

Abstract
Type 2 diabetes mellitus (T2DM) is associated with an increased risk of bone fractures without reduction of bone mineral density. The cholesterol oxide 7-ketocholesterol (7KCHO) has been implicated in numerous diseases such as atherosclerosis, Alzheimer's disease, Parkinson's disease, cancer, age-related macular degeneration and T2DM. In the present study, 7KCHO decreased the viability of MC3T3-E1 cells, increased reactive oxygen species (ROS) production and apoptotic rate, and upregulated the caspase-3/7 pathway. Furthermore, these effects of 7KCHO were abolished by pre-incubation of the cells with N-acetylcysteine (NAC), an ROS inhibitor. Also, 7KCHO enhanced the mRNA expression of two endoplasmic reticulum (ER) stress markers; CHOP and GRP78, in MC3T3-E1 cells. Pre-incubation of the cells with NAC suppressed the 7KCHO-induced upregulation of CHOP, but not GRP78. In conclusion, we demonstrated that 7KCHO induced apoptosis of MC3T3-E1 cells associated with ROS generation, ER stress, and caspase-3/7 activity, and the effects of 7KCHO were abolished by the ROS inhibitor NAC. These findings may provide new insight into the relationship between oxysterol and pathophysiology of osteoporosis seen in T2DM.
Keywords: 7-ketocholesterol, MC3T3-E1 cells, Reactive oxygen species (ROS), N-acetylcysteine (NAC), Apoptosis
Highlights
-
•
We examined the effects of 7-ketocholesterol (7KCHO) on MC3T3-E1 cells.
-
•
7KCHO increased reactive oxygen species (ROS) and apoptosis.
-
•
7KCHO enhanced CHOP and GRP78 expression.
-
•
N-acetylcysteine suppressed 7KCHO-induced ROS, apoptosis and CHOP expression.
1. Introduction
Osteoporosis is a common disease characterized by systemic impairment of bone mass, strength, and microarchitecture, thereby increasing bone fragility [1]. In the Chiba bone survey [2], a logistic regression analysis revealed that bone fracture was closely associated with not only low bone mass but also age, fall, family histories of kyphosis and hip fracture, type 2 diabetes mellitus (T2DM), kidney disease, menopause, as well as lifestyle factors of dieting, exercise and alcohol.
Several studies have shown that T2DM is associated with an increased fracture risk without reduction of bone mineral density (BMD) [3], [4], [5], [6]. This paradox between higher BMD and increased fracture risk in T2DM may be explained by poor bone quality. Endo et al. [7] reported that serum 7-ketocholesterol (7KCHO) was significantly higher in subjects with T2DM (n = 137, 33.8 ng/ml) compared to non-diabetic healthy subjects (n = 89, 16.1 ng/ml). We also have reported that 7KCHO has an apoptosis-inducing effect on vascular smooth muscle cells [8], [9], [10]. 7KCHO is a naturally occurring cholesterol oxide formed by auto-oxidation of cholesterol and cholesterol-fatty acid esters [11]. This oxysterol is known to be highly inflammatory both in vitro [12], [13] and in vivo [14]. Its inflammatory and cytotoxic properties have been implicated in the pathogenesis of numerous aging-related diseases [14] including atherosclerosis [12], [15], [16], Alzheimer's disease [13], [17], cancer [18], Parkinson's disease [17] and age-related macular degeneration [17], [19].
However, there is so far no evidence regarding the effect of 7KCHO on osteoporosis. This study was conducted to determine the influence of 7KCHO on the osteoblastic cell line MC3T3-E1 in the absence or presence of the antioxidant N-acetylcysteine (NAC), an acetylated amino acid and a precursor of glutathione. Furthermore, we investigated the effect of 7KCHO on endoplasmic reticulum (ER) stress in MC3T3-E1 cells.
2. Materials and methods
2.1. Cell culture and reagents
Mouse monoclonal osteoblastic cell line MC3T3-E1 derived from newborn mouse calvaria was obtained from RIKEN Cell Bank (Tsukuba, Japan). The cells were cultured in a growth medium of α-minimum essential medium (α-MEM) (Gibco, Grand Island, USA) supplemented with 10% fetal bovine serum (FBS), 100 mg/dl glucose and antibiotics (100 U/ml of penicillin G and 100 mg/ml of streptomycin) at 37 °C in a humidified atmosphere of 5% CO2, and were maintained at 90–95% confluence unless otherwise indicated. Then cells were pre-incubated overnight with or without different concentrations (2.5 or 5.0 mM) of NAC, followed by incubation with different concentrations (0, 12.5, 25 and 50 μM) of 7KCHO for defined times. Each experiment repeated at least three times, and data are the results of three independent experiments.
2.2. Cell viability assay
The viability of MC3T3-E1 cells treated with different concentrations of 7KCHO was determined using a WST-8 cell counting kit (Dojindo Laboratories, Kumamoto, Japan). The cells were seeded in triplicate into 24-well plates at a density of 1.0 × 104 cells per well. After reaching confluence, the medium was removed, and the cells were incubated for 24 h in α-MEM/10% FBS containing different concentrations (0, 12.5, 25 and 50 μM) of 7KCHO. Then, the medium in each well was replaced with 1000 μl of α-MEM/0% FBS, and 100 μl of a working solution of WST-8 was added. After incubation for 1 h at 37 °C in a humidified atmosphere of 5% CO2, 100 μl of the medium was transferred to a well of a 96-well plate and absorbance was measured at 450 nm using a micro-spectrophotometer (GloMax Multi Detection System; Promega BioSystems Sunnyvale, Sunnyvale, CA, USA). The percentage of viable cells was calculated by comparison to that of control well.
2.3. Detection of reactive oxygen species (ROS) production
After culture in 6-well plates under varying experimental conditions, cells were incubated with 3 μM 2′,7′-dichorodihydrofluorescein diacetate (Invitrogen Corp. Carlsbad, CA, USA) for 30 min. After incubation, cells were washed with phosphate buffered saline (PBS), trypsinized, and resuspended in PBS solution. Then, samples were run on a Becton Dickinson FACScalibur (Immunocytometry Systems, San Jose, CA, USA) equipped with a 15 mW, 488 nm argon laser and filter configuration. Cell samples (20,000 cells) were analyzed using Cell Quest Pro software (BD Biosciences).
2.4. Analysis of apoptosis
After culture in 6-well plates under varying experimental conditions, cellular DNA was quantitated using Sigma-Aldrich propidium iodide staining solution. Cells were trypsinized, washed with PBS and fixed with cold 70% ethanol. The fixed cells were treated with 0.01% RNase (10 mg/ml, Sigma, St. Louis, MO, USA) for 10 min at 37 °C and then stained with 0.05% propidium iodide for 20 min in the dark. Cell cycle distribution was determined using a Becton Dickinson FACScalibur, and the percentage of cells in sub G0/G1 phase was determined.
MC3T3-E1 cells incubated in 96-well plates were washed twice with PBS, and caspase-3/7 activity was determined using the Caspase-Glo® 3/7 Assay (Promega, Fitchburg, WI, USA) according to the manufacturer's protocol. Caspase-3/7 activity was corrected for mean cell number calculated for each group.
2.5. Reverse transcriptase polymerase chain reaction
Total cellular RNA was extracted from MC3T3-E1 cells using an RNeasy kit (Qiagen, Courtaboeuf, France), and complementary DNA was synthesized using a reverse transcription PCR kit (TaKaRa, Tokyo, Japan) according to the manufacturer's instructions. The RNA concentrations were determined by measuring absorbance at 260 nm. Then, reverse transcription PCR was performed using 2 μg of reverse transcribed total RNA. Expression of the housekeeping gene 18S was used as an internal standard. CHOP and GRP78 levels were detected using primers for CHOP (sense 5′-AATAACAGCCGGAACCTGAGGA-3′, antisense 5′-ACTCAGCTGCCATGACTGCAC-3′), GRP 78 (sense 5′-GAACACTGTGGTACCCACCAAGAA-3′, antisense 5′-TCCAGTCAGATCAAATGTACCCAGA-3′), and 18S (sense 5′-TTCTGGCCAACGGTCTAGACAAC-3′, antisense 5′-CCAGTGGTCTTGGTGTGCTGA-3′). Polymerase chain reaction was run on a Stratagene Mx3005P (Agilent Technologies, Santa Clara, CA, USA).
2.6. Statistical analysis
All data are expressed as mean ± standard deviation (SD). Statistical analyses were performed using SPSS software (version 11.5, Chicago, IL, USA). Treatment effects were evaluated using one-way ANOVA followed by Bonferroni multiple comparison test or Student's t-test, and p values < 0.05 were considered significant.
3. Results
3.1. Effects of 7KCHO on MC3T3-E1 cell viability
Fig. 1 shows cell viability of MC3T3-E1 cells at 24 h after the addition of 7KCHO (0, 12.5, 25 or 50 μM). Addition of 7KCHO significantly reduced viability of MC3T3-E1 cells in a dose-dependent manner.
Fig. 1.
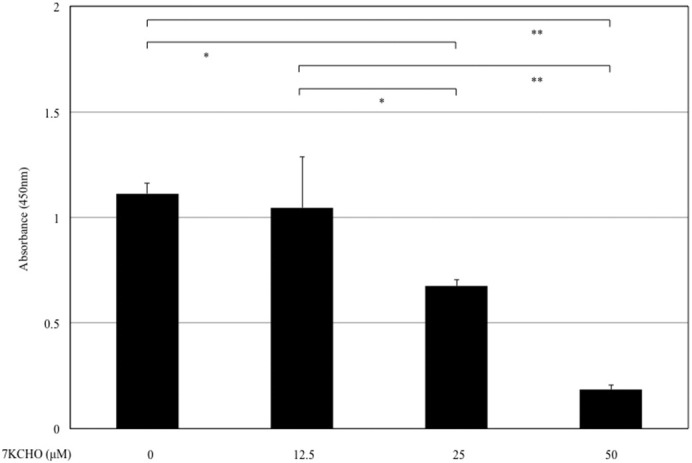
Effect of 7-ketocholesterol (7KCHO) on viability of MC3T3-E1 cells. MC3T3-E1 cells were seeded in 24-well plates (1.0 × 104 cells per well) and cultured until reaching 90–95% confluence. 7KCHO at indicated concentrations were added. Cell viability was assayed using a WST-8 cell counting kit at 24 h after addition of 7KCHO. Data are presented as mean ± SD from three independent experiments. *p < 0.05, **p < 0.01; one-way ANOVA followed by Bonferroni multiple comparison test.
3.2. Effect of 7KCHO on intracellular ROS production in MC3T3-E1 cells with or without NAC
The histograms in Fig. 2 show intracellular ROS production of MC3T3-E1 cells at 24 h after the addition of 7KCHO (12.5, 25 or 50 μM). Addition of 7KCHO caused an increase in intracellular ROS production in a dose-dependent manner, as indicated by a progressive rightward shift of the histogram from control with increasing 7KCHO concentration (Fig. 2A). Pre-incubation of the cells with NAC (5.0 mM) suppressed ROS production induced by 7KCHO (Fig. 2B).
Fig. 2.
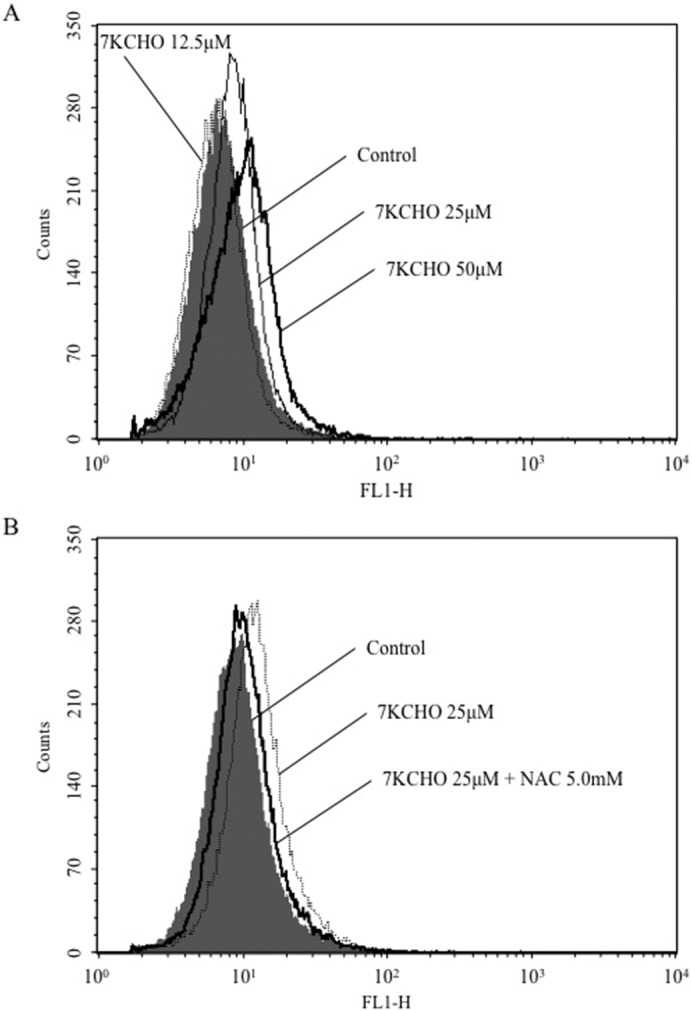
Effect of 7-ketocholesterol (7KCHO) on production of reactive oxygen species (ROS) in MC3T3-E1 cells, with or without N-acetylcysteine (NAC).
(A and B) MC3T3-E1 cells were seeded in 6-well plates (1.0 × 104 cells per well) and cultured until reaching 90–95% confluence. The cells were pre-incubated with or without NAC for 24 h, and 7KCHO at indicated concentrations was added. ROS production was analyzed using 2′,7′-dichorodihydrofluorescein diacetate followed by flow cytometry. Changes in ROS are shown in FL1 histograms.
3.3. Effect of 7KCHO on caspase activity in MC3T3-E1 cells with or without NAC
A luminescent assay was performed to measure caspase-3/7 activity. The addition of 7KCHO (50 μM) significantly increased caspase-3/7 activity (Fig. 3A). Pre-incubation of the cells with NAC (5.0 mM) significantly suppressed the caspase-3/7 activity upregulated by 7KCHO (Fig. 3B).
Fig. 3.

Effects of 7-ketocholesterol (7KCHO) on caspase-3/7 activity and quantitative analysis of apoptosis in MC3T3-E1 cells, with or without N-acetylcysteine (NAC).
(A and B) Caspase-3/7 activity was assayed by luminescent assay. MC3T3-E1 cells were seeded into 96-well plates (1.0 × 104 cells per well), and cultured until reaching 90–95% confluence. The cells were pre-incubated with or without NAC (2.5 or 5.0 mM) for 24 h, and 7KCHO at indicated concentrations were added. Luciferase activity was measured according to the protocol from Promega. (C and D) MC3T3-E1 cells were seeded into 6-well plates (1.0 × 104 cells per well), and cultured until reaching 90–95% confluence. The cells were pre-incubated with or without NAC (2.5 or 5.0 mM) for 24 h, and 7KCHO at indicated concentrations were added. Cells were stained with 0.05% propidium iodide after cell lysis and analyzed by flow cytometry. Apoptotic rate is the percentage of nuclei in the sub-G1 population representing DNA fragmentation as shown in FL2 histograms. Data are presented as mean ± SD from three independent experiments. *p < 0.05, **p < 0.01; one-way ANOVA followed by Bonferroni multiple comparison test.
3.4. Effects of 7KCHO on apoptosis in MC3T3-E1 cells with or without NAC
Analysis of DNA fragmentation using propidium iodide fluorescence was conducted to evaluate the apoptosis-inducing effect of 7KCHO in MC3T3-E1 cells pre-incubated with or without NAC. The addition of 7KCHO (50 μM) significantly increased apoptotic rate (Fig. 3C). Pre-incubation of cells with NAC (5.0 mM) significantly suppressed the apoptotic rate upregulated by 7KCHO (Fig. 3D).
3.5. Effects of 7KCHO on CHOP and GRP78 mRNA expression in MC3T3-E1 cells with or without NAC
Reverse transcription PCR analysis showed that CHOP and GRP78 mRNA expression in MC3T3-E1 cells was significantly enhanced by 7KCHO (25 μM) (Fig. 4A and B). Pre-incubation of the cells with NAC (5.0 mM) suppressed the 7KCHO-upregulated CHOP mRNA expression but not 7KCHO-upregulated GRP78 mRNA expression (Fig. 4C and D).
Fig. 4.

Effects of 7-ketocholesterol (7KCHO) on CHOP and GRP78 mRNA expression in MC3T3-E1 cells, with or without N-acetylcysteine (NAC).
MC3T3-E1 cells were seeded into 6-well plates (1.0 × 104 cells per well), and cultured until reaching 90–95% confluence. The cells were pre-incubated with or without NAC (5.0 mM) for 24 h, and 7KCHO at indicated concentrations was added. CHOP and GRP78 expression was determined by reverse transcription PCR. (A and C) Production of CHOP in the presence of 7KCHO with or without NAC. (B and D) Production of GRP in the presence of 7KCHO with or without NAC. Data are presented as mean ± SD from three independent experiments. †p < 0.05, ‡p < 0.01; Student's t-test. *p < 0.05, **p < 0.01; one-way ANOVA followed by Bonferroni multiple comparison test.
4. Discussion
Previous studies have shown the relationship of T2DM with increased risk for bone fractures. However, the effect of oxysterol, the key mediator involved in the pathophysiology of T2DM, on bone metabolism is not fully understood. In this study, 7KCHO decreased cell viability, increased ROS production and apoptotic rate, and upregulated caspase-3/7 activity in MC3T3-E1 cells. Furthermore, these effects of 7KCHO were abolished by pre-incubation of the cells with NAC. The present report is the first to demonstrate the effects of 7KCHO on MC3T3-E1 cells.
First we measured the effects of 7KCHO on MC3T3-E1 cells. 7KCHO reduced the viability of MC3T3-E1 cells probably by increasing apoptosis through increased ROS generation and upregulation of caspase-3/7-dependent pathway. These effects were inhibited by in the presence of the ROS inhibitor NAC. Ding et al. [20] also measured oxidative stress-induced ROS levels in MC3T3-E1 cells. They used hydrogen peroxide (H2O2) to induce oxidative stress, and reported that apoptosis was induced by manipulating ROS elevation through exposure of MC3T3-E1 cells to hydrogen peroxide H2O2. ROS is generated in cells when challenged with various stresses, and ROS production is a common phenomenon of cellular metabolism [21]. However, abnormal ROS production leading to oxidative stress has been recognized as a major initiating factor for osteoporosis [22], [23], [24]. A previous study showed that reduced bone formation was commonly associated with increased oxidative stress in aged male and female mice [25]. An in vitro study even suggested that ROS reduces bone formation via inhibition of proliferation and calcification processes of osteoblastic cells [26].
ROS was an important component of the events leading to protein misfolding in the ER and ER stress [27]. For this reason, we next measured ER stress markers. In this study, 7KCHO increased the production of two ER stress markers; CHOP and GRP78. Increased production of CHOP induced by 7KCHO was suppressed by pre-incubation of the cells with NAC (5.0 mM). On the other hand, pre-incubation with NAC (5.0 mM) tended to suppress 7KCHO-induced increase in GRP78 production, but the difference was not significant. Huang et al. [28] reported that 7KCHO-induced inflammation was mediated mostly through the TLR4 receptor with some cross-activation of EGFR-related pathways both in vitro and in vivo, and also appeared to involve a robust ER stress response. They also suggested that nuclear factor-kappa B (NF-κB) partially mediated these responses since its inhibition attenuated the induction of CHOP and GRP78. As CHOP is well-known to be involved in ER stress-induced apoptosis [29], the ER stress response may be an indicator of 7KCHO-induced MC3T3-E1 cell apoptosis. On the other hand, GRP78 upregulation is believed to increase the capacity to buffer stress insults generated by the ER. Furthermore, due to its anti-apoptotic property, expression of GRP78 represents an important pro-survival response. GRP78 is one of the most important ER molecular chaperones. It is induced in response to ER stress and serves to refold misfolded or incompletely assembled proteins [30], [31]. Also GRP78 is known to inhibit caspase-mediated cell death by forming complexes with procaspase-7 and procaspase-12. The expression of GRP78 protects against various types of cell death induced by ER stress. If induction of GRP78 fails, cells enter ER stress-induced apoptosis [32], [33], [34]. These results suggest that 7KCHO induces apoptosis of MC3T3-E1 cells through ROS-dependent activation of CHOP, and GRP78 may increase to counteract excessive induction of CHOP. Guo et al. [35] also reported that both CHOP and GRP78 increased by thapsigargin, an inhibitor of Ca2 + ATPase in the ER, causing osteoblasts ER stress.
However, our study has limitations. Firstly, the growth medium contained normal glucose level. Secondly, we could not demonstrate statistically significant data at the concentration equivalent to human serum 7KCHO, and we had no choice but to adapt data of 25 and 50 μM. Further study is required to examine ROS and apoptosis under hyperglycemic condition, and also required to examine under the lower concentration of 7KCHO.
In conclusion, we demonstrated in vitro that 7KCHO induced apoptosis of MC3T3-E1 cells associated with increased ROS generation and ER stress and upregulated caspase-3/7 activity. These effects of 7KCHO were inhibited by the ROS inhibitor NAC. These findings may provide new insight into the relationship between oxysterol and pathophysiology of osteoporosis seen in T2DM.
Declaration of conflicting interests
The authors declare no conflicts of interest with respect to the research, authorship, and/or publication of this article.
Acknowledgment
We would like to thank all the staff members in our Center, who contributed to this study.
References
- 1.Seeman E., Delmas P.D. Bone quality–the material and structural basis of bone strength and fragility. N. Engl. J. Med. 2006;354:2250–2261. doi: 10.1056/NEJMra053077. [DOI] [PubMed] [Google Scholar]
- 2.Tatsuno I., Terano T., Nakamura M., Suzuki K., Kubota K., Yamaguchi J., Yoshida T., Suzuki S., Tanaka T., Shozu M. Lifestyle and osteoporosis in middle-aged and elderly women: Chiba bone survey. Endocr. J. 2013;60:643–650. doi: 10.1507/endocrj.ej12-0368. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz A.V. Diabetes mellitus: does it affect bone? Calcif. Tissue Int. 2003;73:515–519. doi: 10.1007/s00223-003-0023-7. [DOI] [PubMed] [Google Scholar]
- 4.Hofbauer L.C., Brueck C.C., Singh S.K., Dobnig H. Osteoporosis in patients with diabetes mellitus. J. Bone Miner. Res. 2007;22:1317–1328. doi: 10.1359/jbmr.070510. [DOI] [PubMed] [Google Scholar]
- 5.de II L., van der Klift M., de Laet C.E., van Daele P.L., Hofman A., Pols H.A. Bone mineral density and fracture risk in type-2 diabetes mellitus: the Rotterdam Study. Osteoporos. Int. 2005;16:1713–1720. doi: 10.1007/s00198-005-1909-1. [DOI] [PubMed] [Google Scholar]
- 6.Lunt M., Masaryk P., Scheidt-Nave C., Nijs J., Poor G., Pols H., Falch J.A., Hammermeister G., Reid D.M., Benevolenskaya L., Weber K., Cannata J., O'Neill T.W., Felsenberg D., Silman A.J., Reeve J. The effects of lifestyle, dietary dairy intake and diabetes on bone density and vertebral deformity prevalence: the EVOS study. Osteoporos. Int. 2001;12:688–698. doi: 10.1007/s001980170069. [DOI] [PubMed] [Google Scholar]
- 7.Endo K., Oyama T., Saiki A., Ban N., Ohira M., Koide N., Murano T., Watanabe H., Nishii M., Miura M., Sekine K., Miyashita Y., Shirai K. Determination of serum 7-ketocholesterol concentrations and their relationships with coronary multiple risks in diabetes mellitus. Diabetes Res. Clin. Pract. 2008;80:63–68. doi: 10.1016/j.diabres.2007.10.023. [DOI] [PubMed] [Google Scholar]
- 8.Miyashita Y., Shirai K., Ito Y., Watanabe J., Urano Y., Murano T., Tomioka H. Cytotoxicity of some oxysterols on human vascular smooth muscle cells was mediated by apoptosis. J. Atheroscler. Thromb. 1997;4:73–78. doi: 10.5551/jat1994.4.73. [DOI] [PubMed] [Google Scholar]
- 9.Nagayama D., Ishihara N., Bujo H., Shirai K., Tatsuno I. Effects of serotonin on expression of the LDL receptor family member LR11 and 7-ketocholesterol-induced apoptosis in human vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2014;446:906–910. doi: 10.1016/j.bbrc.2014.03.031. [DOI] [PubMed] [Google Scholar]
- 10.Urano Y., Shirai K., Watanabe H., Miyashita Y., Hashiguchi S. Vascular smooth muscle cell outgrowth, proliferation, and apoptosis in young and old rats. Atherosclerosis. 1999;146:101–105. doi: 10.1016/s0021-9150(99)00139-2. [DOI] [PubMed] [Google Scholar]
- 11.Dzeletovic S., Babiker A., Lund E., Diczfalusy U. Time course of oxysterol formation during in vitro oxidation of low density lipoprotein. Chem. Phys. Lipids. 1995;78:119–128. doi: 10.1016/0009-3084(95)02489-6. [DOI] [PubMed] [Google Scholar]
- 12.Brown A.J., Jessup W. Oxysterols and atherosclerosis. Atherosclerosis. 1999;142:1–28. doi: 10.1016/s0021-9150(98)00196-8. [DOI] [PubMed] [Google Scholar]
- 13.Vejux A., Lizard G. Cytotoxic effects of oxysterols associated with human diseases: induction of cell death (apoptosis and/or oncosis), oxidative and inflammatory activities, and phospholipidosis. Mol. Asp. Med. 2009;30:153–170. doi: 10.1016/j.mam.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 14.Amaral J., Lee J.W., Chou J., Campos M.M., Rodriguez I.R. 7-Ketocholesterol induces inflammation and angiogenesis in vivo: a novel rat model. PLoS One. 2013;8 doi: 10.1371/journal.pone.0056099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Reyk D.M., Brown A.J., Hult'en L.M., Dean R.T., Jessup W. Oxysterols in biological systems: sources, metabolism and pathophysiological relevance. Redox Rep. 2006;11:255–262. doi: 10.1179/135100006X155003. [DOI] [PubMed] [Google Scholar]
- 16.Hitsumoto T., Takahashi M., Iizuka T., Shirai K. Clinical significance of serum 7-ketocholesterol concentrations in the progression of coronary atherosclerosis. J. Atheroscler. Thromb. 2009;16:363–370. doi: 10.5551/jat.no703. [DOI] [PubMed] [Google Scholar]
- 17.Poli G., Biasi F., Leonarduzzi G. Oxysterols in the pathogenesis of major chronic diseases. Redox Biol. 2013;1:125–130. doi: 10.1016/j.redox.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang S.F., Chou Y.C., Mazumder N., Kao F.J., Nagy L.D., Guengerich F.P., Huang C., Lee H.C., Lai P.S., Ueng Y.F. 7-Ketocholesterol induces P-glycoprotein through PI3K/mTOR signaling in hepatoma cells. Biochem. Pharmacol. 2013;86:548–560. doi: 10.1016/j.bcp.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodriguez I.R., Larrayoz I.M. Cholesterol oxidation in the retina: implications of 7KCh formation in chronic inflammation and age-related macular degeneration. J. Lipid Res. 2010;51:2847–2862. doi: 10.1194/jlr.R004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding G., Zhao J., Jiang D. Allicin inhibits oxidative stress-induced mitochondrial dysfunction and apoptosis by promoting PI3K/AKT and CREB/ERK signaling in osteoblast cells. Exp. Ther. Med. 2016;11:2553–2560. doi: 10.3892/etm.2016.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kiffin R., Bandyopadhyay U., Cuervo A.M. Oxidative stress and autophagy. Antioxid. Redox Signal. 2006;8:152–162. doi: 10.1089/ars.2006.8.152. [DOI] [PubMed] [Google Scholar]
- 22.Cervellati C., Bonaccorsi G., Cremonini E., Bergamini C.M., Patella A., Castaldini C., Ferrazzini S., Capatti A., Picarelli V., Pansini F.S., Massari L. Bone mass density selectively correlates with serum markers of oxidative damage in post-menopausal women. Clin. Chem. Lab. Med. 2013;51:333–338. doi: 10.1515/cclm-2012-0095. [DOI] [PubMed] [Google Scholar]
- 23.Manolagas S.C. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr. Rev. 2010;31:266–300. doi: 10.1210/er.2009-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baek K.H., Oh K.W., Lee W.Y., Lee S.S., Kim M.K., Kwon H.S., Rhee E.J., Han J.H., Song K.H., Cha B.Y., Lee K.W., Kang M.I. Association of oxidative stress with postmenopausal osteoporosis and the effects of hydrogen peroxide on osteoclast formation in human bone marrow cell cultures. Calcif. Tissue Int. 2010;87:226–235. doi: 10.1007/s00223-010-9393-9. [DOI] [PubMed] [Google Scholar]
- 25.Almeida M., Han L., Martin-Millan M., Plotkin L.I., Stewart S.A., Roberson P.K., Kousteni S., O'Brien C.A., Bellido T., Parfitt A.M., Weinstein R.S., Jilka R.L., Manolagas S.C. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J. Biol. Chem. 2007;282:27285–27297. doi: 10.1074/jbc.M702810200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hosoya S., Suzuki H., Yamamoto M., Kobayashi K., Abiko Y. Alkaline phosphatase and type I collagen gene expressions were reduced by hydroxyl radical-treated fibronectin substratum. Mol. Genet. Metab. 1998;65:31–34. doi: 10.1006/mgme.1998.2734. [DOI] [PubMed] [Google Scholar]
- 27.Malhotra J.D., Kaufman R.J. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid. Redox Signal. 2007;9:2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 28.Huang J.D., Amaral J., Lee J.W., Rodriguez I.R. 7-Ketocholesterol-induced inflammation signals mostly through the TLR4 receptor both in vitro and in vivo. PLoS One. 2014;9 doi: 10.1371/journal.pone.0100985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sano R., Reed J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta. 2013;1833:3460–3470. doi: 10.1016/j.bbamcr.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaufman R.J. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13:1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- 31.Sherman M.Y., Goldberg A.L. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron. 2001;29:15–32. doi: 10.1016/s0896-6273(01)00177-5. [DOI] [PubMed] [Google Scholar]
- 32.Fu Y., Li J., Lee A.S. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res. 2007;67:3734–3740. doi: 10.1158/0008-5472.CAN-06-4594. [DOI] [PubMed] [Google Scholar]
- 33.Reddy R.K., Mao C., Baumeister P., Austin R.C., Kaufman R.J., Lee A.S. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. J. Biol. Chem. 2003;278:20915–20924. doi: 10.1074/jbc.M212328200. [DOI] [PubMed] [Google Scholar]
- 34.Wang M., Ye R., Barron E., Baumeister P., Mao C., Luo S., Fu Y., Luo B., Dubeau L., Hinton D.R., Lee A.S. Essential role of the unfolded protein response regulator GRP78/BiP in protection from neuronal apoptosis. Cell Death Differ. 2010;17:488–498. doi: 10.1038/cdd.2009.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo Y.S., Sun Z., Ma J., Cui W., Gao B., Zhang H.Y., Han Y.H., Hu H.M., Wang L., Fan J., Yang L., Tang J., Luo Z.J. 17Beta-estradiol inhibits ER stress-induced apoptosis through promotion of TFII-I-dependent Grp78 induction in osteoblasts. Lab. Investig. 2014;94:906–916. doi: 10.1038/labinvest.2014.63. [DOI] [PubMed] [Google Scholar]