoking, diabetes mellitus, obesity) and high blood pressure as the four major causes (reviewed by Nieto, 1998). In the early 1970s, several studies demonstrated that viral infections could induce damage to the endothelium and activate acute inflammatory mediators. Together with formation of foam cells, these events could lead to creation of thrombi and unstable atheromatous plaques (Fabricant et al. 1978; Hajjar et al. 1986; Etingin et al. 1990; Laitinen et al. 1997). It has also been demonstrated that cross reactivity between bacterial heat‐shock proteins and human Hsp60 could initiate an autoimmune response that culminates in atherosclerosis (Wick et al. 1997; Kol et al. 1998).
While the role of oral haemolytic streptococci in the aetiology of subacute bacterial endocarditis was well established, the role of periodontal disease in its aetiopathogenesis was not as well known until the late 1990s, when correlations were observed between tooth loss and cardiovascular disease (Mattila et al. 1989; Mattila, 1993; Mattila et al. 1998). Since two infectious diseases, caries and periodontitis, can result in tooth loss, the relative contributions of these two oral infections were examined by Grau et al. (2004), who found that in subjects with periodontitis the risk for stroke was 400% greater than in those with caries. Desvarieux et al. (2003) found a correlation between intimal thickening of the carotid artery (a metric of atherosclerosis) and periodontal pathogens but not health‐compatible organisms in over 600 individuals. DNA from the oral pathogens Tannerella forsythia, F. nucleatum, Prevotella intermedia, Porphyromonas gingivalis and A. actinomycetemcomitans has been identified in carotid atheromas (Cairo et al. 2004), providing further evidence of bacterial translocation. Systemic antibody levels to periodontal pathogens correlated with the incidence of coronary heart disease and subclinical atherosclerosis (Beck et al. 2005; Pussinen et al. 2006) Animal studies on apolipoprotein E‐knockout (ApoE−/−) mice have demonstrated a role for dendritic cells in translocation of oral bacteria to the vasculature. Furthermore, treatment of chronic periodontitis was shown to decrease systemic markers of inflammation and improve endothelial dysfunction (Tonetti et al. 2007). The joint workshop of the European Federation of Periodontology and American Academy of Periodontology (EFP/AAP) concluded that while there is evidence to support the hypothesis that translocated oral microbiota may induce systemic inflammation that influences atherothrombogenesis, and that this biological mechanism is supported by in vitro experiments, animal models and clinical studies, ‘intervention trials to date are not adequate to draw further conclusions’ (Tonetti et al. 2013). The present consensus thus appears to support a temporal relationship between periodontal and cardiovascular diseases, with oral bacteria playing either a direct or an indirect role in disease causation.
Periodontal disease and pregnancy outcomes
The infection hypothesis of adverse pregnancy outcomes postulates that preterm delivery of a low birth‐weight infant may occur as a result of either a local or systemic maternal infection. In the early 1990s, bacterial vaginosis was shown to contribute to preterm delivery of a low birth‐weight infant (Kurki et al. 1992; Hillier et al. 1995). The dental community was already aware that the abundances of certain bacteria, notably the group of organisms collectively known as black pigmented bacteria, are higher in subgingival crevice during pregnancy, possibly because of the increased availability of oestradiol (Kornman & Loesche, 1980). Together, these two findings opened up several lines of research to examine the role of periodontal pathogens on neonatal health. The results have extremely equivocal (Mitchell‐Lewis et al. 2001; Sanchez et al. 2004; Dasanayake et al. 2005; Noack et al. 2005; Michalowicz et al. 2006; Offenbacher et al. 2006; Seymour et al. 2007; Novak et al. 2008). Pre‐term birth and low birth weight have been associated with high levels of Tannerella forsythia, Campylobacter rectus, Prevotella intermedia, Prevotella nigrescens and Porphyromonas gingivalis in maternal subgingival plaque (Mitchell‐Lewis et al. 2001; Sanchez et al. 2004; Offenbacher et al. 2006). Porphyromonas gingivalis has been detected in both amniotic fluid and subgingival plaque of 31% of women with threatened pre‐term labour (Leon et al. 2007). Recent evidence has identified F. nucleatum in the placental microbiome (Aagaard et al. 2014) and has implicated this organism in the aetiopathogenesis of pre‐term deliveries (Han et al. 2004). Two mechanisms have been proposed and tested to explain the correlation between adverse pregnancy outcomes and periodontal infection.
-
(i)
Intrauterine inflammation. It has been shown that periodontal bacteria elicit high levels of prostaglandin E2 (PGE2) and cytokines in circulation and in the placenta (Madianos et al. 2001; Liu et al. 2007). Taken together with the fact that periodontal therapy reduces this pathogen load and is accompanied by a 3.8‐fold decrease in pre‐term births (Offenbacher et al. 2006), these results suggest that circulating periodontal pathogens may trigger an inflammatory response in the uterus, which could contribute to pre‐term birth.
-
(ii)
Fetal response to maternal pathogens. Aagaard et al. (2014) using a deep‐sequencing strategy, have demonstrated that the placenta is not a sterile environment, as was previously believed, but in fact plays host to several oral organisms. Pre‐term babies, but not full‐term infants, demonstrated higher levels of circulating antibodies to C. rectus (an oral pathobiont that has been demonstrated to cross the placental barrier) (Madianos et al. 2001), suggesting another possible mechanism by which certain oral bacteria contribute to pre‐term birth.
However, studies using targeted assays to examine the presence of selected oral organisms, including a large‐scale investigation on 823 pregnant women, found no association between presence or levels of subgingival periodontal pathogens and adverse pregnancy outcomes (Noack et al. 2005; Michalowicz et al. 2006; Novak et al. 2008). Furthermore, periodontal therapy did not result in improvement in pregnancy outcomes. However, in light of evidence that the placental microbiome is already established in early pregnancy, it would be surprising indeed if periodontal therapy during the second trimester did change pregnancy outcomes. It is important to examine this temporal relationship using prospective studies that provide periodontal therapy before the beginning of pregnancy.
Periodontitis and rheumatoid arthritis
Rheumatoid arthritis (RA) is a disease that occurs when normal immune function becomes dysregulated, leading to the production of self‐reactive antibodies (autoantibodies) which ultimately result in a chronic autoimmune inflammatory disease that is characterized by inflammation of synovial cavities, and progressive degeneration of cartilage and bone.
This debilitating disease affects more than 30% of individuals over 65 years of age. Infectious agents, such as the Epstein–Barr virus, cytomegalovirus, certain species of Proteus, and Escherichia coli have been implicated in the pathogenesis of this disease for a long time, and molecular mimicry (especially between bacterial heat‐shock proteins and human Hsp60) has been suggested as a mechanism for creating the autoantibodies (Auger & Roudier, 1997; Kamphuis et al. 2005). Two lines of evidence have emerged to support a role for the gut microbiome in the etiology of this disease:
-
(i)
Animal studies using germ‐free and gnotobiotic animals (those that are colonized by selected, specific species) have revealed that a dysbiosis in the gut microbiome resulting in an increase in the levels of pathobionts and a decrease in commensals, predisposes arthritis‐prone mice to inflammatory joint disease (Wu et al. 2010; Scher et al. 2016), This finding lends support to the molecular mimicry hypothesis which implicates the microbial production of cross‐reactive epitopes in the creation of a pathogenic immune response against self‐antigens.
-
(ii)
There is evidence for a gut–joint axis from human studies, which demonstrated that the presence of organisms like Tropheryma whipplei in the intestine is sufficient to cause joint disease in susceptible individuals (Moos & Schneider, 2011). Further, bacterial sequences that mimic key motifs in the RA‐related human antigens have been identified at significantly higher levels in both gut and oral microbiomes of individuals with RA.
It has been known for some time that an autoimmune response to citrullinated proteins underlies the aetiology of RA. Citrullination is a physiological process that is important for neuronal development and chromatin remodelling; however, it is also upregulated during apoptosis, intracellular stress and inflammation, events that are typically seen during a response to a bacterial infection. Peptidyl deiminase is an enzyme that is involved in deimination of arginine residues (citrullination). P. gingivalis, an oral pathogen, has been implicated in the aetiology of this disease for over two decades, since it is the only organism known to produce peptidyl deiminase. This organism citrullinates fibrinogen, enolase, vimentin and collagen II (Marotte et al. 2006; Lundberg et al. 2010; Gilliam et al. 2011; Kinloch et al. 2011). It has been shown that infection with P. gingivalis precedes onset of RA and that autoantibodies to citrullinated protein (ACPA) titres are higher in aggressive periodontitis (Hendler et al. 2010). Moreover, patients with both RA and periodontitis are more likely to be ACPA positive (Dissick et al. 2010).
While there is an emerging body of evidence to suggest an association between RA and periodontal pathogens, non‐causal confounding factors cannot be ignored. Both diseases have several aspects in common, notably an inflammatory phenotype that is characterized by high levels of cytokines, matrix‐metalloproteinases, neutrophil‐derived mediators and oxidative stress. Also, several contributory factors, especially smoking and lower socio‐economic status, are common to both diseases. Polymorphisms in interleukin genes and Fc‐γ receptor, as well as over‐expression of the MHC class II HLA‐DRB1 allele, are implicated in the aetiopathogenesis of both diseases (Marotte et al. 2006; Song et al. 2013; Mikuls et al. 2014).
Periodontitis and diabetes
Studies on the inter‐relationship between diabetes and periodontitis began over half a century ago, when it was seen that Pima Indians with Type 2 diabetes had more widespread periodontitis, which was also more severe when compared to normoglycemic individuals. Periodontitis became known as the sixth complication of diabetes (Loe, 1993), and several lines of evidence demonstrated that the advanced glycation end products (AGEs) influence immune‐inflammatory homeostasis in the periodontium. AGEs are formed when lipids and proteins combine with reducing sugars (all monosaccharides and some di‐ and oligosaccharides), and undergo a series of irreversible molecular rearrangements. The gingival epithelium, endothelium, immune cells and fibroblasts all carry receptors for AGE, known as RAGE. The AGE–RAGE interactions lead to impaired barrier function, among other defects. It is held that these AGE–RAGE interactions are responsible for lowered immunity, higher cellular oxidant stress, lowered wound healing potential, and pro‐inflammatory phenotypes that increase the risk for periodontitis. Treatment of periodontal disease was shown to reduce glycaemic levels, improve glycaemic control and decrease the amount of hypoglycaemic medication required to titrate blood glucose levels (reviewed by Lalla & Papapanou, 2011; Taylor et al. 2013).
While all of these investigations were unanimous about the changes in subgingival microenvironment wrought by hyperglycaemia, the studies that explored the effect of this glucose‐rich, pro‐oxidant, protein‐rich and anaerobic environment on the periodontal microbiome were not as conclusive. While some early studies found an increase in selected bacterial species in diabetics, they either did not report the periodontal status of the individuals, or did not have a control group, or did not report the statistical test used to validate their results (reviewed by Ohlrich et al. 2010; Taylor et al. 2013). Therefore, it was assumed that the periodontal destruction in diabetics was largely due to its effects on the host, rather than a greater‐than‐ordinarily virulent microbiome. However, evidence is emerging to indicate that the periodontal microbiome in diabetics is distinct from that of normoglycaemics (Casarin et al. 2013; Zhou et al. 2013). While there is convincing evidence to support the effect of periodontal disease on glycaemic control, the mechanisms underlying this are not well studied. Furthermore, while the effect of AGE–RAGE interactions on the subgingival pathophysiology has been investigated in depth, the effect of diabetes on the oral microbiome definitely warrants further investigation.
Periodontitis and Alzheimer's disease
Alzheimer's disease is a chronic neurodegenerative disorder that leads to progressive cognitive deterioration, and is the leading cause of dementia in individuals over 65 years of age. It has been known for several decades that infections by viruses, notably, human herpes simplex virus 1 (HSV‐1), and bacteria such as Helicobacter pylori, Chlamydophila pneumoniae and Borrelia burgdorferi may affect the neuronal axis through central nervous system (CNS) infection, inflammation or by creating autoimmune antibodies that target the brain. The effect of these organisms on initiation or exacerbation of Alzheimer's disease has also been investigated, and there is a robust body of evidence from both human and animal studies to support the infectious aetiology of Alzheimer's disease (Jamieson et al. 1992; Balin et al. 1998; Malaguarnera et al. 2004; Letenneur et al. 2008).
Periodontal pathogens such as Treponema denticola and Porphyromonas gingivalis have been identified in the cerebrospinal fluid and neuronal ganglia (Riviere et al. 2002; Poole et al. 2013). Animal studies have demonstrated that in susceptible hosts, P. gingivalis crosses the blood–brain barrier, and leads to complement C3 activation with bystander neuronal injury (Poole et al. 2013). This has been proposed as a mechanism by which periodontal disease may contribute to initiation or progression of Alzheimer's disease.
In summary, a century of research and clinical correlations have identified a role for periodontal diseases in influencing systemic disease. While the very nature of multifactorial, chronic diseases has made it difficult to establish a definitive causal role for periodontal pathobionts in systemic infection, the body of literature supporting an aetiopathological role for these organisms is too substantial to be ignored as merely coincidental. Therefore, well‐controlled, large‐scale prospective study designs or highly representative animal‐model studies are much needed to explore the relationship between a dysbiotic oral microbiome and systemic disease at a mechanistic level.
Additional information
Competing interests
No competing interests declared.
Funding
The author was supported by a National Institute of Dental and Craniofacial Research (NIDCR) grant (R01‐DE022579).
Biography
Purnima Kumar is an associate professor of Periodontology at The Ohio State University. Her lab is focused on gaining insight into the effects of gene‐environment interactions in shaping the oral microbial ecosystem, the factors that lead to dysbiosis within these communities and the downstream local and systemic consequences of these dysbiotic events.

This is an Editor's Choice article from the 15 January 2017 issue.
References
- Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J & Versalovic J (2014). The placenta harbors a unique microbiome. Sci Transl Med 6, 237ra65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aas JA, Paster BJ, Stokes LN, Olsen I & Dewhirst FE (2005). Defining the normal bacterial flora of the oral cavity. J Clin Microbiol 43, 5721–5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auger I & Roudier J (1997). A function for the QKRAA amino acid motif: Mediating binding of DnaJ to DnaK – Implications for the association of rheumatoid arthritis with HLA‐DR4. J Clin Invest 99, 1818–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balin BJ, Gerard HC, Arking EJ, Appelt DM, Branigan PJ, Abrams JT, Whittum‐Hudson JA & Hudson AP (1998). Identification and localization of Chlamydia pneumoniae in the Alzheimer's brain. Med Microbiol Immunol 187, 23–42. [DOI] [PubMed] [Google Scholar]
- Baltch AL, Pressman HL, Schaffer C, Smith RP, Hammer MC, Shayegani M & Michelsen P (1988). Bacteremia in patients undergoing oral procedures. Study following parenteral antimicrobial prophylaxis as recommended by the American Heart Association, 1977. Arch Intern Med 148, 1084–1088. [DOI] [PubMed] [Google Scholar]
- Bearfield C, Davenport ES, Sivapathasundaram V & Allaker RP (2002). Possible association between amniotic fluid micro‐organism infection and microflora in the mouth. BJOG 109, 527–533. [DOI] [PubMed] [Google Scholar]
- Beck JD, Eke P, Lin D, Madianos P, Couper D, Moss K, Elter J, Heiss G & Offenbacher S (2005). Associations between IgG antibody to oral organisms and carotid intima‐medial thickness in community‐dwelling adults. Atherosclerosis 183, 342–348. [DOI] [PubMed] [Google Scholar]
- Beighton D, Hardie JM & Whiley RA (1991). A scheme for the identification of viridans streptococci. J Med Microbiol 35, 367–372. [DOI] [PubMed] [Google Scholar]
- Billings F (1930). Focal infection as the cause of general disease. Bull NY Acad Med 6, 759–773. [PMC free article] [PubMed] [Google Scholar]
- Blew RD (1996). On the definition of ecosystem. Bull Ecol Soc Am 77, 171–173. [Google Scholar]
- Bocca M, Zombolo L, Coscia D & Moniaci D (1989). The correlation between dental pathology and ophthalmic pathology. Minerva Stomatol 38, 1117–1120. [PubMed] [Google Scholar]
- Cairo F, Gaeta C, Dorigo W, Oggioni MR, Pratesi C, Pini Prato GP & Pozzi G (2004). Periodontal pathogens in atheromatous plaques. A controlled clinical and laboratory trial. J Periodontal Res 39, 442–446. [DOI] [PubMed] [Google Scholar]
- Carroll GC & Sebor RJ (1980). Dental flossing and its relationship to transient bacteremia. J Periodontol 51, 691–692. [DOI] [PubMed] [Google Scholar]
- Casarin RC, Barbagallo A, Meulman T, Santos VR, Sallum EA, Nociti FH, Duarte PM, Casati MZ & Goncalves RB (2013). Subgingival biodiversity in subjects with uncontrolled type‐2 diabetes and chronic periodontitis. J Periodontal Res 48, 30–36. [DOI] [PubMed] [Google Scholar]
- Cecil RL & Angevine DM (1938). Clinical and experimental observations of focal infection, with an analysis of 200 cases of rheumatoid arthritis. Ann Intern Med 12, 577–584. [Google Scholar]
- Collins LM & Dawes C (1987). The surface area of the adult human mouth and thickness of the salivary film covering the teeth and oral mucosa. J Dent Res 66, 1300–1302. [DOI] [PubMed] [Google Scholar]
- Dasanayake AP, Li Y, Wiener H, Ruby JD & Lee MJ (2005). Salivary Actinomyces naeslundii genospecies 2 and Lactobacillus casei levels predict pregnancy outcomes. J Periodontol 76, 171–177. [DOI] [PubMed] [Google Scholar]
- Debelian GJ, Olsen I & Tronstad L (1995). Bacteremia in conjunction with endodontic therapy. Endod Dent Traumatol 11, 142–149. [DOI] [PubMed] [Google Scholar]
- Desvarieux M, Demmer RT, Rundek T, Boden‐Albala B, Jacobs DR Jr, Papapanou PN & Sacco RL (2003). Relationship between periodontal disease, tooth loss, and carotid artery plaque: the Oral Infections and Vascular Disease Epidemiology Study (INVEST). Stroke 34, 2120–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiulio DB, Romero R, Amogan HP, Kusanovic JP, Bik EM, Gotsch F, Kim CJ, Erez O, Edwin S & Relman DA (2008). Microbial prevalence, diversity and abundance in amniotic fluid during preterm labor: a molecular and culture‐based investigation. PLoS One 3, e3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dissick A, Redman RS, Jones M, Rangan BV, Reimold A, Griffiths GR, Mikuls TR, Amdur RL, Richards JS & Kerr GS (2010). Association of periodontitis with rheumatoid arthritis: a pilot study. J Periodontol 81, 223–230. [DOI] [PubMed] [Google Scholar]
- Dussault G & Sheiham A (1982). Medical theories and professional development. The theory of focal sepsis and dentistry in early twentieth century Britain. Soc Sci Med 16, 1405–1412. [DOI] [PubMed] [Google Scholar]
- Etingin OR, Silverstein RL, Friedman HM & Hajjar DP (1990). Viral activation of the coagulation cascade: molecular interactions at the surface of infected endothelial cells. Cell 61, 657–662. [DOI] [PubMed] [Google Scholar]
- Fabricant CG, Fabricant J, Litrenta MM & Minick CR (1978). Virus‐induced atherosclerosis. J Exp Med 148, 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish EW (1940). The teeth as a source of focal sepsis. Postgrad Med J 16, 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francke OC (1973). William Hunter's “oral sepsis” and American odontology. Bull Hist Dent 21, 73–79. [PubMed] [Google Scholar]
- Gibbons RJ, Socransky SS, Dearaujo WC & Vanhoute J (1964). Studies of the predominant cultivable microbiota of dental plaque. Arch Oral Biol 9, 365–370. [DOI] [PubMed] [Google Scholar]
- Gilliam BE, Reed MR, Chauhan AK, Dehlendorf AB & Moore TL (2011). Evidence of fibrinogen as a target of citrullination in IgM rheumatoid factor‐positive polyarticular juvenile idiopathic arthritis. Pediatr Rheumatol Online J 9, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau AJ, Becher H, Ziegler CM, Lichy C, Buggle F, Kaiser C, Lutz R, Bultmann S, Preusch M & Dorfer CE (2004). Periodontal disease as a risk factor for ischemic stroke. Stroke 35, 496–501. [DOI] [PubMed] [Google Scholar]
- Hajjar DP, Fabricant CG, Minick CR & Fabricant J (1986). Virus‐induced atherosclerosis. Herpesvirus infection alters aortic cholesterol metabolism and accumulation. Am J Pathol 122, 62–70. [PMC free article] [PubMed] [Google Scholar]
- Han YW, Redline RW, Li M, Yin L, Hill GB & McCormick TS (2004). Fusobacterium nucleatum induces premature and term stillbirths in pregnant mice: implication of oral bacteria in preterm birth. Infect Immun 72, 2272–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartzell TB & Henrici AT (1916). The dental path: Its importance as an avenue to infection. Public Health J 7, 254–259. [Google Scholar]
- Heimdahl A, Hall G, Hedberg M, Sandberg H, Soder PO, Tuner K & Nord CE (1990). Detection and quantitation by lysis‐filtration of bacteremia after different oral surgical procedures. J Clin Microbiol 28, 2205–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendler A, Mulli TK, Hughes FJ, Perrett D, Bombardieri M, Houri‐Haddad Y, Weiss EI & Nissim A (2010). Involvement of autoimmunity in the pathogenesis of aggressive periodontitis. J Dent Res 89, 1389–1394. [DOI] [PubMed] [Google Scholar]
- Hernichel‐Gorbach E, Kornman KS, Holt SC, Nichols F, Meador H, Kung JT & Thomas CA (1994). Host responses in patients with generalized refractory periodontitis. J Periodontol 65, 8–16. [DOI] [PubMed] [Google Scholar]
- Hillier SL, Nugent RP, Eschenbach DA, Krohn MA, Gibbs RS, Martin DH, Cotch MF, Edelman R, Pastorek JG, Rao AV, McNellis D, Regan JA, Carey JC & Klebanoff MA (1995). Association between bacterial vaginosis and preterm delivery of a low‐birth‐weight infant. N Engl J Med 333, 1737–1742. [DOI] [PubMed] [Google Scholar]
- Hunter W (1900). Oral sepsis as a cause of disease. Br Med J 2, 215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingle JI, Bakland LK & Baumgartner JC (2008). Ingle's Endodontics 6 BC Decker, Hamilton, Ontario. [Google Scholar]
- Jamieson GA, Maitland NJ, Wilcock GK, Yates CM & Itzhaki RF (1992). Herpes simplex virus type 1 DNA is present in specific regions of brain from aged people with and without senile dementia of the Alzheimer type. J Pathol 167, 365–368. [DOI] [PubMed] [Google Scholar]
- Kamphuis S, Kuis W, de Jager W, Teklenburg G, Massa M, Gordon G, Boerhof M, Rijkers GT, Uiterwaal CS, Otten HG, Sette A, Albani S & Prakken BJ (2005). Tolerogenic immune responses to novel T‐cell epitopes from heat‐shock protein 60 in juvenile idiopathic arthritis. Lancet 366, 50–56. [DOI] [PubMed] [Google Scholar]
- Kell D, Potgieter M & Pretorius E (2015). Individuality, phenotypic differentiation, dormancy and ‘persistence’ in culturable bacterial systems: commonalities shared by environmental, laboratory, and clinical microbiology. F1000Res 4, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinloch AJ, Alzabin S, Brintnell W, Wilson E, Barra L, Wegner N, Bell DA, Cairns E & Venables PJ (2011). Immunization with Porphyromonas gingivalis enolase induces autoimmunity to mammalian α‐enolase and arthritis in DR4‐IE‐transgenic mice. Arthritis Rheum 63, 3818–3823. [DOI] [PubMed] [Google Scholar]
- Klotz O (1913). Arterial lesions associated with rheumatic fever. J Path Bact 18, 259–269. [Google Scholar]
- Kol A, Sukhova GK, Lichtman AH & Libby P (1998). Chlamydial heat shock protein 60 localizes in human atheroma and regulates macrophage tumor necrosis factor‐α and matrix metalloproteinase expression. Circulation 98, 300–307. [DOI] [PubMed] [Google Scholar]
- Kornman KS & Loesche WJ (1980). The subgingival microbial flora during pregnancy. J Periodontal Res 15, 111–122. [DOI] [PubMed] [Google Scholar]
- Kumar PS, Griffen AL, Barton JA, Paster BJ, Moeschberger ML & Leys EJ (2003). New bacterial species associated with chronic periodontitis. J Dent Res 82, 338–344. [DOI] [PubMed] [Google Scholar]
- Kumar PS, Matthews CR, Joshi V, de Jager M & Aspiras M (2011). Tobacco smoking affects bacterial acquisition and colonization in oral biofilms. Infect Immun 79, 4730–4738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurki T, Sivonen A, Renkonen OV, Savia E & Ylikorkala O (1992). Bacterial vaginosis in early‐pregnancy and pregnancy outcome. Obstet Gynecol 80, 173–177. [PubMed] [Google Scholar]
- Laitinen K, Laurila A, Pyhala L, Leinonen M & Saikku P (1997). Chlamydia pneumoniae infection induces inflammatory changes in the aortas of rabbits. Infect Immun 65, 4832–4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalla E & Papapanou PN (2011). Diabetes mellitus and periodontitis: a tale of two common interrelated diseases. Nat Rev Endocrinol 7, 738–748. [DOI] [PubMed] [Google Scholar]
- Leon R, Silva N, Ovalle A, Chaparro A, Ahumada A, Gajardo M, Martinez M & Gamonal J (2007). Detection of Porphyromonas gingivalis in the amniotic fluid in pregnant women with a diagnosis of threatened premature labor. J Periodontol 78, 1249–1255. [DOI] [PubMed] [Google Scholar]
- Letenneur L, Peres K, Fleury H, Garrigue I, Barberger‐Gateau P, Helmer C, Orgogozo JM, Gauthier S & Dartigues JF (2008). Seropositivity to herpes simplex virus antibodies and risk of Alzheimer's disease: a population‐based cohort study. PLoS One 3, e3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levison ME (1994). Pneumonia, including necrotizing pulmonary infections (lung abscess) In Harrison's Principles of Internal Medicine, 13th edn, ed. Isselbacher KJ, Braunwald E, Wilson JD. et al, pp. 1197–1206. McGraw Hill, New York. [Google Scholar]
- Li X, Kolltveit KM, Tronstad L & Olsen I (2000). Systemic diseases caused by oral infection. Clin Microbiol Rev 13, 547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liljemark WF, Bloomquist CG, Uhl LA, Schaffer EM, Wolff LF, Pihlstrom BL & Bandt CL (1984). Distribution of oral Haemophilus species in dental plaque from a large adult population. Infect Immun 46, 778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden GJ, Herzberg MC & working group 4 of the joint EFP/AAP workshop (2013). Periodontitis and systemic diseases: a record of discussions of working group 4 of the Joint EFP/AAP Workshop on Periodontitis and Systemic Diseases. J Clin Periodontol 40 Suppl 14, S20–S23. [DOI] [PubMed] [Google Scholar]
- Listgarten MA (1986). Pathogenesis of periodontitis. J Clin Periodontol 13, 418–430. [DOI] [PubMed] [Google Scholar]
- Liu H, Redline RW & Han YW (2007). Fusobacterium nucleatum induces fetal death in mice via stimulation of TLR4‐mediated placental inflammatory response. J Immunol 179, 2501–2508. [DOI] [PubMed] [Google Scholar]
- Loe H (1993). Periodontal disease. The sixth complication of diabetes mellitus. Diabetes Care 16, 329–334. [PubMed] [Google Scholar]
- Loesche WJ (1982). Dental Caries: A Treatable Infection. Charles C. Thomas, Springfield, IL. [Google Scholar]
- Lofthus JE, Waki MY, Jolkovsky DL, Otomo‐Corgel J, Newman MG, Flemmig T & Nachnani S (1991). Bacteremia following subgingival irrigation and scaling and root planing. J Periodontol 62, 602–607. [DOI] [PubMed] [Google Scholar]
- Lorenz KA & Weiss PJ (1994). Capnocytophagal pneumonia in a healthy man. West J Med 160, 79–80. [PMC free article] [PubMed] [Google Scholar]
- Lundberg K, Wegner N, Yucel‐Lindberg T & Venables PJ (2010). Periodontitis in RA—the citrullinated enolase connection. Nat Rev Rheumatol 6, 727–730. [DOI] [PubMed] [Google Scholar]
- Madianos PN, Lieff S, Murtha AP, Boggess KA, Auten RL Jr, Beck JD & Offenbacher S (2001). Maternal periodontitis and prematurity. Part II: Maternal infection and fetal exposure. Ann Periodontol 6, 175–182. [DOI] [PubMed] [Google Scholar]
- Mai X, LaMonte MJ, Hovey KM, Nwizu N, Freudenheim JL, Tezal M, Scannapieco F, Hyland A, Andrews CA, Genco RJ & Wactawski‐Wende J (2014). History of periodontal disease diagnosis and lung cancer incidence in the Women's Health Initiative Observational Study. Cancer Causes Control 25, 1045–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaguarnera M, Bella R, Alagona G, Ferri R, Carnemolla A & Pennisi G (2004). Helicobacter pylori and Alzheimer's disease: a possible link. Eur J Intern Med 15, 381–386. [DOI] [PubMed] [Google Scholar]
- Marotte H, Farge P, Gaudin P, Alexandre C, Mougin B & Miossec P (2006). The association between periodontal disease and joint destruction in rheumatoid arthritis extends the link between the HLA‐DR shared epitope and severity of bone destruction. Ann Rheum Dis 65, 905–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason MR, Preshaw PM, Nagaraja HN, Dabdoub SM, Rahman A & Kumar PS (2014). The subgingival microbiome of clinically healthy current and never smokers. ISME J 9, 268–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattila KJ (1993). Dental infections as a risk factor for acute myocardial infarction. Eur Heart J 14 Suppl K, 51–53. [PubMed] [Google Scholar]
- Mattila KJ, Nieminen MS, Valtonen VV, Rasi VP, Kesaniemi YA, Syrjala SL, Jungell PS, Isoluoma M, Hietaniemi K & Jokinen MJ (1989). Association between dental health and acute myocardial infarction. BMJ 298, 779–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattila KJ, Valtonen VV, Nieminen MS & Asikainen S (1998). Role of infection as a risk factor for atherosclerosis, myocardial infarction, and stroke. Clin Infect Dis 26, 719–734. [DOI] [PubMed] [Google Scholar]
- Michalowicz BS, Hodges JS, DiAngelis AJ, Lupo VR, Novak MJ, Ferguson JE, Buchanan W, Bofill J, Papapanou PN, Mitchell DA, Matseoane S & Tschida PA (2006). Treatment of periodontal disease and the risk of preterm birth. N Engl J Med 355, 1885–1894. [DOI] [PubMed] [Google Scholar]
- Mikuls TR, Payne JB, Yu F, Thiele GM, Reynolds RJ, Cannon GW, Markt J, McGowan D, Kerr GS, Redman RS, Reimold A, Griffiths G, Beatty M, Gonzalez S, Bergman DA, Hamilton BC, Erickson AR, Sokolove J, Robinson W, Walker C, Chandad F & O'Dell JR (2014). Periodontitis and Porphyromonas gingivalis in patients with rheumatoid arthritis. Arthritis Rheum 66, 1090–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller WD (1880). The Micro‐Organisms of the Human Mouth: The Local and General Diseases Which are Caused by Them. S. S. White, Philadelphia. [Google Scholar]
- Mitchell‐Lewis D, Engebretson SP, Chen J, Lamster IB & Papapanou PN (2001). Periodontal infections and pre‐term birth: early findings from a cohort of young minority women in New York. Eur J Oral Sci 109, 34–39. [DOI] [PubMed] [Google Scholar]
- Moos V & Schneider T (2011). Changing paradigms in Whipple's disease and infection with Tropheryma whipplei . Eur J Clin Microbiol Infect Dis 30, 1151–1158. [DOI] [PubMed] [Google Scholar]
- Murray CA & Saunders WP (2000). Root canal treatment and general health: a review of the literature. Int Endod J 33, 1–18. [DOI] [PubMed] [Google Scholar]
- Nieto FJ (1998). Infections and atherosclerosis: new clues from an old hypothesis? Am J Epidemiol 148, 937–948. [DOI] [PubMed] [Google Scholar]
- Noack B, Klingenberg J, Weigelt J & Hoffmann T (2005). Periodontal status and preterm low birth weight: a case control study. J Periodontal Res 40, 339–345. [DOI] [PubMed] [Google Scholar]
- Novak MJ, Novak KF, Hodges JS, Kirakodu S, Govindaswami M, Diangelis A, Buchanan W, Papapanou PN & Michalowicz BS (2008). Periodontal bacterial profiles in pregnant women: response to treatment and associations with birth outcomes in the obstetrics and periodontal therapy (OPT) study. J Periodontol 79, 1870–1879. [DOI] [PubMed] [Google Scholar]
- Offenbacher S (1996). Periodontal diseases: pathogenesis. Ann Periodontol 1, 821–878. [DOI] [PubMed] [Google Scholar]
- Offenbacher S, Heasman PA & Collins JG (1993). Modulation of host PGE2 secretion as a determinant of periodontal disease expression. J Periodontol 64, 432–444. [DOI] [PubMed] [Google Scholar]
- Offenbacher S, Lin D, Strauss R, McKaig R, Irving J, Barros SP, Moss K, Barrow DA, Hefti A & Beck JD (2006). Effects of periodontal therapy during pregnancy on periodontal status, biologic parameters, and pregnancy outcomes: a pilot study. J Periodontol 77, 2011–2024. [DOI] [PubMed] [Google Scholar]
- Ohlrich EJ, Cullinan MP & Leichter JW (2010). Diabetes, periodontitis, and the subgingival microbiota. J Oral Microbiol 2, doi: 10.3402/jom.v2i0.5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paju S & Scannapieco FA (2007). Oral biofilms, periodontitis, and pulmonary infections. Oral Dis 13, 508–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paster BJ, Boches SK, Galvin JL, Ericson RE, Lau CN, Levanos VA, Sahasrabudhe A & Dewhirst FE (2001). Bacterial diversity in human subgingival plaque. J Bacteriol 183, 3770–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson GR, Hitti J, Paul K, Hirschi R, Weibel M, Rothen M & Persson RE (2008). Tannerella forsythia and Pseudomonas aeruginosa in subgingival bacterial samples from parous women. J Periodontol 79, 508–516. [DOI] [PubMed] [Google Scholar]
- Poole S, Singhrao SK, Kesavalu L, Curtis MA & Crean S (2013). Determining the presence of periodontopathic virulence factors in short‐term postmortem Alzheimer's disease brain tissue. J Alzheimers Dis 36, 665–677. [DOI] [PubMed] [Google Scholar]
- Pussinen PJ, Alfthan G, Jousilahti P, Paju S & Tuomilehto J (2006). Systemic exposure to Porphyromonas gingivalis predicts incident stroke. Atherosclerosis 193, 222–228. [DOI] [PubMed] [Google Scholar]
- Rammelkamp CH; Committee on Prevention of Rheumatic Fever and Bacterial Endocarditis (1957). Prevention of rheumatic fever and bacterial endocarditis through control of streptococcal infections. Circulation 15, 154–158. [Google Scholar]
- Riviere GR, Riviere KH & Smith KS (2002). Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer's disease. Oral Microbiol Immunol 17, 113–118. [DOI] [PubMed] [Google Scholar]
- Roberts GJ, Holzel HS, Sury MR, Simmons NA, Gardner P & Longhurst P (1997). Dental bacteremia in children. Pediatr Cardiol 18, 24–27. [DOI] [PubMed] [Google Scholar]
- Roberts HL (1921). Focal infection. Br J Dermatol 33, 353–373. [Google Scholar]
- Rosenow EC (1930). Elective localization of streptococci. BMJ 1, 1100–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell SL, Boylan RJ, Kaslick RS, Scannapieco FA & Katz RV (1999). Respiratory pathogen colonization of the dental plaque of institutionalized elders. Spec Care Dentist 19, 128–134. [DOI] [PubMed] [Google Scholar]
- Sanchez AR, Kupp LI, Sheridan PJ & Sanchez DR (2004). Maternal chronic infection as a risk factor in preterm low birth weight infants: the link with periodontal infection. J Int Acad Periodontol 6, 89–94. [PubMed] [Google Scholar]
- Scannapieco FA ( 2006). Pneumonia in nonambulatory patients. The role of oral bacteria and oral hygiene. J Am Dent Assoc 137 Suppl, 21S–25S. [DOI] [PubMed] [Google Scholar]
- Scannapieco FA & Binkley CJ (2012). Modest reduction in risk for ventilator‐associated pneumonia in critically ill patients receiving mechanical ventilation following topical oral chlorhexidine. J Evid Based Dent Pract 12, 15–17. [DOI] [PubMed] [Google Scholar]
- Scannapieco FA, Stewart EM & Mylotte JM (1992). Colonization of dental plaque by respiratory pathogens in medical intensive care patients. Crit Care Med 20, 740–745. [DOI] [PubMed] [Google Scholar]
- Scher JU, Littman DR & Abramson SB (2016). Microbiome in inflammatory arthritis and human rheumatic diseases. Arthritis Rheumatol 68, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour GJ, Ford PJ, Cullinan MP, Leishman S & Yamazaki K (2007). Relationship between periodontal infections and systemic disease. Clin Microbiol Infect 13 Suppl 4, 3–10. [DOI] [PubMed] [Google Scholar]
- Shinzato T & Saito A (1995). The Streptococcus milleri group as a cause of pulmonary infections. Clin Infect Dis 21 Suppl 3, S238–S243. [DOI] [PubMed] [Google Scholar]
- Shorter E (2011). A brief history of placebos and clinical trials in psychiatry. Can J Psychiatry 56, 193–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair DG & Evans TW (1994). Nosocomial pneumonia in the intensive care unit. Br J Hosp Med 51, 177–180. [PubMed] [Google Scholar]
- Socransky SS & Haffajee AD (2005). Periodontal microbial ecology. Periodontol 2000 38, 135–187. [DOI] [PubMed] [Google Scholar]
- Song GG, Bae SC, Kim JH & Lee YH (2013). Interleukin‐4, interleukin‐4 receptor, and interleukin‐18 polymorphisms and rheumatoid arthritis: a meta‐analysis. Immunol Invest 42, 455–469. [DOI] [PubMed] [Google Scholar]
- Taylor JJ, Preshaw PM & Lalla E (2013). A review of the evidence for pathogenic mechanisms that may link periodontitis and diabetes. J Clin Periodontol 40 Suppl 14, S113–S134. [DOI] [PubMed] [Google Scholar]
- Terpenning M, Bretz W, Lopatin D, Langmore S, Dominguez B & Loesche W (1993). Bacterial colonization of saliva and plaque in the elderly. Clin Infect Dis 16 Suppl 4, S314–S316. [DOI] [PubMed] [Google Scholar]
- Toews GB ( 1986). Nosocomial pneumonia. Am J Med Sci 291, 355–367. [DOI] [PubMed] [Google Scholar]
- Tonetti MS, D'Aiuto F, Nibali L, Donald A, Storry C, Parkar M, Suvan J, Hingorani AD, Vallance P & Deanfield J (2007). Treatment of periodontitis and endothelial function. N Engl J Med 356, 911–920. [DOI] [PubMed] [Google Scholar]
- Tonetti MS, Van Dyke TE & working group 1 of the joint EFP/AAP workshop (2013). Periodontitis and atherosclerotic cardiovascular disease: consensus report of the Joint EFP/AAP Workshop on Periodontitis and Systemic Diseases. J Periodontol 84, S24–S29. [DOI] [PubMed] [Google Scholar]
- Vaizey JM & Clark‐Kennedy AE (1939). Dental sepsis: anaemia, dyspepsia, and rheumatism. BMJ 1, 1269–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessely S ( 2009). Surgery for the treatment of psychiatric illness: the need to test untested theories. J R Soc Med 102, 445–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick G, Romen M, Amberger A, Metzler B, Mayr M, Falkensammer G & Xu Q (1997). Atherosclerosis, autoimmunity, and vascular‐associated lymphoid tissue. FASEB J 11, 1199–1207. [DOI] [PubMed] [Google Scholar]
- Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, Littman DR, Benoist C & Mathis D (2010). Gut‐residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 32, 815–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama T, Hashimoto K, Fukuda H, Ishida M, Arai H, Sekizawa K, Yamaya M & Sasaki H (1996). Oral hygiene reduces respiratory infections in elderly bed‐bound nursing home patients. Arch Gerontol Geriatr 22, 11–19. [DOI] [PubMed] [Google Scholar]
- Yuan A, Yang PC, Lee LN, Chang DB, Kuo SH & Luh KT (1992). Actinobacillus actinomycetemcomitans pneumonia with chest wall involvement and rib destruction. Chest 101, 1450–1452. [DOI] [PubMed] [Google Scholar]
- Zhou M, Rong R, Munro D, Zhu C, Gao X, Zhang Q & Dong Q (2013). Investigation of the effect of type 2 diabetes mellitus on subgingival plaque microbiota by high‐throughput 16S rDNA pyrosequencing. PLoS One 8, e61516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zijlstra EE, Swart GR, Godfroy FJ & Degener JE (1992). Pericarditis, pneumonia and brain abscess due to a combined Actinomyces–Actinobacillus actinomycetemcomitans infection. J Infect 25, 83–87. [DOI] [PubMed] [Google Scholar]
- Zinkernagel AS, Gmur R, Fenner L, Schaffner A, Schoedon G & Schneemann M (2003). Marginal and subgingival plaque–a natural habitat of Tropheryma whipplei? Infection 31, 86–91. [DOI] [PubMed] [Google Scholar]