

Abstract
Transport of fluid and electrolytes in the intestine allows for appropriate adjustments in luminal fluidity while reclaiming water used in digesting and absorbing a meal, and is closely regulated. This article discusses various endogenous and exogenous mechanisms whereby transport is controlled in the gut, placing these in the context of the ideas about the neurohumoral control of alimentary physiology that were promulgated by William Bayliss and Ernest Starling. The article considers three themes. First, mechanisms that intrinsically regulate chloride secretion, centred on the epidermal growth factor receptor (EGFr), are discussed. These may be important in ensuring that excessive chloride secretion, with the accompanying loss of fluid, is not normally stimulated by intestinal distension as the meal passes through the gastrointestinal tract. Second, mechanisms whereby probiotic microorganisms can impart beneficial effects on the gut are described, with a focus on targets at the level of the epithelium. These findings imply that the commensal microbiota exert important influences on the epithelium in health and disease. Finally, mechanisms that lead to diarrhoea in patients infected with an invasive pathogen, Salmonella, are considered, based on recent studies in a novel mouse model. Diarrhoea is most likely attributable to reduced expression of absorptive transporters and may not require the influx of neutrophils that accompanies infection. Overall, the goal of the article is to highlight the many ways in which critical functions of the intestinal epithelium are regulated under physiological and pathophysiological conditions, and to suggest possible targets for new therapies for digestive disease states.
Introduction
This brief article summarizes the presentation I delivered as the Bayliss–Starling Prize Lecturer of The Physiological Society in London on 2 December 2015. I was hugely honoured to be the recipient of this lectureship, not least because, as a GI physiologist, I have referred to Bayliss and Starling's seminal discovery of secretin, and elaboration of the concept of a hormone, in my introductory lectures to medical students for more than 20 years. In fact, their critical work was published in full in the pages of this journal (Bayliss & Starling, 1902), and its importance, as well as the excitement it generated, was beautifully underscored by Barry Hirst in his Classical Perspective on the original paper (Hirst, 2004).
My own work has focused on developing a comprehensive understanding of the transport and barrier functions of the intestinal epithelium, both a source and a target of the hormones envisaged by Bayliss and Starling. We have focused, in large part, on the signalling mechanisms intrinsic to the epithelium that exert a finely tuned control on epithelial function in health and disease. Thus, not only do epithelial cells respond to classical hormones and neurotransmitters, but they also contain sufficient machinery to determine their own minute‐to‐minute destiny and respond to alterations in the luminal environment with appropriate adjustments in fluid and electrolyte transport. In fact, the epithelium, a single layer of columnar cells stretching from the stomach to the anus, faces specific challenges in performing its physiological function of nutrient absorption while excluding pathogens and toxins. The intestinal epithelium is also characterized by its life‐long association, beginning at birth, with a richly diverse commensal microbiota (Lozupone et al. 2012). Interactions between the epithelium and these microorganisms almost certainly exert functional effects on the epithelium itself as well as underlying cell types. The differentiated cell types of the epithelium, including absorptive enterocytes, goblet, Paneth and endocrine cells, arise from stem cells localized to the crypts of both the small and large intestines (VanDussen et al. 2012). The epithelium must also engage constantly in processes of fluid and electrolyte transport to provide appropriate levels of luminal fluidity while at the same time ensuring whole body fluid and electrolyte homeostasis. In fact, the healthy gut encounters 8–9 litres of fluid per day in processing meals, with the majority of this coming from the intestine itself and organs that drain into it, rather than from oral intake (Barrett & Keely, 2000). The vast majority of this fluid input is reabsorbed in conjunction with active absorption of electrolytes and digested nutrients. Nevertheless, all segments of the gut continue to engage in fluid secretion, driven predominantly by the secretion of chloride ions and often in response to reflexes that are triggered by mucosal distension (Frieling et al. 1992; Barrett & Keely, 2000). As such, ongoing bursts of secretion may represent a mechanism whereby the mucosa is lubricated to prevent epithelial damage as a food bolus passes. We also know that alterations in epithelial transport function may underlie diarrhoeal symptoms in a variety of settings (Barrett & Keely, 2000).
Three predominant transport mechanisms are recognized as being critical for the movement of fluid into and out of the gut in the period between meals, when nutrients are not present. Electroneutral NaCl absorption is mediated by the coupled activity of an apical chloride/bicarbonate exchanger known as DRA (for down‐regulated in adenoma, also referred to as Slc26a3) and the NHE‐3 isoform of the sodium/hydrogen exchanger family (Melvin et al. 1999; Zachos et al. 2005; Kato & Romero, 2011). The activity of these apical transporters is driven indirectly by the electrochemical gradients established by a basolateral sodium/potassium ATPase, and chloride is thought to exit the basolateral membrane via ClC‐2 chloride channels, with basolateral Kir 7.1 potassium channels also functioning to maintain the driving force for chloride exit. The net effect is that sodium and chloride ions are transferred in equal amounts from the lumen to the bloodstream via a transcellular route, with water following paracellularly to ensure osmotic balance. The electroneutral NaCl absorptive mechanism is present in both the small intestine and colon, perhaps with some slight regional variations in the precise molecular identity of the constituent membrane transporters (Simpson et al. 2007; Talbot & Lytle, 2010).
In the distal colon, sodium ions are also reclaimed via an electrogenic mechanism whereby Na+ enters across apically localized epithelial sodium (ENaC) channels, and exits through the basolateral sodium/potassium ATPase (Kunzelmann & Mall, 2002). Chloride as a counterion, and water, are thus stimulated to follow paracellularly. There is also some evidence to suggest that water may travel, at least in part, via a transcellular route mediated by specific aquaporins (Laforenza, 2012). The electrogenic pathway for sodium absorption can be considered to function as a salvage mechanism that ensures the final dehydration of the stool. Its importance is illustrated by observations that ENaC expression is significantly reduced in both animal models of colitis as well as human patients with inflammatory bowel disease, which is considered to contribute significantly to the occurrence of diarrhoea in these settings (Greig & Sandle, 2000; Greig et al. 2004; McCole et al. 2005).
On the secretory side of the equation, active electrogenic chloride secretion is conducted largely (although not exclusively) by cells in the crypts throughout the small intestine and colon (Welsh et al. 1982; Jakab et al. 2011, 2013). The chloride secretory mechanism rests heavily on the apical chloride channel known as the cystic fibrosis transmembrane conductance regulator (CFTR), although other accessory chloride channels, such as TMEM16A/anoctamin 1, may also participate to a lesser degree depending on the secretagogue involved (Barrett & Keely, 2000). At the basolateral membrane, chloride is taken up in association with sodium and potassium ions via the cotransporter NKCC1 (Slc12a2), whose activity is driven secondarily by the sodium/potassium ATPase. In actively secreting cells, potassium is also recycled across the basolateral membrane via calcium‐ and cAMP‐dependent potassium channels (KCNN4 and KCNQ1/KCNE3, respectively) to sustain the driving force for apical chloride exit. Thus, chloride is transferred from the bloodstream to the intestinal lumen, with water and sodium ions following paracellularly and/or transcellularly.
Having set the stage with this general introduction to intestinal transport physiology, I will now turn to three ‘stories’ from my laboratory that illustrate how transport function is subject to regulation by endogenous and exogenous factors, and the links from these processes to our understanding of both inflammatory and infectious diarrhoea.
Intrinsic regulation of epithelial chloride secretion
As mentioned above, while absorption is the predominant vector in health, both small and large intestines must also engage in secretory transport on an ongoing basis. Not only may this be important for lubrication and adjusting luminal fluidity such that the contents of the intestine can be propelled along its length, but there are also suggestions that active secretion of chloride ions, and thus fluid, into the crypts is important to flush these blind‐ended structures of bacteria and other toxins, maintaining crypt sterility and protecting the crucial stem cell niche (Asfaha et al. 2001; Keely et al. 2012). There are two main pathways whereby chloride secretion can be triggered (Barrett & Keely, 2000). The first of these is produced by agonists that cause increases in the levels of intracellular cyclic nucleotides, and results in large secretory responses that are sustained for the duration of agonist exposure. The second pathway is mediated by elevations in cytosolic free calcium. Calcium‐dependent chloride secretory responses are small and transient, even if the calcium signal is maintained, and are involved in the reflex secretion that results from deformation of the mucosa, amongst other responses. We have speculated that the transient nature of these secretory responses subserves their physiological function, ensuring that lubrication occurs only briefly as the bolus passes and avoiding excessive fluid loss (Barrett & Keely, 2000). In a series of publications, we explored the basis of this ‘physiological braking’ that limits the extent of calcium‐dependent chloride secretion.
Serendipity, in the form of a new PhD student, Jorge Uribe, who had an interest in peptide growth factors, led us to study whether epidermal growth factor (EGF) had any effect on epithelial chloride secretion. In fact, while EGF did not serve as an agonist of secretion, we observed that it inhibited subsequent responses to calcium‐dependent agonists, such as the acetylcholine analogue, carbachol, as well as histamine and thapsigargin (Uribe et al. 1996a). We mapped the signalling pathways that mediated the ability of EGF to inhibit secretion (Fig. 1), and showed that they involved recruitment of the enzyme phosphatidylinositol 3‐kinase (PI3K) to the basolaterally localized EGF receptor (EGFr), where PI3K could act on its substrates in the plasma membrane to generate 3‐phosphorylated lipids such as phosphatidylinositol (3,4)‐bisphosphate (PI(3,4)P2) (Uribe et al. 1996b). In turn, these lipid messengers activated the epsilon isoform of protein kinase C (PKCε) that directly or indirectly could inhibit basolateral potassium channels (Chow et al. 2000). Because potassium ions must be recycled via these channels to avoid dissipation of the driving force for apical chloride exit during active transcellular chloride secretion, the net effect of EGF was to inhibit this transport mechanism. Moreover, because ligands of EGFr are abundant in the intestine, they may provide for a mechanism whereby calcium‐dependent chloride secretion is intrinsically limited (Uribe & Barrett, 1997; Beck & Podolsky, 1999).
Figure 1. Distinct pathways for intrinsic regulation of calcium‐dependent chloride secretion in intestinal epithelial cells.
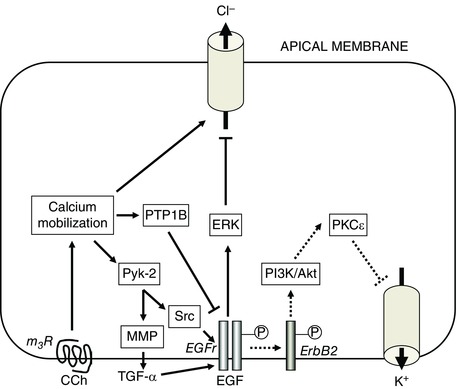
Calcium‐dependent secretion is evoked by agonists such as carbachol (CCh), which binds to m3 muscarinic receptors (m3R) on the basolateral membranes of intestinal epithelial cells and stimulates the mobilization of calcium from intercellular stores. This causes activation of apical chloride channels and (not shown) basolateral potassium channels. As shown by the continuous arrows on the left side of the figure, CCh also recruits additional downstream effectors, including the receptors for epidermal growth factor (EGFr) that ultimately terminate chloride secretion via an inhibitory effect on apical chloride channels. CCh concomitantly stimulates protein tyrosine phosphatase 1B (PTP1B) that dephosphorylates EGFr, thereby constraining the downstream signalling pathways that EGFr can recruit. On the other hand, as shown by the dotted arrows in the right side of the figure, EGF binds to EGFr and activates a distinct signalling cascade that inhibits chloride secretion via an inhibitory effect on a basolateral potassium channel. Unlike CCh, EGF does not act initially as an agonist of the secretory process. For further details, see text. MMP, matrix metalloproteinase; PI3K, phosphatidylinositol 3‐kinase; PKC, protein kinase C.
However, the findings described above did not clarify why secretory responses to agonists such as carbachol are intrinsically limited, including in intestinal epithelial cell lines that lack exogenous factors. Using an experimental protocol in which the T84 human colonic epithelial cell line was treated sequentially with carbachol and thapsigargin, we observed that the ability of carbachol to reduce the secretory response to a second stimulus was sensitive to inhibition of tyrosine kinase activity, and, surprisingly, accompanied by phosphorylation of EGFr (Keely et al. 1998). Our findings therefore added to a concept that was emerging at that time holding that not only could receptor tyrosine kinases such as EGFr be activated by their ligands, but also that they could be ‘transactivated’ downstream of occupancy of G‐protein‐coupled receptors, such as the m3 muscarinic receptor by which carbachol activates chloride secretion (Piiper & Zeuzem, 2004). We went on to show that carbachol activates the matrix metalloproteinase‐mediated cleavage of the membrane‐bound form of the EGFr ligand, transforming growth factor‐α (TGF‐α). This, in turn, mediated EGFr activation, recruitment of downstream effectors such as the extracellular signal‐regulated (ERK)1/2 kinases, and inhibition of chloride secretion via an inhibitory effect on apical chloride channels (Barrett et al. 1998; McCole et al. 2002) (Fig. 1).
Our data, however, also raised an interesting paradox. We were aware that the ability of EGF and carbachol to reduce secretory responses to a subsequent calcium‐dependent agonist were additive, and thus involved presumably distinct mechanisms. Moreover, EGF targeted basolateral potassium channels and was not an agonist of chloride secretion, whereas carbachol both stimulated and then inhibited chloride secretion, with the latter action directed at an apical chloride channel (Barrett et al. 1998). Nevertheless, our data implied that the inhibitory action of carbachol occurred via EGFr activation (Keely et al. 1998). It was therefore important to dissect why transactivation of EGFr, as opposed to activation of the receptor by EGF itself, recruited divergent downstream mechanisms (Fig. 1). Part of the answer to this puzzle lay in the knowledge that the cytoplasmic tail of EGFr contains many tyrosine residues that are dynamically phosphorylated, and that specific signalling outcomes may be linked to the patterns of such residues that are phosphorylated by any given stimulus. Further, EGFr (also known as ErbB1) is one member of the ErbB family of tyrosine kinase receptors, which can dimerize and/or heterodimerize, thereby triggering divergent recruitment of signalling outcomes. We showed EGF and TGF‐α stimulated the phosphorylation of six specific tyrosine residues (845, 992, 1068, 1086, 1148 and 1173) on EGFr, heterodimerization of EGFr with ErbB2, and recruitment of PI3K (albeit with some differences in potency) (McCole et al. 2007) (Fig. 2). Carbachol, on the other hand, failed to phosphorylate Tyr992 and Tyr1068, despite the fact that it transactivated EGFr via the release of TGF‐α (McCole et al. 2002, 2007). We went on to show that, in addition to triggering TGF‐α‐mediated signalling, carbachol also stimulates protein tyrosine phosphatase 1B (PTP1B), which dephosphorylates Tyr992 and Tyr1068 (McCole et al. 2007) (Figs 1 and 2). In cells in which PTP1B was knocked down, the signalling responses to carbachol reverted to those produced by EGF or TGF‐α alone.
Figure 2. The downstream consequences of activation of the epidermal growth factor receptor (EGFr) in intestinal epithelial cells differ depending on the way in which it is activated.
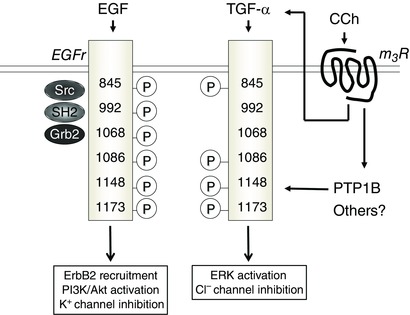
When EGFr is directly activated by EGF, it becomes phosphorylated at at least six cytoplasmic tyrosine residues that recruit distinct binding partners (left side of figure). When EGFr is transactivated secondary to the binding of carbachol (CCh) to the m3 muscarinic receptor (m3R), which occurs via the release of transforming growth factor‐α (TGF‐α), two cytoplasmic tyrosine residues (Y992 and Y1068) are simultaneously dephosphorylated by the ability of CCh also to activate protein tyrosine phosphatase 1B (PTP1B) (right side of figure). This alters the signalling intermediates and consequences of EGFr activation.
In essence, therefore, chloride secretory responses in the gut that are triggered by calcium are intrinsically restrained by a variety of signalling mechanisms that centre on EGFr, but involve distinct pathways and targets (Fig. 1). We can speculate that the ability of agonists such as carbachol to trigger PTP1B may allow calcium‐dependent chloride secretion to be limited without a risk of excessive mitogenic signalling, while also ensuring that diarrhoeal responses do not occur every time food passes along the alimentary canal. Conversely, the ability of growth factors such as EGF and TGF‐α, whose expression is increased in the setting of mucosal injury, may limit the energetically costly process of chloride secretion and redirect resources to repair of the epithelial barrier (Beck & Podolsky, 1999).
Impact of probiotics on transport and barrier function
As noted above, the intestine, and particularly the colon, is characterized by its life‐long relationship with a complex commensal microbial community known as the luminal microbiota. In health, the estimated ∼1000 species that make up the microbiota co‐exist peacefully with the intestine without triggering mucosal injury or inappropriate activation of the mucosal immune system. Conversely, a variety of disease states are associated with so‐called dysbiosis – an imbalance between beneficial and harmful microbes in the gut and a loss of microbial diversity (Brüls & Weissenbach, 2011; Yang & Jobin, 2014). Probiotics are commensal microorganisms that have been selected for their apparent health benefits as well as their ability to bypass inherent barriers (e.g. gastric acid) to reach the distal intestine when ingested orally (Shanahan, 2009). They are usually safe and well‐accepted by patients, have been shown at least anecdotally to be of benefit in a number of gastrointestinal as well as other extra‐intestinal disease states, and have long been used in food preparation as well as traditional medicines. They are assumed, at least in part, to exert therapeutic actions by reversing dysbiosis, but their precise mechanisms of action were not well understood (Shanahan, 2009; Shanahan & Quigley, 2014). We and others have hypothesized that the epithelium might represent a key locus of activity, because intestinal epithelial cells are the first point of interaction of commensals with the host.
The recruitment of a trained microbiologist, Silvia Resta‐Lenert, to the lab allowed us explore how probiotics might impact epithelial function. We studied the combination of two prototypic probiotics, Streptococcus thermophilis and Lactobacillus acidophilus, since emerging literature as well as preliminary data implied that many probiotics are more effective when given as a cocktail than alone. We also began our studies by examining whether these probiotics could prevent adverse effects on epithelial function attributable to infection with invasive bacteria such as Salmonella typhimurium and enteroinvasive E. coli, since it had been suggested that probiotics or other commensals could compete with pathogenic microorganisms to colonize the gut in vivo. In fact, pretreatment with the probiotics prevented defects in both transport and barrier function induced by the pathogens in a variety of epithelial cell lines (Resta‐Lenert & Barrett, 2003). These beneficial effects required the interaction of live probiotics with the epithelial cells, since they could not be reproduced by heat‐killed bacteria, spent media or bacterial DNA. However, they were also associated with a reduction in pathogen adherence to, and invasion into, the epithelial cell monolayers, so we worried that the functional effects were simply a consequence of the lower burden of infection. However, the probiotics alone had an independent effect to increase transepithelial resistance over a 24 h period. Furthermore, the probiotics were also able to prevent the reductions in chloride secretion and barrier function that were produced by inflammatory cytokines such as interferon‐γ and tumour necrosis factor, and they also reverse the ability of the cytokines to reduce expression of CFTR and NKCC1 (Resta‐Lenert & Barrett, 2006). Finally, the probiotics interrupted proinflammatory signalling invoked by the cytokines because they prevented the degradation of the inhibitory factor of nuclear factor kappa‐B (NF‐kappaB) IκB, thereby dampening the NFκB pathway (Resta‐Lenert & Barrett, 2006).
We further wished to examine the efficacy of the probiotics in vivo in a mouse model of colitis. We chose the mdr1a knock‐out mouse for these studies because colitis is believed to result from a defect in epithelial barrier function rather than an immune defect (Panwala et al. 1998; Resta‐Lenert et al. 2005). Daily treatment of mdr1a −/− mice with probiotics by gavage, beginning prior to the onset of spontaneous colitis, preserved weight gain in these animals whereas untreated mutant mice showed attenuated weight gain compared to wild‐type mice as colitis became established at 4–8 weeks of age. Further, the probiotics also attenuated the fall in epithelial barrier function that was seen as colitis developed, partially reversed the reduction in forskolin‐stimulated colonic transport responses in the mutant mice, and reduced the proportion of mice displaying diarrhoeal symptoms. Furthermore, not only could the probiotics reduce or delay the onset of colitic symptoms in the mutant mice, but they were also effective in treating colitis that was already established, albeit with somewhat reduced efficacy compared to the pretreatment model (Resta‐Lenert & Barrett, 2009).
We conclude that the epithelium is probably a critical site for the actions of probiotics, at least using the organisms we studied. They can block adhesion and invasion by invasive pathogens, antagonize the effects of inflammatory mediators, improve barrier dysfunction in vitro and in vivo, and reverse transport dysfunction associated with infection and inflammation (Fig. 3). Overall, these properties of probiotics may contribute to their efficacy in a variety of digestive diseases (Resta‐Lenert & Barrett, 2009). Furthermore, selection of probiotics on the basis of their ability to protect epithelial function may optimize their effectiveness.
Figure 3. Summary of putative beneficial effects of probiotics that are targeted to the intestinal epithelium or its interactions with luminal pathogens.
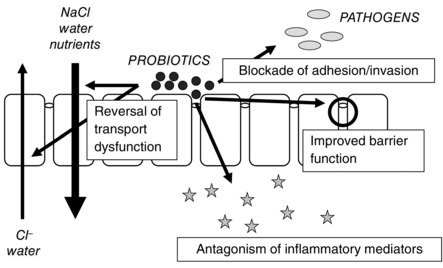
Probiotics can directly impede the adhesion of pathogens to the epithelial surface and thus their ability to invade, restore barrier properties of the epithelium, antagonize the effect of inflammatory mediators such as cytokines on epithelial integrity, or restore epithelial transport functions that have been deranged by either infection or inflammation.
Mechanism of diarrhoeal disease in the setting of infection with Salmonella
The final story relates to our efforts to establish the mechanisms whereby invasive pathogens, and specifically non‐typhoidal Salmonella spp., induce diarrhoeal symptoms in vivo. Foodborne infections with Salmonella have been with us for millennia, and they continue to represent one of the most prevalent causes of infectious diarrhoeal diseases as well as imposing the greatest disease burden (Batz et al. 2013; Batz et al. 2014; Havelaar et al. 2015; Cummings et al. 2016). Even in developed countries, the disease may be serious enough to lead to death, particularly in vulnerable populations, and yet the mechanisms underpinning the development of diarrhoea have remained poorly understood. In part, this may have reflected the lack of a tractable small animal model in which to study diarrhoea in vivo. Oral infection of standard laboratory mice with Salmonella typhimurium results in a systemic disease rather than the bowel‐delimited infection with diarrhoea that is typical of the disease in humans (Santos et al. 2001). This has been ascribed to the fact that most inbred mouse strains harbour a mutation in Nramp1, an iron transporter that is critical to containing the infection in macrophages (Vassiloyanakopoulos et al. 1998; Loomis et al. 2014). My collaborators, Josh Fierer and Don Guiney, discovered that congenic mice expressing wild‐type Nramp1 developed diarrhoeal symptoms, measured as an increase in stool water, if they were pretreated with the antibiotic kanamycin and subsequently infected orally with Salmonella (Woo et al. 2008). We therefore decided to use these mice to explore the pathophysiological correlates of their diarrhoea.
We harvested colonic segments from the mice, mounted them in Ussing chambers, and examined their transport responses to forskolin and carbachol. Somewhat to our surprise, both basal and forskolin‐stimulated ion transport was reduced in infected animals compared with kanamycin‐treated controls, without an effect on the response to carbachol, in both proximal and distal colon (Marchelletta et al. 2013). Furthermore, bacterial invasion was required to produce these functional effects because they were not present when mice were infected with mutant bacteria lacking the ability efficiently to invade the epithelium (invA and sssV mutants). At least in part, the deficit in ion transport appeared to be attributable to an internalization of CFTR in the surface colonic epithelial cells of infected mice, without a change in expression of the transporter. On the other hand, in the distal colon, expression of the β‐subunit of ENaC was reduced by infection with wild‐type but not invasion‐deficient Salmonella, as examined by Western blotting, RT‐PCR and confocal immunohistochemistry. This reduction in the capacity for distal electrogenic sodium absorption would be expected to contribute to diarrhoeal responses, but only if excessive fluid was present in the distal colonic lumen. Therefore, we also examined the presence of ion transporters in the proximal colon following infection. Expression of DRA in surface epithelial cells was dramatically reduced by infection, again in an invasion‐dependent fashion (Marchelletta et al. 2013). On the other hand, NHE‐3, which partners with DRA to provide for electroneutral absorption of NaCl in the small and large intestine, was unaffected by infection. Nevertheless, these transporters function in a coupled fashion and the loss even of only DRA would be expected to significantly reduce NaCl, and thus fluid, uptake in the gut. This effect, moreover, would be expected to be bolstered by our finding that infection also resulted in mislocalization of the sodium/potassium ATPase that provides the driving force for electroneutral NaCl absorption as well as other active transport mechanisms in the gut. Confocal microscopy revealed that infection with wild‐type Salmonella resulted in a redistribution of sodium/potassium ATPase from its normal location in the basolateral membrane of colonic epithelial cells to the apical membrane, perhaps also implying that the ‘fence’ function of the epithelial tight junctions is compromised in the setting of infection (Marchelletta et al. 2013).
Our current work focuses on the underlying mechanisms whereby Salmonella induces changes in the expression and localization of ion transporters in the colon. A clue came from the observation that the compartment where high levels of basolateral NKCC1 staining is seen in the colonic crypts was expanded in infected mice, without a significant change in expression of the cotransporter (Marchelletta et al. 2013). We hypothesized that the loss of some absorptive transporters from surface colonocytes might similarly reflect epithelial immaturity secondary to increased epithelial turnover in infected mice. Indeed, measurements of crypt depth and Ki67 staining for proliferative cells revealed that the proliferative compartment is significantly expanded in mice infected with wild‐type Salmonella, but not invasion‐deficient mutants (Marchelletta et al. 2013). We are currently exploring whether Salmonella influences cell fate decisions in the colon such that differentiation into secretory lineages, such as goblet cells, is favoured in infected mice at the expense of absorptive cells.
We have also explored whether the neutrophilic infiltration that has long been associated with Salmonella infection is required for diarrhoeal symptoms. Indeed, reports have long suggested that neutrophils contribute significantly to disease pathogenesis (Maenza et al. 1970; Giannella, 1979). However, when we reproduced our infection model in mice lacking the receptor for interleukin‐8 (IL‐8), the diarrhoeal response continued unabated despite a complete lack of infiltrating neutrophils in infected tissues (Marchelletta et al. 2015). Furthermore, the absence of the IL‐8 receptor failed to prevent the loss of DRA expression and the crypt hyperplasia that were associated with infection (Marchelletta et al. 2015).
In summary, therefore, Salmonella infection in vivo is accompanied by changes in ion transport responses in the colon as well as reduced expression of some, but not all, absorptive transporters. The physiological effect appears to be a reduction in the capacity of surface epithelial cells to absorb NaCl or Na+ in the proximal and distal colon, respectively (Fig. 4). We propose that this is the primary mechanism whereby diarrhoea arises. At the same time, secretory function of the colonic crypts is also reduced, which would be expected to result in defective flushing action, and perhaps is permissive for continued bacterial invasion (Keely et al. 2012) (Fig. 4). Ultimately, understanding how Salmonella impacts transport function may improve our ability to treat the associated diarrhoea. As our food system becomes more consolidated, the risks for foodborne illnesses such as that induced by Salmonella may rise (Havelaar et al. 2015). Indeed, just last year, the chief executive of a large peanut butter company was sentenced to 28 years in prison for knowingly shipping product that was tainted with Salmonella (https://www.washingtonpost.com/national/health‐science/former‐peanut‐executive‐sentenced‐to‐28‐years‐in‐prison‐for‐outbreak‐that‐killed‐nine‐people‐sickened‐hundreds/2015/09/21/aba7500e‐60a7‐11e5‐8e9e‐dce8a2a2a679_story.html?tid = a_inl). His company was a major supplier to schools, hospitals and nursing homes, which house individuals who are probably the most susceptible to life‐threatening Salmonella infections. Almost a thousand people were made ill by the resulting outbreak of salmonellosis, and at least nine died.
Figure 4. Comparison of colonic transport properties in health and in the setting of infection with non‐typhoidal Salmonella spp.
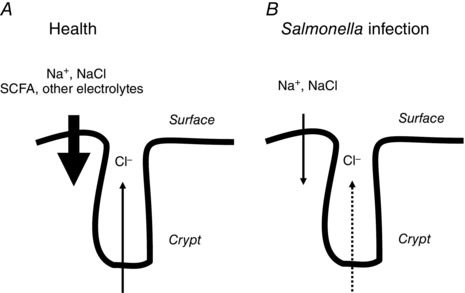
A, in health, absorption of electrolytes and short chain fatty acids (SCFA) by surface epithelial cells quantitatively outweighs lower levels of chloride secretion arising predominantly from the crypts. Thus, while fluid secretion is ongoing, the net effect is that water is reclaimed from the luminal contents. B, in the setting of Salmonella infection, there is loss of expression of DRA and ENaC from the surface epithelial cells of the proximal and distal colon, respectively. This probably accounts for fluid loss and diarrhoeal symptoms. At the same time, chloride secretion by the crypts is also reduced, as indicated by the dotted arrow. The associated reduction in the flushing action that is normally driven by crypt secretion of chloride may result in a loss of crypt sterility and increased infection.
Conclusions
The vignettes presented hopefully convince the reader that intestinal homeostasis requires precise control of epithelial transport and barrier functions. Thus, interplay amongst a large number of endogenous and exogenous factors controls the epithelium both minute by minute (as needed to respond to a meal) as well as over longer time scales. An important exogenous factor that regulates the epithelium is the immense and dynamic microbiota that establishes an intimate and life‐long relationship with the epithelial surface. Some beneficial effects of the microbiota, particularly for epithelial physiology, can be harnessed by the provision of probiotics. On the other hand, with its large surface area and physiological imperative to permit uptake of substances from the external environment, the epithelium is also vulnerable to orally delivered pathogens, many of which cause diarrhoea. It is our hope that an improved understanding of the response of the epithelium to such pathogens, as well as overall hormonal mechanisms that regulate epithelial function, may yield novel targets for the therapy of diseases that remain a scourge worldwide (Havelaar et al. 2015).
Additional information
Competing interests
None declared.
Funding
The studies described here have been made possible by grant support received from the National Institute of Allergy and Infectious Diseases (AI077661), National Institute of Diabetes, Digestive and Kidney Diseases (DK28305), and the National Center for Complementary and Alternative Medicine (AT01180) (all affiliated with the National Institutes of Health, USA) as well as the Crohn's and Colitis Foundation of America.
Acknowledgements
I am grateful to Ms Glenda Wheeler for assistance with manuscript submission as well as the following current and former members of my laboratory who contributed to the studies described: Lone Bertelsen, Melanie Gareau, Elaine Hanson, Stephen Keely, Rachel Klinkenburg, Beomjae Lee, Ronald Marchelletta, Declan McCole, Colin Reardon, Elise Roel, Jane Smitham, Silvia Resta‐Lenert, Jorge Uribe, Anouk van Berkel and Roos Visser. The contributions of our collaborators in this work – Robert Coffey, Joshua Fierer, Donald Guiney, Sharon Okamoto, Mark Donowitz and Nick Zachos – are also gratefully acknowledged.
Biography
Kim E. Barrett is Distinguished Professor of Medicine and Dean of the Graduate Division at the University of California, San Diego. She is a gastrointestinal physiologist who has devoted her career to gaining an understanding of the functioning of the intestinal epithelium as well as its dysfunction in disease states such as inflammatory bowel diseases and infectious diarrhoea. She has also been highly active in professional societies and in scholarly publishing. She is a past President of the American Physiological Society, former Editor‐in‐Chief of the American Journal of Physiology‐Cell Physiology, and current Editor‐in‐Chief of The Journal of Physiology.

References
- Asfaha S, MacNaughton WK, Appleyard CB, Chadee K & Wallace JL (2001). Persistent epithelial dysfunction and bacterial translocation after resolution of intestinal inflammation. Am J Physiol Gastrointest Liver Physiol 281, G635–G644. [DOI] [PubMed] [Google Scholar]
- Barrett KE & Keely SJ (2000). Chloride secretion by the intestinal epithelium: molecular basis and regulatory aspects. Annu Rev Physiol 62, 535–572. [DOI] [PubMed] [Google Scholar]
- Barrett KE, Smitham J, Traynor‐Kaplan A & Uribe JM (1998). Inhibition of Ca2+‐dependent Cl− secretion in T84 cells: membrane target(s) of inhibition is agonist specific. Am J Physiol 274, C958–C965. [DOI] [PubMed] [Google Scholar]
- Batz M, Hoffmann S & Morris JG Jr (2014). Disease‐outcome trees, EQ‐5D scores, and estimated annual losses of quality‐adjusted life years (QALYs) for 14 foodborne pathogens in the United States. Foodborne Pathog Dis 11, 395–402. [DOI] [PubMed] [Google Scholar]
- Batz MB, Henke E & Kowalcyk B (2013). Long‐term consequences of foodborne infections. Infect Dis Clin North Am 27, 599–616. [DOI] [PubMed] [Google Scholar]
- Bayliss WM & Starling EH (1902). The mechanism of pancreatic secretion. J Physiol 28, 325–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck PL & Podolsky DK (1999). Growth factors in inflammatory bowel disease. Inflamm Bowel Dis 5, 44–60. [DOI] [PubMed] [Google Scholar]
- Brüls T & Weissenbach J (2011). The human metagenome: our other genome? Hum Mol Genet 20, R142–R148. [DOI] [PubMed] [Google Scholar]
- Chow JY, Uribe JM & Barrett KE (2000). A role for protein kinase Cε in the inhibitory effect of epidermal growth factor on calcium‐stimulated chloride secretion in human colonic epithelial cells. J Biol Chem 275, 21169–21176. [DOI] [PubMed] [Google Scholar]
- Cummings PL, Kuo T, Javanbakht M, Shafir S, Wang M & Sorvillo F (2016). Salmonellosis hospitalizations in the United States: Associated chronic conditions, costs, and hospital outcomes, 2011, trends 2000–2011. Foodborne Pathog Dis 13, 40–48. [DOI] [PubMed] [Google Scholar]
- Frieling T, Wood JD & Cooke HJ (1992). Submucosal reflexes: distension‐evoked ion transport in the guinea pig distal colon. Am J Physiol 263, G91–G96. [DOI] [PubMed] [Google Scholar]
- Giannella RA (1979). Importance of the intestinal inflammatory reaction in salmonella‐mediated intestinal secretion. Infect Immun 23, 140–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig E & Sandle GI (2000). Diarrhea in ulcerative colitis. The role of altered colonic sodium transport. Ann NY Acad Sci 915, 327–332. [DOI] [PubMed] [Google Scholar]
- Greig ER, Boot‐Handford RP, Mani V & Sandle GI (2004). Decreased expression of apical Na+ channels and basolateral Na+, K+‐ATPase in ulcerative colitis. J Pathol 204, 84–92. [DOI] [PubMed] [Google Scholar]
- Havelaar AH, Kirk MD, Torgerson PR, Gibb HJ, Hald T, Lake RJ, Praet N, Bellinger DC, de Silva NR, Gargouri N, Speybroeck N, Cawthorne A, Mathers C, Stein C, Angulo FJ & Devleesschauwer B; World Health Organization Foodborne Disease Burden Epidemiology Reference Group (2015). World Health Organization global estimates and regional comparisons of the burden of foodborne disease in 2010. PLoS Med 12, e1001923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst BH (2004). Secretin and the exposition of hormonal control. J Physiol 560, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakab RL, Collaco AM & Ameen NA (2011). Physiological relevance of cell‐specific distribution patterns of CFTR, NKCC1, NBCe1, and NHE3 along the crypt‐villus axis in the intestine. Am J Physiol Gastrointest Liver Physiol 300, G82–G98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakab RL, Collaco AM & Ameen NA (2013). Characterization of CFTR High Expresser cells in the intestine. Am J Physiol Gastrointest Liver Physiol 305, G453–G465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato A & Romero MF (2011). Regulation of electroneutral NaCl absorption by the small intestine. Annu Rev Physiol 73, 261–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keely S, Kelly CJ, Weissmueller T, Burgess A, Wagner BD, Robertson CE, Harris JK & Colgan SP (2012). Activated fluid transport regulates bacterial‐epithelial interactions and significantly shifts the murine colonic microbiome. Gut Microbes 3, 250–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keely SJ, Uribe JM & Barrett KE (1998). Carbachol stimulates transactivation of epidermal growth factor receptor and mitogen‐activated protein kinase in T84 cells. Implications for carbachol‐stimulated chloride secretion. J Biol Chem 273, 27111–27117. [DOI] [PubMed] [Google Scholar]
- Kunzelmann K & Mall M (2002). Electrolyte transport in the mammalian colon: mechanisms and implications for disease. Physiol Rev 82, 245–289. [DOI] [PubMed] [Google Scholar]
- Laforenza U (2012). Water channel proteins in the gastrointestinal tract. Mol Aspects Med 33, 642–650. [DOI] [PubMed] [Google Scholar]
- Loomis WP, Johnson ML, Brasfield A, Blanc MP, Yi J, Miller SI, Cookson BT & Hajjar AM (2014). Temporal and anatomical host resistance to chronic Salmonella infection is quantitatively dictated by Nramp1 and influenced by host genetic background. PLoS One 9, e111763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK & Knight R (2012). Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCole DF, Keely SJ, Coffey RJ & Barrett KE (2002). Transactivation of the epidermal growth factor receptor in colonic epithelial cells by carbachol requires extracellular release of transforming growth factor‐α. J Biol Chem 277, 42603–42612. [DOI] [PubMed] [Google Scholar]
- McCole DF, Rogler G, Varki N & Barrett KE (2005). Epidermal growth factor partially restores colonic ion transport responses in mouse models of chronic colitis. Gastroenterology 129, 591–608. [DOI] [PubMed] [Google Scholar]
- McCole DF, Truong A, Bunz M & Barrett KE (2007). Consequences of direct versus indirect activation of epidermal growth factor receptor in intestinal epithelial cells are dictated by protein‐tyrosine phosphatase 1B. J Biol Chem 282, 13303–13315. [DOI] [PubMed] [Google Scholar]
- Maenza RM, Powell DW, Plotkin GR, Formal SB, Jervis HR & Sprinz H (1970). Experimental diarrhea: salmonella enterocolitis in the rat. J Infect Dis 121, 475–485. [DOI] [PubMed] [Google Scholar]
- Marchelletta RR, Gareau MG, McCole DF, Okamoto S, Roel E, Klinkenberg R, Guiney DG, Fierer J & Barrett KE (2013). Altered expression and localization of ion transporters contribute to diarrhea in mice with Salmonella‐induced enteritis. Gastroenterology 145, 1358–1368.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchelletta RR, Gareau MG, Okamoto S, Guiney DG, Barrett KE & Fierer J (2015). Salmonella‐induced diarrhea occurs in the absence of IL‐8 receptor (CXCR2)‐dependent neutrophilic inflammation. J Infect Dis 212, 128–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melvin JE, Park K, Richardson L, Schultheis PJ & Shull GE (1999). Mouse down‐regulated in adenoma (DRA) is an intestinal Cl−/HCO3 − exchanger and is up‐regulated in colon of mice lacking the NHE3 Na+/H+ exchanger. J Biol Chem 274, 22855–22861. [DOI] [PubMed] [Google Scholar]
- Panwala CM, Jones JC & Viney JL (1998). A novel model of inflammatory bowel disease: mice deficient for the multiple drug resistance gene, mdr1a, spontaneously develop colitis. J Immunol 161, 5733–5744. [PubMed] [Google Scholar]
- Piiper A & Zeuzem S (2004). Receptor tyrosine kinases are signaling intermediates of G protein‐coupled receptors. Curr Pharm Des 10, 3539–3545. [DOI] [PubMed] [Google Scholar]
- Resta‐Lenert S & Barrett KE (2003). Live probiotics protect intestinal epithelial cells from the effects of infection with enteroinvasive Escherichia coli (EIEC). Gut 52, 988–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resta‐Lenert S & Barrett KE (2006). Probiotics and commensals reverse TNF‐α‐ and IFN‐γ‐induced dysfunction in human intestinal epithelial cells. Gastroenterology 130, 731–746. [DOI] [PubMed] [Google Scholar]
- Resta‐Lenert S, Smitham J & Barrett KE (2005). Epithelial dysfunction associated with the development of colitis in conventionally housed mdr1a−/− mice. Am J Physiol Gastrointest Liver Physiol 289, G1176–G1176. [DOI] [PubMed] [Google Scholar]
- Resta‐Lenert SC & Barrett KE (2009). Modulation of intestinal barrier properties by probiotics: role in reversing colitis. Ann NY Acad Sci 1165, 175–182. [DOI] [PubMed] [Google Scholar]
- Santos RL, Zhang S, Tsolis RM, Kingsley RA, Adams LG & Bäumler AJ (2001). Animal models of Salmonella infections: enteritis versus typhoid fever. Microbes Infect 3, 1335–1344. [DOI] [PubMed] [Google Scholar]
- Shanahan F (2009). Therapeutic implications of manipulating and mining the microbiota. J Physiol 587, 4175–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanahan F & Quigley EMM (2014). Manipulation of the microbiota for treatment of IBS and IBD – challenges and controversies. Gastroenterology 146, 1554–1563. [DOI] [PubMed] [Google Scholar]
- Simpson JE, Schweinfest CW, Shull GE, Gawenis LR, Walker NM, Boyle KT, Soleimani M & Clarke LL (2007). PAT‐1 (Slc26a6) is the predominant apical membrane Cl−/HCO3 − exchanger in the upper villous epithelium of the murine duodenum. Am J Physiol Gastrointest Liver Physiol 292, G1079–G1088. [DOI] [PubMed] [Google Scholar]
- Talbot C & Lytle C (2010). Segregation of Na/H exchanger‐3 and Cl/HCO3 exchanger SLC26A3 (DRA) in rodent cecum and colon. Am J Physiol Gastrointest Liver Physiol 299, G358–G367. [DOI] [PubMed] [Google Scholar]
- Uribe JM & Barrett KE (1997). Nonmitogenic actions of growth factors: an integrated view of their role in intestinal physiology and pathophysiology. Gastroenterology 112, 255–268. [PubMed] [Google Scholar]
- Uribe JM, Gelbmann CM, Traynor‐Kaplan AE & Barrett KE (1996a). Epidermal growth factor inhibits Ca2+‐dependent Cl− transport in T84 human colonic epithelial cells. Am J Physiol 271, C914–C922. [DOI] [PubMed] [Google Scholar]
- Uribe JM, Keely SJ, Traynor‐Kaplan AE & Barrett KE (1996b). Phosphatidylinositol 3‐kinase mediates the inhibitory effect of epidermal growth factor on calcium‐dependent chloride secretion. J Biol Chem 271, 26588–26595. [DOI] [PubMed] [Google Scholar]
- VanDussen KL, Carulli AJ, Keeley TM, Patel SR, Puthoff BJ, Magness ST, Tran IT, Maillard I, Siebel C, Kolterud A, Grosse AS, Gumucio DL, Ernst SA, Tsai YH, Dempsey PJ & Samuelson LC (2012). Notch signaling modulates proliferation and differentiation of intestinal crypt base columnar stem cells. Development 139, 488–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassiloyanakopoulos AP, Okamoto S & Fierer J (1998). The crucial role of polymorphonuclear leukocytes in resistance to Salmonella dublin infections in genetically susceptible and resistant mice. Proc Natl Acad Sci USA 95, 7676–7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh MJ, Smith PL, Fromm M & Frizzell RA (1982). Crypts are the site of intestinal fluid and electrolyte secretion. Science 218, 1219–1221. [DOI] [PubMed] [Google Scholar]
- Woo H, Okamoto S, Guiney D, Gunn JS & Fierer J (2008). A model of Salmonella colitis with features of diarrhea in SLC11A1 wild‐type mice. PLoS One 3, e1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y & Jobin C (2014). Microbial imbalance and intestinal pathologies: connections and contributions. Dis Model Mech 7, 1131–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachos NC, Tse M & Donowitz M (2005). Molecular physiology of intestinal Na+/H+ exchange. Annu Rev Physiol 67, 411–443. [DOI] [PubMed] [Google Scholar]