

Abstract
Xanthomonas oryzae pv. oryzae ( Xoo) is a serious pathogen of rice causing bacterial leaf blight disease. Resistant varieties and breeding programs are being hampered by the emergence of highly virulent strains. Herein we report population based whole genome sequencing and analysis of 100 Xoo strains from India. Phylogenomic analysis revealed the clustering of Xoo strains from India along with other Asian strains, distinct from African and US Xo strains. The Indian Xoo population consists of a major clonal lineage and four minor but highly diverse lineages. Interestingly, the variant alleles, gene clusters and highly pathogenic strains are primarily restricted to minor lineages L-II to L-V and in particularly to lineage L-III. We could also find the association of an expanded CRISPR cassette and a highly variant LPS gene cluster with the dominant lineage. Molecular dating revealed that the major lineage, L-I is youngest and of recent origin compared to remaining minor lineages that seems to have originated much earlier in the past. Further, we were also able to identify core effector genes that may be helpful in efforts towards building durable resistance against this pathogen.
Rice is the staple food for more than half of the human population. The gram-negative bacterium, Xanthomonas oryzae pv. oryzae (Xoo) is a serious problem in rice cultivation. Xoo infections are not only endemic to Asia and West Africa, but also have been reported from Australia and Latin America1. The infection of the pathogen in the xylem tissues of rice leads to leaf blight symptoms, that were first characterized back in the late 19th century2,3. Introduction of Resistance (R) genes into rice cultivars has been considered to be the best management option for Xoo, and in this direction, at least 40 different R genes of rice have been identified till date against Xoo4. But every R gene is not efficient against every race of Xoo, due to the co-evolution of the pathogen along with the host5.
India is the second largest producer and also a major centre of diversity of rice. There are reports of some of the strains that can breakdown most of the major R genes deployed for resistance to Xoo in India5,6,7,8,9,10,11. Hence, a comprehensive understanding of the genetic diversity of the population of Xoo from India and its relationship with strains from the rest of the world is necessary. However, earlier efforts in this direction have been primarily limited to non-sequence based hyper-variable markers and few housekeeping genes12. Apart from resolving the relationship, there is also a need to study evolution of gene(s) that are known to be important for virulence, pathogenicity and fitness.
Advent of genomics era has revolutionized the field of bacteriology. Now by genome sequencing, we can generate and access complete genotype of an organism at an unprecedented rate and scale. Genome sequences of Xoo strains from other part of Asia, Africa and USA are already available. Apart from type strain of species X. oryzae, no other strain from India has been sequenced. Herein, we carried out whole genome sequencing of 100 Xoo strains, collected from 19 rice cultivating states in India in the last two decades. The pathotype information for 46 of these strains is available and they have been classified into eleven pathotypes that were assigned based on their reaction towards ten major resistance genes of rice5.
Apart from understanding the relationship of the Indian strains to those present worldwide, the present study allowed us to gain insights into the origin of lineages, pathotypes and highly pathogenic strains from India. Further, we were also able to analyze the evolutionary history of genes known to be important for virulence and pathogenesis. The study also provided novel insights into the origin of the closely related pathovar Xanthomonas oryzae pv. oryzicola ( Xoc) that specifically infects parenchymatous tissue. This mega-genomic resource would be invaluable in surveillance of the pathogen and future comparative studies of this phytopathogen.
Results
Whole genome sequencing and phylogenomic status of Xoo strains
We sequenced the whole genomes of 100 Xoo strains and one Xoc strain BXOR1. The raw reads for all strains were de novo assembled into genomes with <500 contigs and >100x coverage. The Xoo strains have an N50 value of ~18–24 kb while BXOR1 assembly has N50 value 46.7 kb. All the sequenced strains show conservation in genome size and number of genes. Assembly statistics and annotation features of these genomes are listed in Supplementary Table S1.
Sequenced Xoo strains have Average Nucleotide Identity (ANI) values >99% with the Xanthomonas oryzae type strain 35933 (XO35933) which are above the cut-off of 96% for delineation of novel species13. We constructed a phylogenomic marker genes based tree of sequenced strains along with their relatives whose sequences are available publically14. It suggested that the 106 Xoo strains that include 100 Indian Xoo strains from the present study, type strain belonging to India and five strains from other parts of Asia from the public domain form a distinct cluster that is closer to pathovar Xoc than African Xoo and USA Xo strains. Interestingly, Xoc strains appear to be a variant lineage of Xoo population (Fig. 1).
Figure 1. Phylogenomic markers based tree of Xanthomonas oryzae strains.
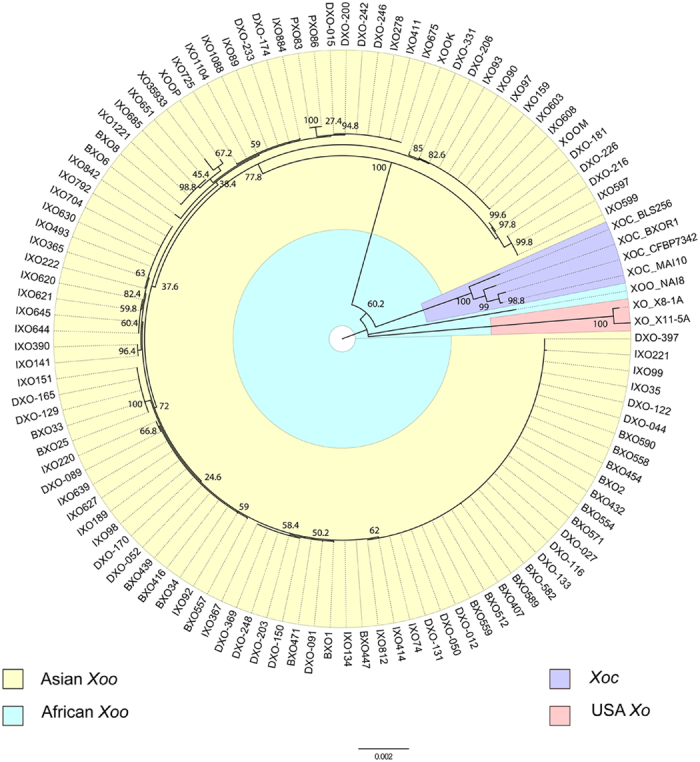
31 phylogenomic marker genes were extracted from the 113 genomes, concatenated and aligned using ClustalW algorithm. Maximum likelihood tree of conserved phylogenomic marker genes was constructed using General Time Reversible model (Gamma distributed with Invariant sites (G + I)). Bootstrap values shown on the nodes are percentage of 500 replicates. The scale bar (0.002) indicates the number of nucleotide substitutions per site. Clades from different geographical locations are coloured differently.
Clonal analysis reveals a major clonal lineage and minor diverse lineages
As the 106 Xoo strains formed a clade, distinct from African, USA and Xoc strains, this major group may be freely recombining and exchanging genes. Hence, we carried out an in-depth phylogenetic analysis specifically using regions not affected by recombination (see methods). Whole genome based tree (Fig. 2) showed the presence of five distinct lineages with lineage L-I encompassing >50% (55/100) of strains, a predominant lineage with high clonality. The rest of the four lineages, constitute the other half of Xoo population and are highly diverse than lineage L-I.
Figure 2. ClonalFrameML tree obtained from genomic sequences of 100 Indian Xoo strains and six Xoo strains from other parts of Asia.

Genomes of 106 strains were aligned and core genome was analysed using ClonalFrameML to obtain a tree considering recombination. Different lineages inferred are coloured differently. On the tree in circular way, information on seven different genes/clusters is marked. Moving outward in the circles, the strains are marked with having BXO8 type cellobiosidase allele (1), BXO8 type LPS cassette (2), genes having non-synonymous changes in raxX (3), raxST (4), raxA (5), raxB (6) and diameter of outermost circle indicating the variation in the number of CRISPR spacers.
It is interesting to note that lineages L-I and L-II are exclusively constituted by Indian Xoo strains. Philippine strains Xoo PXO86 (PXO86), Xoo PXO83 (PXO83) and Xoo PXO99A (XOOP) belong to lineage L-III. Japanese Xoo strain, Xoo MAFF 311018 (XOOM) belongs to lineage L-IV, while Korean strain Xoo KACC 10331 (XOOK) is an out-group to the clade consisting of lineages L-I to L-III. We also looked at the geographical distribution of various lineages in India (Fig. 3). While southern and eastern regions of the country are mainly dominated by lineages L-I, L-II and L-III, all five lineages were found in the northern region. The lineage L-II is restricted to the eastern parts of the country, excepting for one isolate from the northern region.
Figure 3. Map of India indicating the geographical origin of Xanthomonas oryzae pv. oryzae strains under study.
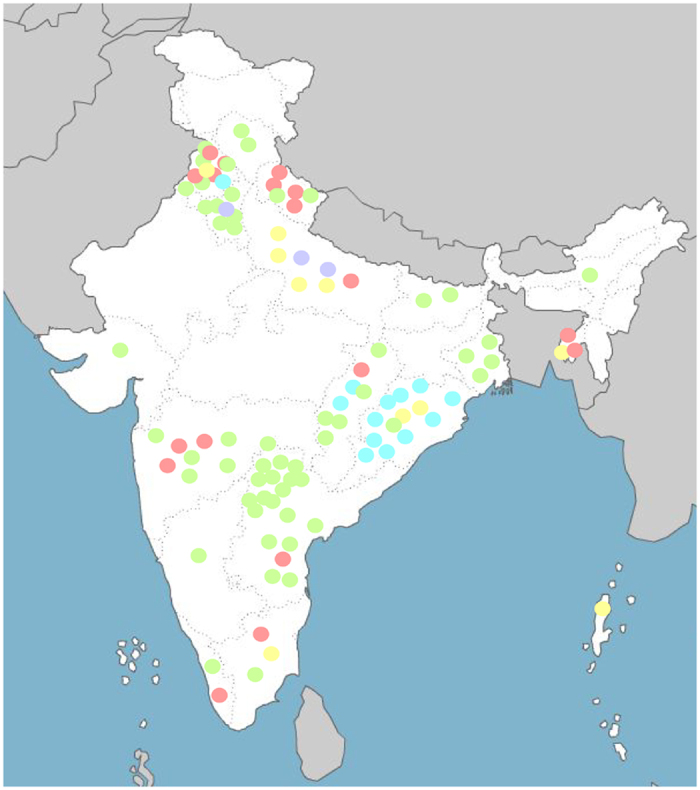
Strains belonging to different lineages are colored differently; Lineages L-I (in green), L–II (in blue), L-III (in pink), L-IV (in yellow) and L-V (in purple) (Map has been adapted from http://d-maps.com/carte.php?num_car=4182&lang=en).
Role of recombination in shaping the diversity of Xoo lineages
Since the ClonalFrameML tree is based on clonal regions of the genome, the tree and branch length relationship and diversity we observe, is based on mutation rate alone. The analysis showed that the ratio of recombination to mutation (R/θ) is 0.2586, the average length of the recombined fragment (δ) is 576 bp and average distance between donor and recipient (ν) is 0.015. Thus, the mutations are ~3.86 times more frequent than recombination, while the impact of recombination over mutation is 2.2 times higher towards the evolution of these strains.
Graphical representation of recombinational events in these Xoo strains is shown in Fig. 4. The graph shows a higher density of variation in lineages L-II to L-V as compared to lineage L-I, which has a comparatively lesser number of recombinational events as well as substitutions. Lineage L-II shows a higher density of yellow-red vertical bars which is consistent with its (R/θ) value (0.1734), implying ~5.76 times more mutational occurrences as compared to recombinational events. For lineage L-III recombinational events are higher as well as the substitutions, with a final impact of recombination 2.4 times higher than mutations. Thus, the strains in lineage L-I are less diverse than the strains found in all other lineages. Interestingly, three highly diverse strains DXO-216, IXO599 and IXO597 that are from lineage L-V are more basal and probably constitute the ancestral lineage.
Figure 4. Graphical representation of recombinational events in the Xanthomonas oryzae pv. oryzae strains.

The image shows the phylogenomic relationship of the strains along with sites of recombination and substitutions. Each clade is compared to its most recent common ancestor for variations and represented in different colored bars. Recombination events are marked by dark blue horizontal bars and substitutions by vertical lines. Light blue vertical sites refer to no substitution, white sites to the non-homoplasic substitutions whereas any other colour refers to homoplasic substitutions, with increase in redness from white to red marks increase in homoplasy.
Clonal analysis reveals ancestral and lineage associated pathotypes
For 46 strains that are part of this study, detailed pathotype information is available and they belong to eleven pathotypes5. The distribution of these strains in the phylogenetic tree was assessed. Two strains (IXO1088 and IXO1104) belonging to the most virulent pathotype XI, which can break down all 10 resistance genes, are restricted to highly diverse lineage L-III. Similarly, strains belonging to pathotype III-V are restricted to lineage L-I, while strains belonging to pathotype VI, II and IX are restricted to lineages L-II, L-III and L-IV respectively. Four pathotypes (I, VII, VIII and X) do not show any association or restriction to a particular lineage. Hence, these four may be ancestral, while others may be of recent origin.
Interestingly, the five lineages also differed in their reaction towards two major resistant genes, xa5 and xa13. These are recessive resistant genes, where the recessive allele has mutations in the coding region (xa5) or promoter region (xa13) that makes them recalcitrant for promoting bacterial growth and proliferation15,16. Looking at the phylogeny of Xoo strains pathotyped earlier5, it showed that strains compatible with xa5 are grouped in lineages L-I and L-III, while the strains compatible with xa13 are clustered in lineages L-II, L-IV and L-V.
Molecular dating of Xoo lineages
In order to estimate the age of establishment of Xoo lineages in India, we deployed Bayesian method approach. We first tested for the presence of a sufficient temporal signal in the dataset using root-to-tip regression approach implemented in TempEst17. Then we checked for both the strict and relaxed clock model to know which fits better for our data (Supplementary Table S2). The relaxed clock model analysis results showed higher likelihood log values with both harmonic mean and stepping stone methods for our data. Finding a better performance for relaxed clock model, we used this model to estimate the age of various nodes to determine the emergence time for Xoo strains (Supplementary Fig. S1). We used a mutation rate estimated in Xanthomonas pathogens earlier (2 × 10−5 mutations per gene per year)18 to define the priors in the analysis. The analysis suggested the emergence of these strains from a common ancestor around 0.97 [95% HPD (highest-probability density): 0.922–1.00]) Myr ago and emergence of lineage L-I around 0.3 [95% HPD: 0.267–0.354] Myr ago.
Variation across candidate virulence genes and hypervariable loci
Plant cells recognize various microbial signature molecules such as flagellin, lipopolysaccharide, etc. that act as Microbial/Pathogen Associated Molecular Patterns (MAMP/PAMP)19. To circumvent this recognition and hence triggered immunity, bacteria release specialized protein molecules directly into the host cell known as type III effectors using its well-evolved type III secretion system20. In turn, plants have also evolved specialized Resistance (R) genes that act in response to effector molecules21. Herein, we analyzed variation in well-known genes/cassettes that are associated with either damage associated molecular pattern (DAMP) (e.g. cellobiosidase gene) or PAMP (e.g. raxX, fliC, lipopolysaccharide cassette) and type III effectome that is known to counteract PAMP triggered immunity (PTI).
Cellobiosidase
Cellobiosidase, secreted by type II secretion system, is a major pathogenicity determinant of Xoo22. The phylogenetic tree of the cbsA gene of 100 Indian Xoo strains and six other Asian strains is not in congruence with the genome based phylogeny (Fig. 5A). There are two different alleles of cbsA (referred as BXO1 type and BXO8 type), marked on Fig. 2. Further looking at the amino acid level in the catalytic domain (33–457 aa), it showed the changes in 12 amino acid residue positions in both alleles (Fig. 5B). We further checked for the dN/dS ratio, which is 0.667, comparatively much higher than for a housekeeping gene rpoB, for which it is 0.001. We also checked for the selection pressure on amino acid residues using Selecton server23. Figure 5C shows the different amino acid residues and their positions, which are under high selection pressure.
Figure 5. Phylogenetic analysis of cellobiosidase encoding gene.

Cellobiosidase protein sequences of 100 Indian Xoo strains and six Xoo strains from other parts of Asia were aligned and phylogenomic tree was constructed using Neighbour Joining method. Bootstrap values shown on the nodes are percentage of 500 replicates. The scale bar (0.002) indicates the number of amino acid substitutions per site (A), variation in the amino acid residues of catalytic domain are listed in tabular form (B) and selection pressure for each residue position is depicted with colour variations where an increase in yellow colour represents increase in the positive selection pressure on the residue (C).
fliC gene
The fliC gene encodes for flagellin which serves as a PAMP24. We also looked for the variation at this locus in the Xoo strains and analysis revealed that fliC gene is highly conserved amongst the strains. The protein sequences encoded by this gene in 106 Xoo strains are highly identical except for changes at two amino acid residue in XOOK. We also found the presence of two copies of fliC gene in XOOP, encoding for identical copies of protein.
raxX and raxSTAB
The gene raxX is a recently discovered PAMP in Xoo that encodes for a peptide which is recognized by resistance gene Xa2125. Mutation in raxX restores the ability to cause disease in Xa21 containing host plants. To explore the raxX sequences in Indian Xoo strains, we constructed a RaxX protein tree (Supplementary Fig. S2). The tree revealed that three of the Xoo strains (IXO651, IXO685, and IXO1221) have highly variant raxX allele closer to Xoc strains, rather than other Xoo strains. These three strains are already reported to break down the Xa21 mediated resistance in the rice5,25. These strains are also grouped together in the genome based tree and differ by ~20–40 SNPs. Interestingly the amino acid positions P44 and P48 which are known to be important for Xa21 mediated immunity25, are variant in these strains in reference to BXO1. These non-conservative variations, where a hydrophobic amino acid (proline) is replaced by a hydrophilic amino acid (serine or threonine), are also similar to Xoc strains (Table 1).
Table 1. RaxX allele variations in the Xanthomonas oryzae pv. oryzae strains.
| Strains | 3 | 4 | 9 | 17 | 20 | 27 | 44 | 46 | 48 | 55 | 57 |
| BXO1 | H | S | T | R | G | P | P | A | P | R | P |
| IXO884 | Q | ||||||||||
| IXO1088/IXO1104/DXO331/XOOP | A | ||||||||||
| IXO651/IXO685/IXO1221 | L | W | R | S | P | T | P | N | |||
| BXOR1/MAI10/CFBP7342 | L | S | P | T | P | N | |||||
| BLS256 | L | L | S | P | T | P | N |
Various amino acid residues in different strains are compared and variations are listed with reference to BXO1 strain.
As raxST, raxA and raxB are present in the genome as a single operon, adjacent to raxX gene, protein trees for RaxST, RaxA and RaxB were obtained and strains having variations in the four proteins in reference to BXO1 allele are marked in Fig. 2. Similar to the raxX sequences, the raxST gene in IXO651, IXO685and IXO1221 is also having high similarity to Xoc than other Xoo strains. Interestingly except for one strain DXO-165 in lineage L-I, all the non-synonymous changes in rax genes have taken place on diverse lineages L-II to L-V. We also looked into variation in five other genes (raxC, raxH raxP, raxQ and raxR) related to this cluster. The variations are marked in the Supplementary Fig. S3, which are mostly clustered in lineages L-III to L-V, except for raxQ which showed variation in lineage L-II.
Lipopolysaccharide
Lipopolysaccharide (LPS) is a constituent of the outer membrane of gram-negative bacteria and LPS gene clusters are hypervariable because of horizontal gene transfer. Published studies in Xoo have reported two different LPS cassette types; BXO1 type and BXO8 type26. Interestingly all the strains of lineage L-I have BXO1 type LPS cassette, while 13 strains sequenced in this study (BXO8, IXO1221, IXO651, IXO685, IXO597, IXO599, DXO-216, IXO390, IXO141, IXO621, IXO644, IXO645 and IXO620) and XO35933 that belong to diverse lineages, showed the presence of BXO8 type LPS cassette (Fig. 2).
Type III Effectors
Type III Effectors (T3Es) of Xanthomonas play an indispensable role in disease development. Owing to the repetitive nature of TAL (transcription activator-like) effectors, it is difficult to study them in draft genomes. Hence we focused only on the non-TAL effectors of Xoo. We checked for the presence and conservation of T3E repertoire in Xoo strains by analyzing the 24 non-TAL T3Es, which are listed in www.xanthomonas.org to be associated with Xoo. Out of the 24 non-TAL T3Es, only five effectors (AvrBs2, XopI, XopQ, XopR and XopV) make the core effectome of all 113 Xo strains (including Indian and few Asian, African and USA strains) of Xanthomonas oryzae and are conserved throughout, while XopAA is also conserved in all Asian Xoo strains studied here. A detailed list of presence/absence and changes in the T3Es in all 113 strains is provided in Supplementary Table S3. Figure 6 shows a heatmap for T3E conservation in the 113 Xo strains.
Figure 6. Heatmap depicting the conservation of 24 type III effectors in Xanthomonas oryzae strains.

Presence of an effector is marked with black colour, absence in white and effectors with partial sequences/contig break/disruption or frameshift mutation are marked in grey. In the upper row with the name of type III effector, the effectors that are conserved in all strains are marked with red font and an effector conserved only in 106 Asian strains (100 Indian Xoo strains sequenced and six Xoo strains from other parts of Asia) is marked with cyan colour. Strain names on the right side are arranged according the phylogenomics relationship.
Lineage associated variation in number of repeats at a CRISPR locus
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) represent the acquired immunity by bacteria, where acquired spacer regions from phages or plasmid sequences act as the inheritable memory and help in recognition of cognate protospacers in the invasive elements limiting their attack27,28. Hence an evolved CRISPR cassette can provide immunity from phages that use LPS as a receptor and also restrict the rate of horizontal gene transfer through extra-chromosomal elements like plasmids. Hence it is pertinent to study variation in CRISPR in Xoo population. Two strains, BXO8 and XO35933 did not show any spacer regions, but the entire locus containing cas3, cas5, cas8c, cas7, cas4, cas1 and cas2 is present in both draft genomes. Remaining Xoo strains showed the presence of CRISPR locus with a highly variable number of repeats and spacer regions (Fig. 2) that belong to CRISPR type I. The CRISPR sequences and number of spacers are listed in Supplementary Table S4.
The direct repeats are 31 bp in length and number of spacer sequences varies from 0–108. There is an association of higher and uniform number of repeats with members of the major lineage L-I, while members of minor lineages have less or highly variable number of repeats. Highly virulent pathotype XI strains IXO1088 and IXO1104 showed the presence of maximum number of spacers (108). CRISPR sequences for both the strains are highly similar. We also compared the alignment of repeats of CRISPR sequences of IXO1088 with Philippine race (XOOP), Korean race (XOOK), Japanese race (XOOM) and Supplementary Fig. 4 shows the alignment of spacer sequences in the four strains.
Plasmid detection and analysis
We also checked for the presence of plasmid in the raw reads and 11/100 strains showed the presence of plasmid sequences in the data. Interestingly, seven of the strains are from major lineage L-I, while remaining four strains are from minor lineages. With BLAST analysis of plasmids assembled using plasmidSpades29, we could find four different type of plasmids in these strains; Xanthomonas albilineans str. GPE PC73, plasmid plasmIII30 (BXO1, IXO35, IXO704, IXO842), Xanthomonas campestris pv. campestris B1459, plasmid I31 (IXO35, IXO74, IXO97, DXO-050, DXO-091, DXO-133, DXO-206), Xanthomonas citri subsp. citri strain 306, plasmid pXAC6432 (IXO134), Burkholderia vietnamiensis G4, plasmid pBVIE0433 (DXO-206). A compiled list of the length, assembly statistics of these plasmids and their BLAST result is provided in Supplementary Table S5. Further BLAST analysis of the plasmids identified in the 100 genomes in genomic sequences showed the presence of Xanthomonas albilineans str. GPE PC73, plasmid plasmIII in BXO447 and Burkholderia vietnamiensis, G4 plasmid pBVIE04 in BXO1 strain also.
Discussion
Being a staple food of half of the world’s population, rice improvement and protection is of paramount importance. Further, the history of domestication of rice parallels with the birth of human civilization and advancement. Hence detailed understanding of phylogeny and evolution of its pathogens is important. Even though, India is a major region of diversity of rice and Xoo, whole genome based studies are markedly lacking. The present population genomics study clearly revealed that Indian Xoo strains along with few Asian strains form a lineage that is distinct from USA and African strains. Further, the Indian Xoo population exhibits epidemiological structure with a predominant clonal lineage and few minor diverse lineages. The analysis also suggested that Xoc may be a variant lineage that emerged from the Xoo population.
New studies using genomic data reveal that rate of evolution can be highly variable in pathogenic bacteria34. Hence it is necessary to test both strict and relaxed clock model with methods that allow incorporations of uncertainties in the inferences. This is particularly relevant for a pathogen of a staple crop like rice that is cultivated in millions of hectares and with multiple cropping seasons within a year in tropical regions. Accordingly our testing revealed that a relaxed clock model is more suitable in Xoo and there have been two waves of selection. One population consisting of strains belonging to a major clonal lineage is younger and of recent origin while the other population consisting of minor recombining lineages is diverging from a much longer time.
Recombination analysis on the Xoo population has identified a clonal lineage L-I, which is predominant in India and lineage L-III, which is highly diverse and consists of strains of pathotypes I, II and XI. Grouping of these three pathotypes together marks this lineage as the most virulent lineage among all that were identified in this study, as very few resistance genes are effective against these pathotypes5. Interestingly, pathotype X consists of strains that are incompatible with all the R genes used for pathotyping5. They were considered to have lost their virulence. As these strains are present in multiple lineages, it appears that loss of virulence has occurred multiple times. Interestingly the Indian Xoo strains show compatibility to rice genotypes containing either xa5 or xa13, but not to both (except IXO1088 and IXO1104 belonging to pathotype XI)5. This implies that the switching of capability to break xa5 from xa13 has occurred twice in Xoo population, once in the ancestor of lineage L-III and once in the ancestor of lineage L-I.
Studying the phylogeny of determinants of pathogenicity, virulence and fitness is as important as understanding the strains phylogeny. Population genomic studies in particular provide opportunity to study variation in genes that encode proteins that act as PAMP/DAMP and also hypervariable genomic regions. One of the well-known PAMP in phytopathogen is flagellin which is under purifying selection in Xoo. Earlier studies have shown that a mutation in the gene coding for the cell wall degrading enzyme cellobiosidase leads to high virulence deficiency in Xoo22. Purified cellobiosidase is shown to induce innate immune responses and programmed cell death in rice tissues22. Our study revealed the presence of two alleles of cbsA (BXO1/BXO8 type) and one allele (BXO1 type) is replacing the other (BXO8 type) over time. Interestingly, the gene encoding for cbsA has acquired a frameshift mutation in Xoc and hence was reported to be missing in Xoc35. This suggests the strong selection pressure on cbsA gene in Xoo strains and pathovars.
In Xoo, several genes have been recognized that are required for Xa21 activity of rice, which is the most effective R gene against the Indian population of Xoo5. They are known as rax (required for AvrXa21 activity), and a gene known as raxX encodes for a 60 aa peptide, which is tyrosine-sulfated by raxST and recognized by Xa21 in rice for its activity25. Interestingly the limited SNPs amongst three strains IXO651, IXO685 and IXO1221 that have a variant raxX allele enabling them to evade the Xa21 immunity, suggests their clonal nature and their recent origin. In this context, it is important to note that Xa21 has been formally deployed only in the last 10–15 years. The IXO651 and IXO685 strains were isolated in 2006 from widely separated locations in India, and IXO1221 was isolated in 2011. It is unclear whether these strains have evolved in response to the recent deployment of this gene or whether they had arisen in response to some other selection pressure. Irrespective of the nature of selection, the raxX polymorphisms can be used to develop PCR based assays for detecting Xoo strains that are highly virulent on Xa21 containing cultivars.
Similar to the Xa21 mediated activity, flagellin sensing activity is also conserved in the rice plants, but as shown earlier the flagellin molecule produced in rice infecting Xanthomonas strains has a variant structure to avoid this detection36. While another Xanthomonas pathogen X. campestris has shown high level of polymorphism at this locus37, we could not find much differences in the rice pathogens. A very high level of similarity at this locus in all Xoo strains is suggestive of the high conservation of this newly gained trait in the population.
Lipopolysaccharide is well known to act as PAMP, virulence determinant and an elicitor of defense responses in plants38,39,40,41,42,43. Xanthomonas LPS locus is known to be hypervariable in nature26 and presence of two cassette types in Xoo population further supports this fact. BXO1 type LPS cassette is present in the majority of the population, while BXO8 type is present not only in the ancestral Xoo lineage and hyper virulent strains IXO651, IXO685 and IXO1221, but also in the old strain X. oryzae type strain XO35933. One remarkable finding is that the non-canonical BXO8 cassette is strikingly absent in lineage L-I. Besides orthologues of BXO8 type LPS cassette has also been identified in type strains of X. axonopodis, X. citri and Xoc strains26. Presence of a same type of LPS cassette in multiple pathovars and species, while restriction of the BXO1 type LPS cassette to Xoo strains only raises the possibility that the BXO8 type of cassette is the ancestral cassette for X. oryzae, while the BXO1 type cassette must have been acquired during later stages of Xoo evolution. LPS does not only act as PAMP, but is also known to be a receptor of bacteriophages44 and hence the variation can also be due to selection pressure from the phages.
T3Es are injected by pathogenic bacteria into the host cells to take control of host machinery. T3Es are known to be important to counteract PAMP induced defense responses by the host45. Thus the core effectome of the population can be an important resource for molecular and traditional breeding strategies to tackle the disease. In our study on Xoo, we have been able to identify T3Es (AvrBs2, XopI, XopQ, XopR and XopV) that are core to the whole Xoo population studied. Out of the five genes we identified as core, three were also listed as core T3Es in an earlier study based on PCR and dot-blot hybridization12. Interestingly, out of the five effectors common to all rice pathogens, 4 are also shared with beans pathogens46, 3 with tomato/pepper pathogens47,48 and one XopV is also shared by cassava pathogens49 and none with X. campestris pathovars50.
Various T3Es of Xanthomonas has been shown to interact in a distinct way inside the host plant. While some of them are involved in suppressing the innate immunity pathways of the plant51, others play a role in suppressing the defense mechanisms activated inside the host due to damage occurred by various cell wall degrading enzymes of bacteria52. Some T3Es also have target receptors inside the host that play intricate role in interfering with various signaling pathways53. Amongst the five T3Es identified to be conserved in this study, AvrBs2 is known to be required for the full virulence of bacteria54, XopQ is involved in suppressing the damage associated rice immune responses52, XopR plays a role in inhibiting the basal defense responses55, while detailed function of XopI and XopV still needs to be elucidated. Future similar population genomic based comparison of effectome of all Xanthomonas pathogens will be an intriguing area of research.
While CRISPR provides immunity to bacteria from phages, at the same time it will limit acquisition of novel genes through bacteriophages and plasmids. This has major implication on the genome dynamics and virulence of a pathogenic bacterium like Xoo. Hence it is important to check the presence and distribution of plasmid sequence in Xoo lineages. Interestingly, Asian Xoc strains and all African and USA Xo strains lack CRISPR loci as well as the CRISPR-associated genes56. Comparison of CRISPR cassette of Indian, Philippine, Korean and Japanese race showed that Indian race is closer to Philippine race as compared to Korean and Japanese.
It is striking to note that only in the major L-I lineage, most of the strains harbor a CRISPR cassette with >80 spacers. Interestingly, most of the strains encoding plasmid(s) are from lineage L-I. This suggests a highly evolved CRISPR locus, that might be central to providing immunity against invading phages and plasmids during the population expansion of lineage L-I. Also, only in major lineage L-I, all the strains have a variant BXO1 type of LPS locus, specific to X. oryzae, whose products again might have a role in protection against phages. Association of a large CRISPR cassette and a variant LPS gene cluster gives us a hint on the success of lineage L-I, and also suggests a promising way of controlling lineage L-I using bacteriophages. Such an association of CRISPR cassette with a predominant lineage and hypervirulent strains is intriguing.
Being a pathogen of a major staple crop seems to be a highly intricate evolutionary and ecological process. The predominant population of Xoo is clonal while the hyper variable counterpart forms a minor population. The latter also harbours highly virulent strains and highly variant allele(s)/gene cluster(s). In any case, considering the potential damage the minor lineages and variant strains can inflict, there is a need to track their movement and variation, along with effective deployment of resistance genes. This genomic resource will be invaluable in surveillance of Xoo by designing strain specific primers for quick PCR based diagnostic tools. However, considering the genome dynamics in Xoo, it is necessary to test such primers on much larger and new collection of strains. Specific mutations in the raxX gene are clearly associated with an ability to cause disease on Xa21 containing cultivars. This information can be used to develop PCR based diagnostic tests for detecting such isolates that are highly virulent on Xa21 containing rice lines.
Material and Methods
Genome sequencing
We have used two different sets of strains for this study, 46 strains from the Indian Xoo pathotype diversity study (2004–2009), where 11 different pathotypes were assigned to Xoo based on their reaction towards ten major resistance genes of rice5, and 54 strains from another old collection (1991–2014). We have included 2–5 representatives of each pathotype in the present study. A total of 100 Xoo strains, collected from diverse geographical locations of the country and a strain of Xanthomonas oryzicola pv. oryzicola, BXOR1 (listed in Supplementary Table S1), were grown on peptone sucrose agar (PSA) and genomic DNA was isolated using ZR Fungal/bacterial DNA isolation kit (Zymo Research Corporation, Orange, CA, USA). DNA quality was checked by running DNA samples on the 0.8% agarose gel and quantitation was done using Qubit 2.0 fluorometer (Invitrogen, Carlsbad, CA, USA). For sequencing on Illumina platform, the library was prepared using Nextera XT sample preparation kit (Illumina, Inc., San Diego, CA, USA) with dual indexing. Sample libraries were either normalized with the beads provided in the kit or quantitated by KAPA library quantification kit (KAPA Biosystems) using real time PCR and then loaded onto in-house Illumina MiSeq platform (Illumina, Inc., San Diego, CA, USA). The strains were sequenced using Illumina paired end sequencing technology (2 × 250).
Assembly and Annotation
Assembly of the raw sequences (>100x coverage) was performed using CLC Genomics workbench 6.5 (CLC bio, Aarhus, Denmark) into contigs (<500). Annotation was done using PGAAP pipeline of NCBI. CRISPR sequences were recognized using CRISPR recognition tool57 and CRISPRFinder webserver58. Spacer sequences were compared using blastn. LPS cassette of BXO1 and BXO8 were retrieved from NCBI to find their homologs in Xoo strains. Type III effector (AvrBs2, XopC, XopF, XopG, XopI, XopK, XopL, XopN, XopP, XopQ, XopR, XopT, XopU, XopV, XopW, XopX, XopY, XopZ, XopAA, XopAB, XopAD, XopAE, XopA and HpaA) sequences of XOOM were retrieved from the list provided on www.xanthomonas.org and further used to find the homologs in Xoo strains.
Phylogenetic analysis
Average Nucleotide Identity was calculated using JSpecies v1.2.113. Phylogenomic tree of Indian Xoo strains along with other Asian strains [XOOK59, XOOM60, XOOP61, PXO8662, PXO8363, XO35933 (NCBI accession number: AXVI00000000), XOC_BLS25664], African strains (XOO_NAI865, XOC_MAI1065, XOC_CFBP734262) and the USA strains (X8-1A56, X11-5A56) was obtained using 31 phylogenomic marker genes14, extracted from each genome and concatenated. Mega 7.0 was used for obtaining multiple sequence alignment of the concatenated sequences as well as to obtain phylogenetic tree66. Maximum likelihood tree was constructed using General Time Reversible model (Gamma distributed with Invariant sites (G + I)) method with 500 bootstrap replications.
ClonalFrameML tool (which uses maximum likelihood inference) was deployed for identifying the recombined fragments and applying a correction for recombination in the final phylogeny inferred67. For obtaining tree with correction for recombination, genomes of 106 Xoo strains were aligned using MAUVE and a maximum likelihood tree was obtained using PhyML. MAUVE alignment68 and PhyML tree69 were further used to generate the ClonalFrameML tree and recombination parameters with 100 bootstrap replications (emsim = 100).
For phylogenomic inferences of different genes, protein sequences were aligned for RaxX, RaxST, RaxA, RaxB, RaxC, RaxH, RaxP, RaxQ, RaxR and cellobiosidase using Mega v6.070. A neighbour joining tree was obtained for different protein alignment with 500 bootstrap replications using Mega v6.0.
Molecular clock analysis
TempEst v1.5 was used to check for the temporal signal17. A maximum likelihood tree was associated with isolation dates of the strains using root-to-tip regression approach. A best fitting root option was used and correlation coefficient function was determined to be 0.372. For Bayesian analysis, Xoo genomes of 100 Indian Xoo strains and six Xoo strains from other parts of Asia were aligned using Mauve and the alignment was analysed using Mr. Bayes v3.271. Testing of strict clock model and relaxed clock model [independent gamma rates (igr)] was done using harmonic mean method (ngen = 100000) and stepping stone method (ngen = 255000) and values for harmonic mean and maximum likelihood were compared. For dating analysis, Mr. Bayes was run with GTR substitution model with gamma-distributed rate variation across sites and a proportion of invariable sites for 1 M iterations (ngen = 1000000) and two parallel runs (nrun = 2). Molecular dating was done with clock rate (0.02, 0.004) per million year considering the mutation rate of 2 × 10−5 mutations per gene per year as earlier reported in Xanthomonas18 and assuming the average gene size as 1 kb. After confirming for convergence of two runs, parameters were summarized and results were obtained.
Plasmid Detection
Presence of plasmids was predicted using Spades v3.8.0 with argument – plasmid based on the reads coverage29. The contigs (size >5 kb) predicted as plasmid by plasmidSpades were further tested using blastn in complete plasmid database as well as in nr database. Plasmids identified in any genome were further also manually checked for their presence in other 100 genomes sequenced.
Additional Information
Accession codes: Genomic sequences have been submitted to NCBI GenBank. Genomic sequences of Xanthomonas oryzae pv. oryzae strains are available under accession number JXDM-JXHH and genomic sequences of Xanthomonas oryzae pv. oryzicola strain is available under accession number JXHI. Genome files with annotation of 101 strains are available at https://figshare.com/s/10e290cfe8a5f31858d3.
How to cite this article: Midha, S. et al. Population genomic insights into variation and evolution of Xanthomonas oryzae pv. oryzae. Sci. Rep. 7, 40694; doi: 10.1038/srep40694 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
The work was supported by grants to PBP and RVS under the Plant-Microbe and Soil Interactions (BSC0117) project of the Council of Scientific and Industrial Research (CSIR). RVS was also supported by the J C Bose Fellowship of the Department of Science and Technology, Government of India. SM, SK and AMG are supported by a fellowship from the CSIR and KBa is supported by a fellowship from UGC.
Footnotes
Author Contributions S.M. carried out whole genome sequencing and submission along with the computational analysis of sequence data. K.Ba. participated in constructing a phylogenomic tree using housekeeping genes, SNP and plasmid analysis studies. S.K. participated in molecular dating analysis of the strains. A.M.G., D.M., K.Br., G.S.L., R.M.S. and R.V.S. provided strains along with their pathogenicity and metadata. S.M. and P.B.P. drafted the manuscript along with inputs from D.M., G.S.L., R.M.S. and R.V.S. S.M., R.V.S. and P.B.P. participated in design and interpretation of data.
References
- Nino-Liu D. O., Ronald P. C. & Bogdanove A. J. Xanthomonas oryzae pathovars: model pathogens of a model crop. Mol Plant Pathol 7, 303–324 (2006). [DOI] [PubMed] [Google Scholar]
- Reddy A., Mackenzie D., Rouse D. & Rao A. Relationship of bacterial leaf blight severity to grain yield of rice. Phytopathology 69, 967–969 (1979). [Google Scholar]
- Ou S. Rice Disease (2 nd). Commonwealth Agricultural Bureau, Kew, Surrey (1985).
- Kim S.-M. et al. Identification and fine-mapping of a new resistance gene, Xa40, conferring resistance to bacterial blight races in rice (Oryza sativa L.). Theor Appl Genet 128, 1933–1943 (2015). [DOI] [PubMed] [Google Scholar]
- Mishra D. et al. Pathotype and genetic diversity amongst Indian isolates of Xanthomonas oryzae pv. oryzae. PloS One 8, e81996 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey S., Singh B. & Kumar J. DNA typing and virulence determination of Xanthomonas oryzae pv. oryzae population for the management of bacterial leaf blight of rice in Udham Singh Nagar, India. Eur J Plant Pathol 138, 847–862 (2014). [Google Scholar]
- Lore J. S. et al. Genotypic and pathotypic diversity of Xanthomonas oryzae pv. oryzae, the cause of bacterial blight of rice in Punjab State of India. J Phytopathol 159, 479–487 (2011). [Google Scholar]
- Shanti M. L. et al. Identification of resistance genes effective against rice bacterial blight pathogen in eastern India. Plant Dis 85, 506–512 (2001). [DOI] [PubMed] [Google Scholar]
- Mondal K. K. et al. Pathotyping and genetic screening of type III effectors in Indian strains of Xanthomonas oryzae pv. oryzae causing bacterial leaf blight of rice. Physiol Mol Plant Pathol 86, 98–106 (2014). [Google Scholar]
- Gautam R. K. et al. Analysis of pathogenic diversity of the rice bacterial blight pathogen (Xanthomonas oryzae pv. oryzae) in the Andaman Islands and identification of effective resistance genes. J Phytopathol 163, 423–432 (2015). [Google Scholar]
- Bharathkumar S. et al. Differential disease reaction of rice pathogen Xanthomonas oryzae pv. oryzae prevailing in India on rice cultivars. The Bioscan 9, 1257–1262 (2014). [Google Scholar]
- Hajri A. et al. Multilocus sequence analysis and type III effector repertoire mining provide new insights into the evolutionary history and virulence of Xanthomonas oryzae. Mol Plant Pathol 13, 288–302 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter M. & Rosselló-Móra R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA 106, 19126–19131 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M. & Eisen J. A. A simple, fast, and accurate method of phylogenomic inference. Genome Biol 9, R151 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer A. S. & McCouch S. R. The rice bacterial blight resistance gene xa5 encodes a novel form of disease resistance. Mol Plant Microbe Interact 17, 1348–1354 (2004). [DOI] [PubMed] [Google Scholar]
- Yuan M., Chu Z., Li X., Xu C. & Wang S. Pathogen-induced expressional loss of function is the key factor in race-specific bacterial resistance conferred by a recessive R gene xa13 in rice. Plant Cell Physiol 50, 947–955 (2009). [DOI] [PubMed] [Google Scholar]
- Rambaut A., Lam T. T., Carvalho L. M. & Pybus O. G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol 2, vew007 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mhedbi-Hajri N. et al. Evolutionary history of the plant pathogenic bacterium Xanthomonas axonopodis. PLoS One 8, e58474 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boller T. & He S. Y. Innate immunity in plants: an arms race between pattern recognition receptors in plants and effectors in microbial pathogens. Science 324, 742–744 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- White F. F., Potnis N., Jones J. B. & Koebnik R. The type III effectors of Xanthomonas. Mol Plant Pathol 10, 749–766 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu K. et al. R gene expression induced by a type-III effector triggers disease resistance in rice. Nature 435, 1122–1125 (2005). [DOI] [PubMed] [Google Scholar]
- Jha G., Rajeshwari R. & Sonti R. V. Functional interplay between two Xanthomonas oryzae pv. oryzae secretion systems in modulating virulence on rice. Mol Plant Microbe Interact 20, 31–40 (2007). [DOI] [PubMed] [Google Scholar]
- Doron-Faigenboim A., Stern A., Mayrose I., Bacharach E. & Pupko T. Selecton: a server for detecting evolutionary forces at a single amino-acid site. Bioinformatics 21, 2101–2103 (2005). [DOI] [PubMed] [Google Scholar]
- Gómez-Gómez L. & Boller T. FLS2: an LRR receptor–like kinase involved in the perception of the bacterial elicitor flagellin in Arabidopsis. Mol Cell 5, 1003–1011 (2000). [DOI] [PubMed] [Google Scholar]
- Pruitt R. N. et al. The rice immune receptor XA21 recognizes a tyrosine-sulfated protein from a Gram-negative bacterium. Sci Adv 1, e1500245 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil P. B., Bogdanove A. J. & Sonti R. V. The role of horizontal transfer in the evolution of a highly variable lipopolysaccharide biosynthesis locus in xanthomonads that infect rice, citrus and crucifers. BMC Evol Biol 7, 243 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R. et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712 (2007). [DOI] [PubMed] [Google Scholar]
- Bolotin A., Quinquis B., Sorokin A. & Ehrlich S. D. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiol 151, 2551–2561 (2005). [DOI] [PubMed] [Google Scholar]
- Antipov D. et al. plasmidSPAdes: Assembling Plasmids from Whole Genome Sequencing Data. bioRxiv 048942 (2016). [DOI] [PubMed] [Google Scholar]
- Pieretti I. et al. The complete genome sequence of Xanthomonas albilineans provides new insights into the reductive genome evolution of the xylem-limited Xanthomonadaceae. BMC Genomics 10, 616 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wibberg D. et al. Draft genome of the xanthan producer Xanthomonas campestris NRRL B-1459 (ATCC 13951). J Biotechnol 204, 45–46 (2015). [DOI] [PubMed] [Google Scholar]
- da Silva A. C. et al. Comparison of the genomes of two Xanthomonas pathogens with differing host specificities. Nature 417, 459–463 (2002). [DOI] [PubMed] [Google Scholar]
- Shields M., Reagin M., Gerger R., Campbell R. & Somerville C. TOM, a new aromatic degradative plasmid from Burkholderia ( Pseudomonas) cepacia G4. Appl Environ Microbiol 61, 1352–1356 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biek R., Pybus O. G., Lloyd-Smith J. O. & Didelot X. Measurably evolving pathogens in the genomic era. Trends Ecol Evol 30, 306–313 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieretti I. et al. Genomic insights into strategies used by Xanthomonas albilineans with its reduced artillery to spread within sugarcane xylem vessels. BMC Genomics 13, 658 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S. et al. Rice OsFLS2-mediated perception of bacterial flagellins is evaded by Xanthomonas oryzae pvs. oryzae and oryzicola. Mol Plant 8, 1024–1037 (2015). [DOI] [PubMed] [Google Scholar]
- Sun W., Dunning F. M., Pfund C., Weingarten R. & Bent A. F. Within-species flagellin polymorphism in Xanthomonas campestris pv campestris and its impact on elicitation of Arabidopsis FLAGELLIN SENSING2–dependent defenses. Plant Cell 18, 764–779 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshavarzi M. et al. Basal defenses induced in pepper by lipopolysaccharides are suppressed by Xanthomonas campestris pv. vesicatoria. Mol Plant Microbe Interact 17, 805–815 (2004). [DOI] [PubMed] [Google Scholar]
- Nurnberger T., Brunner F., Kemmerling B. & Piater L. Innate immunity in plants and animals: striking similarities and obvious differences. Immunol Rev 198, 249–266 (2004). [DOI] [PubMed] [Google Scholar]
- Drigues P. et al. Comparative studies of lipopolysaccharide and exopolysaccharide from a virulent strain of Pseudomonas solanacearum and from three avirulent mutants. J Bacteriol 162, 504–509 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham T. L., Sequeira L. & Huang T. S. Bacterial lipopolysaccharides as inducers of disease resistance in tobacco. Appl Environ Microbiol 34, 424–432 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman M. A., Dow J. M., Molinaro A. & Parrilli M. Priming, induction and modulation of plant defence responses by bacterial lipopolysaccharides. J Endotoxin Res 13, 69–84 (2007). [DOI] [PubMed] [Google Scholar]
- Dharmapuri S., Yashitola J., Vishnupriya M. R. & Sonti R. V. Novel genomic locus with atypical G + C content that is required for extracellular polysaccharide production and virulence in Xanthomonas oryzae pv. oryzae. Mol Plant Microbe Interact 14, 1335–1339 (2001). [DOI] [PubMed] [Google Scholar]
- Wright A., McConnell M. & Kanegasaki S. In Virus Receptors 27–57 (Springer, 1980). [Google Scholar]
- Alfano J. R. & Collmer A. Type III secretion system effector proteins: double agents in bacterial disease and plant defense. Annu. Rev. Phytopathol. 42, 385–414 (2004). [DOI] [PubMed] [Google Scholar]
- Aritua V. et al. Genome sequencing reveals a new lineage associated with lablab bean and genetic exchange between Xanthomonas axonopodis pv. phaseoli and Xanthomonas fuscans subsp. fuscans. Front Microbiol 6, 1080 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potnis N. et al. Comparative genomics reveals diversity among xanthomonads infecting tomato and pepper. BMC Genomics 12, 146 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz A. R. et al. Phylogenomics of Xanthomonas field strains infecting pepper and tomato reveals diversity in effector repertoires and identifies determinants of host specificity. Front Microbiol 6, 535 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bart R. et al. High-throughput genomic sequencing of cassava bacterial blight strains identifies conserved effectors to target for durable resistance. Proc Natl Acad Sci USA 109, E1972–1979 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux B. et al. Genomics and transcriptomics of Xanthomonas campestris species challenge the concept of core type III effectome. BMC Genomics 16, 975 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- White F. F., Potnis N., Jones J. B. & Koebnik R. The type III effectors of Xanthomonas. Mol Plant Pathol 10, 749–766 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha D., Gupta M. K., Patel H. K., Ranjan A. & Sonti R. V. Cell wall degrading enzyme induced rice innate immune responses are suppressed by the type 3 secretion system effectors XopN, XopQ, XopX and XopZ of Xanthomonas oryzae pv. oryzae. PLoS One 8, e75867 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay S. & Bonas U. How Xanthomonas type III effectors manipulate the host plant. Curr Opin Microbiol 12, 37–43 (2009). [DOI] [PubMed] [Google Scholar]
- Kearney B. & Staskawicz B. J. Widespread distribution and fitness contribution of Xanthomonas campestris avirulence gene avrBs2. Nature 346, 385–386 (1990). [DOI] [PubMed] [Google Scholar]
- Akimoto-Tomiyama C. et al. XopR, a type III effector secreted by Xanthomonas oryzae pv. oryzae, suppresses microbe-associated molecular pattern-triggered immunity in Arabidopsis thaliana. Mol Plant Microbe Interact 25, 505–514 (2012). [DOI] [PubMed] [Google Scholar]
- Triplett L. R. et al. Genomic analysis of Xanthomonas oryzae isolates from rice grown in the United States reveals substantial divergence from known X. oryzae pathovars. Appl Environ Microbiol 77, 3930–3937 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bland C. et al. CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics 8, 1 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissa I., Vergnaud G. & Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res 35, W52–W57 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B. M. et al. The genome sequence of Xanthomonas oryzae pathovar oryzae KACC10331, the bacterial blight pathogen of rice. Nucleic Acids Res 33, 577–586 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochiai H., Inoue Y., Takeya M., Sasaki A. & Kaku H. Genome sequence of Xanthomonas oryzae pv. oryzae suggests contribution of large numbers of effector genes and insertion sequences to its race diversity. Jpn Agric Res Q 39, 275–287 (2005). [Google Scholar]
- Salzberg S. L. et al. Genome sequence and rapid evolution of the rice pathogen Xanthomonas oryzae pv. oryzae PXO99A. BMC Genomics 9, 204 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booher N. J. et al. Single molecule real-time sequencing of Xanthomonas oryzae genomes reveals a dynamic structure and complex TAL (transcription activator-like) effector gene relationships. Microb Genom 1, 1–22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau J. et al. AnnoTALE: bioinformatics tools for identification, annotation, and nomenclature of TALEs from Xanthomonas genomic sequences. Sci Rep 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanove A. J. et al. Two new complete genome sequences offer insight into host and tissue specificity of plant pathogenic Xanthomonas spp. J Bacteriol 193, 5450–5464 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang J. M. et al. Sensitive detection of Xanthomonas oryzae pathovars oryzae and oryzicola by loop-mediated isothermal amplification. Appl Environ Microbiol 80, 4519–4530 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Stecher G. & Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. msw054 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didelot X. & Wilson D. J. ClonalFrameML: Efficient inference of recombination in whole bacterial genomes. PLoS Comput Biol 11, e1004041–e1004041 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling A. C., Mau B., Blattner F. R. & Perna N. T. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14, 1394–1403 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59, 307–321 (2010). [DOI] [PubMed] [Google Scholar]
- Tamura K., Stecher G., Peterson D., Filipski A. & Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30, 2725–2729 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F. et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61, 539–542 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.