

Abstract
Sweet potato is an important food and bio-energy crop, and investigating the mechanisms underlying salt tolerance will provide information for salt-tolerant breeding of this crop. Here, the root transcriptomes of the salt-sensitive variety Lizixiang and the salt-tolerant line ND98 were compared to identify the genes and pathways involved in salt stress responses. In total, 8,744 and 10,413 differentially expressed genes (DEGs) in Lizixiang and ND98, respectively, were involved in salt responses. A lower DNA methylation level was detected in ND98 than in Lizixiang. In both genotypes, the DEGs, which function in phytohormone synthesis and signalling and ion homeostasis, may underlie the different degrees of salt tolerance. Significant up-regulations of the genes involved in the jasmonic acid (JA) biosynthesis and signalling pathways and ion transport, more accumulation of JA, a higher degree of stomatal closure and a lower level of Na+ were found in ND98 compared to Lizixiang. This is the first report on transcriptome responses to salt tolerance in sweet potato. These results reveal that the JA signalling pathway plays important roles in the response of sweet potato to salt stress. This study provides insights into the mechanisms and genes involved in the salt tolerance of sweet potato.
Salinity stress is becoming a major constraint on crop production; it affects approximately 19.5% of the irrigated soil worldwide, and will result in a loss of up to 50% of cultivable land by the middle of the twenty-first century1,2. High concentrations of salts lead to ion imbalances and hyperosmotic effects and cause a number of physiological disorders in plants, including photosynthesis inhibition, metabolic dysfunctions and cellular structure damage3. Therefore, developing salt-tolerant cultivars has become one of the most important goals in crop breeding.
Phytohormones play important roles in mediating host responses to various abiotic stresses. Increasing evidence has shown that jasmonic acid (JA, or the related compound methyl jasmonate, MeJA) functions in diverse physiological processes in plants, such as the regulation of plant responses to abiotic stresses4,5,6,7,8,9,10,11. The exogenous application of JA to wheat seedlings for three days has been shown to significantly enhance the salt tolerance of these plants by increasing the activities of antioxidant enzymes and the concentrations of antioxidative compounds4. MeJA improves salinity resistance in German chamomile by increasing the activity of antioxidant enzymes5. The endogenous JA content has been shown to be markedly increased under drought and cold stresses but reduced under heat stress in rice6. Expression profiling and physiological characterization on salinity stress response in barley reveal the JA-mediated adaptation to salinity stress7. Zhao et al. showed that a wheat allene oxide cyclase (AOC) gene enhances salinity tolerance via the JA signalling pathway8. The steady-state levels of two key enzymes in JA biosynthesis, lipoxygenase (LOX) and allene oxide synthase (AOS), and the content of JA are higher in salt-tolerant tomato cultivars9. The salt hypersensitivity of the lox3 mutant can be complemented by MeJA10. VaNAC26 enhances drought tolerance by improving JA biosynthesis in Arabidopsis11. Stomatal closure induced by abscisic acid (ABA) and JA is a fundamental physiological response in plants12. Calcium-dependent protein kinase 6 (CPK6) functions as a positive regulator of MeJA signalling to activate ion channels in Arabidopsis guard cells13. Oxylipins serve as signalling molecules to control stomatal closure in a JA-dependent manner to enhance the defence response in Arabidopsis14. ABA is another mediator of plant responses to a variety of stresses, and this hormone plays a critical role in regulating the water status and growth of plants via the induction of genes encoding enzymes and other proteins involved in cellular dehydration tolerance15. Moreover, ABA is involved in adaptations to salt and drought in rice, tobacco and other plants16,17,18. Salicylic acid (SA) plays an indeterminate role in defence mechanisms against abiotic stresses19. The application of exogenous SA has been shown to enhance the drought and salt tolerance of bean and tomato20, whereas pre-treatment with SA has been shown to decrease drought tolerance in maize21.
Illumina paired-end sequencing technology is an important method for accessing genomic data sources for gene discovery and functional studies. High-throughput sequencing of transcriptomes has been performed for many model and non-model plant species, such as Arabidopsis, rice, maize, wheat and cotton3,22,23,24,25. Sweet potato (Ipomoea batatas (L.) Lam.) is an important root and tuber crop and is used as an industrial and bio-energy resource. This crop has considerable potential to grow on saline land26,27, and several salt tolerance genes have been identified in sweet potato28,29,30,31,32. Recently, de novo whole-genome sequencing of the selfed line Mx23Hm and the highly heterozygous line 0431-1 of I. trifida (2n = 2x = 30, one of the likely diploid ancestors of sweet potato) was performed on an Illumina HiSeq platform, but the assembly and annotation of these genomes are still in the early stages33. Yan et al. reported the complete chloroplast genome sequence of sweet potato variety Xushu 1834. However, the sweet potato genome is still unavailable due to its highly heterozygous, generally self-incompatible, outcrossing and autohexaploidy nature. To date, the transcriptome sequencing of sweet potato has provided important transcriptional data for studying storage root formation and starch, carotenoid and anthocyanin biosynthesis and characterizing the associated key genes in this crop27,35,36,37; however, transcriptome sequencing studies of sweet potato under abiotic or biotic stresses are rare.
In the present study, RNA-Seq was used to identify salt tolerance-related genes and to determine their crosstalk in the roots of the salt-sensitive sweet potato variety Lizixiang and the salt-tolerant line ND98 under salt stress. Transcript profile analysis indicated that the JA signalling pathway is involved in the responses of sweet potato to salt stress. Decreased DNA methylation may also contribute to the salt tolerance of this crop. The present study provides a better understanding of how sweet potato responds to salt stress and has potential applications for the genetic improvement of sweet potato on marginal land.
Results
Transcriptome sequencing of Lizixiang and ND98
In vitro-grown Lizixiang and ND98 plants were assessed using RNA-Seq analysis in two independent biological replicates. RNA samples were collected from the roots at 0, 12, and 48 h after exposure to 200 mM NaCl stress for RNA-Seq. In total, 368,444,980 clean reads were obtained from the 12 RNA-Seq datasets, and 230,357 transcripts, with a mean length of 1,298 bp, were de novo assembled using Trinity (Fig. 1A, Supplementary Table S1). The transcripts were further clustered into 63,673 non-redundant transcripts (unigenes) with a mean length of 953.5 bp (Supplementary Table S1). For functional annotation, the sequences of the assembled unigenes were searched among a variety of databases, and 37,016 unigenes were annotated with putative functions based on hits from at least one database (Supplementary Table S2). In addition, when the sequences of the assembled unigenes were compared to the genome sequences of I. trifida (2n = B1B1 = 2x = 30)33, a low pair alignment rate of 51.70% was obtained. This might be because the assembly of the I. trifida (2x) genome is still in the early stages and sweet potato is a highly heterozygous autohexaploid (2n = B1B1B2B2B2B2 = 6x = 90).
Figure 1. Gene expression analysis of the Lizixiang and ND98 root transcriptomes.

(A) Correlation of the global gene expression in Lizixiang and ND98 roots under 0-, 12-, and 48-h salt treatments. (B) Statistics of the gene expression levels (FPKM) in Lizixiang and ND98 samples. L0, Lizixiang 0 h; L12, Lizixiang 12 h; L48, Lizixiang 48 h; N0, ND98 0 h; N12, ND98 12 h; and N48, ND98 48 h.
Identification and functional annotation of differentially expressed genes
Fragments per kilobase of transcript per million mapped reads (FPKM) was used to represent the gene expression levels generated using TopHat. The proportion of genes with FPKM ≥ 1 was 2.5% (671 genes) lower in ND98 than in Lizixiang under normal conditions (0 h), whereas the proportion was 13.9% (3099 genes) and 15.2% (3466 genes) higher in ND98 than in Lizixiang under 12 and 48 h of salt stress, respectively (Fig. 1B). The differentially expressed genes (DEGs) of Lizixiang and ND98 were detected via pair-wise comparisons at each of the two time points. In total, 8,744 and 10,413 DEGs were detected in Lizixiang and ND98, respectively (Table 1, Supplementary Table S3).
Table 1. Numbers of DEGs at different salt-stressed time points in the two genotypes.
| Items | Lizixiang |
ND98 |
|||
|---|---|---|---|---|---|
| No. | % | No. | % | ||
| 12 versus 0 h | up-regulated | 2,833 | 46.74 | 3,500 | 44.11 |
| down-regulated | 3,228 | 53.26 | 4,435 | 55.89 | |
| total | 6,061 | 7,935 | |||
| 48 versus 0 h | up-regulated | 3,410 | 49.13 | 3,255 | 41.60 |
| down-regulated | 3,531 | 50.87 | 4,569 | 58.40 | |
| total | 6,941 | 7,824 | |||
| 48 versus 12 h | up-regulated | 697 | 64.18 | 404 | 39.11 |
| down-regulated | 389 | 35.82 | 629 | 60.89 | |
| total | 1,086 | 1,033 | |||
| Total | 8,744 | 10,413 | |||
Using the K-means method, 8,744 and 10,413 DEGs of Lizixiang and ND98, respectively, were divided into 10 clusters based on their expression patterns (Supplementary Fig. S1). A total of 2,819 DEGs showed the same expression patterns between Lizixiang and ND98, and 7299 DEGs exhibited different expression patterns. Further analysis indicated that the expression levels of 1238 DEGs were significantly higher in ND98 than in Lizixiang for at least one time point during salt treatment; these genes were associated with signal transduction, ion transport, cell wall synthesis, disease resistance, photosynthesis processes, reactive oxygen species (ROS) scavenging and some unknown functions (Supplementary Table S4).
Using Gene Ontology (GO) analysis, the 7299 DEGs with different expression patterns between Lizixiang and ND98 were classified into three categories: biological process, molecular function and cellular component. In the biological process class, 20 biological processes with significant differences in enrichment were identified, including “metabolic processes” (3,542 DEGs), “cellular processes” (3,270 DEGs), “single-organism processes” (2,780 DEGs), “responses to stimuli” (1,207 DEGs) and other processes. In the molecular function class, “binding” (3,338 DEGs) and “electron carrier activity” (2,984 DEGs) were significantly enriched. In the cellular component class, “cell part” was the most significantly enriched GO term (2581 DEGs) (Fig. 2A).
Figure 2. Functional enrichment of genes with different expression patterns between Lizixiang and ND98.

(A) GO functional classification analysis of genes with different expression patterns between the two genotypes (p < 0.05). (B) Top 50 KEGG pathways enriched with genes with different expression patterns between the two genotypes. The pathways related to phytohormone biosynthesis and signalling are indicated with red arrows.
To explore the biological pathways important for sweet potato responses to salt stress, the DEGs with different expression patterns between Lizixiang and ND98 were further annotated to the reference pathways in the Kyoto Encyclopedia of Genes and Genomes (KEGG) using KeggArray software (Fig. 2B). These DEGs were assigned to 258 KEGG pathways. The pathway search results were sorted based on the number of hits, and the ribosome pathway (205 members) and plant hormone signal transduction (163 members) were the most highly represented groups. The genes involved in protein processing in the endoplasmic reticulum (113 members), phenylpropanoid biosynthesis (90 members), plant-pathogen interactions (78 members) and phenylalanine metabolism (64 members) were also hallmarks of sweet potato roots under salt stress (Fig. 2B).
Phytohormone synthesis- and transport-related genes in the roots of both genotypes were identified and categorized into four groups via gene expression profiling (Fig. 3). The first group showed low basal expression in both Lizixiang and ND98 under normal conditions, but these genes were induced under salt stress only in ND98. The second group exhibited high and low basal expression in Lizixiang and ND98, respectively, and these genes were suppressed and induced under salt stress in Lizixiang and ND98, respectively. The genes in the third group were highly expressed only in ND98 under normal conditions, but were not induced under salt stress in either genotype. The genes in the fourth group were highly expressed in both genotypes under normal conditions, but were induced under salt stress only in ND98 (Fig. 3). These results suggest that the two genotypes have different mechanisms underlying the phytohormone synthesis and transduction pathways in response to salt stress.
Figure 3. Heat maps of DEGs with different expression patterns between Lizixiang and ND98 that participate in phytohormone signalling pathways.
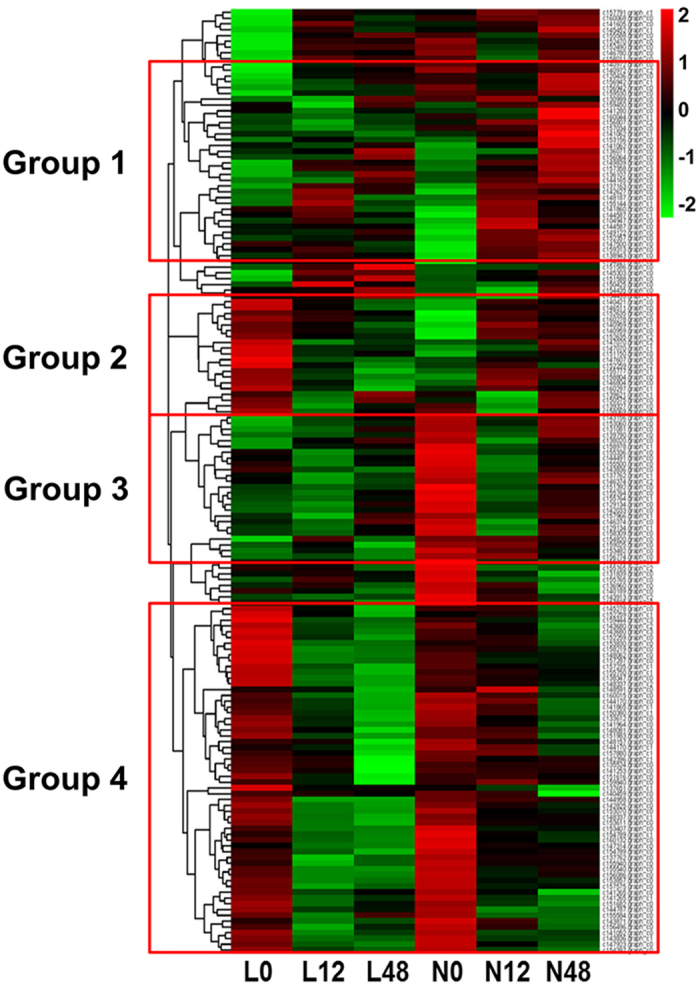
Group 1 genes showed low basal expression in both Lizixiang and ND98 under normal conditions but were induced under salt stress only in ND98. Group 2 genes exhibited high and low basal expression in Lizixiang and ND98, respectively, and were suppressed and induced under salt stress in Lizixiang and ND98, respectively. Group 3 genes were highly expressed only in ND98 under normal conditions but were not induced under salt stress in either genotype. Group 4 genes were highly expressed in both genotypes under normal conditions but were induced under salt stress only in ND98. L0, Lizixiang 0 h; L12, Lizixiang 12 h; L48, Lizixiang 48 h; N0, ND98 0 h; N12, ND98 12 h; and N48, ND98 48 h.
Analysis of DNA methylation
DNA methylation influences global gene expression under abiotic stresses in plants38. All DNA methylation- and demethylation-related genes were identified via gene expression profiling under salt stress (Fig. 4A, Supplementary Table S5). The expression level of the key gene associated with methylation, DNA methyltransferase DNMT1-like enzyme (IbMET1), was higher in ND98 than in Lizixiang, but three other key gene types, domains rearranged methyltransferase 1 (IbDRM1), domains rearranged methyltransferase 2 (IbDRM2) and chromomethylase 2 (IbCMT2), did not show differences between the two genotypes (Supplementary Table S6). Both key demethylation-related genes, namely DNA demethylases demeter-like protein (IbDML3) and DNA repressor of silencing 1 (IbROS1), exhibited higher expression levels in ND98 than in Lizixiang (Fig. 4B, Supplementary Table S6).
Figure 4. Differences in the global genomic DNA methylation in Lizixiang and ND98 under salt stress.

(A) Heat maps showing the expression of the genes related to DNA methylation or demethylation. (B) Differential expression of DNA demethylation-related genes validated using qRT-PCR. IbROS1, c144343.graph_c0; IbDML3, c148603.graph_c0. (C) MSAP profiling analysis showing different patterns of locus-specific DNA methylation in Lizixiang and ND98. The gel shows the results for one primer combination; twenty EcoRI + HpaII/MspI primer combinations were used in total. H, HpaII; M, MspI. Differentially methylated sites between Lizixiang and ND98 are indicated with arrows. L0, Lizixiang 0 h; L12, Lizixiang 12 h; L48, Lizixiang; N0, ND98 0 h; N12, ND98 12 h; and N48, ND98 48 h.
A methylation-sensitive amplified polymorphism (MSAP) analysis was performed to detect methylation variations at specific sites in Lizixiang and ND98 root samples (Fig. 4C). Twenty EcoRI + HpaII/MspI primer combinations yielded 731, 744 and 718 clear and reproducible sites in Lizixiang root samples and 767, 858 and 717 clear and reproducible sites in ND98 root samples at 0, 12 and 48 h, respectively (Table 2). The proportions of unmethylated (pattern HpaII/MspI = 1/1), fully methylated (pattern HpaII/MspI = 0/1) and hemi-methylated (pattern HpaII/MspI = 1/0) CCGG sites in Lizixiang and ND98 root samples are listed in Table 2. The Lizixiang root samples presented 225, 295 and 266 methylated CCGG sites and the ND98 root samples presented 215, 255 and 251 methylated CCGG sites at 0, 12 and 48 h, respectively, revealing higher demethylation in the ND98 samples than in the Lizixiang samples (Table 2).
Table 2. Comparison of DNA methylation levels between Lizixiang and ND98 based on MASP analysis under normal and salt stress conditions.
| Individuals | Total sites | Unmethylated CCGG sites (%) | Methylated CCGG sites (%) |
||
|---|---|---|---|---|---|
| Total sites | Full-methylated internal cytosine sites | Hemi-methylated external cytosine sites | |||
| L0 | 731 | 506 (69.22) | 225 | 120 (53.33) | 105 (46.67) |
| L12 | 744 | 449 (60.34) | 295 | 158 (53.56) | 137 (46.44) |
| L48 | 718 | 452 (62.95) | 266 | 148 (55.64) | 118 (44.36) |
| L0-12-48-Total | 2,193 | 1,407 | 786 | 426 | 360 |
| N0 | 767 | 552 (71.96) | 215 | 128 (59.53) | 87 (40.47) |
| N12 | 858 | 503 (66.35) | 255 | 106 (41.57) | 149 (58.43) |
| N48 | 717 | 466 (64.99) | 251 | 133 (52.99) | 118 (47.01) |
| N0-12-48-Total | 2,342 | 1,521 | 721 | 367 | 354 |
The enhanced expression of DNA methylation- and demethylation-related genes was accompanied by changes in the DNA methylation and demethylation levels. Under salt stress, lower DNA methylation levels were accompanied by a larger proportion of FPKM ≥ 1 genes in ND98 than in Lizixiang (Fig. 1B, Table 2). Both genotypes showed variations in epigenetic modifications, particularly DNA methylation, which in turn influenced the expression of stress tolerance-related genes in response to salt stress.
Response of JA, ABA and SA signalling pathways to salt stress
Genes with different expression patterns between the two genotypes and implicated in different phytohormone signalling pathways were identified under salt stress. The top three categories, in terms of phytohormones, were JA, ABA and SA, accounting for 64.2% of the phytohormone signalling-related genes (Fig. 5A, Supplementary Table S7). The expression of sweet potato homologs of key genes involved in JA, ABA and SA signalling pathways was validated using real-time quantitative PCR (qRT-PCR)8,9,39,40,41,42. The JA biosynthesis and signalling genes lipoxygenase (IbLOX), allene oxide synthase (IbAOS), OPDA reductase 3 (IbOPR3), coronatine-insensitive 1 (IbCOI1) and plant defensin gene 1.2 (IbPDF1.2) were significantly induced under salt stress in ND98, and the expression levels of these genes were significantly higher in ND98 than in Lizixiang after exposure to salt stress (Fig. 5B). The expression levels of ABA biosynthesis genes zeaxanthin epoxidase (IbZEP), aldehyde oxidase (IbAO) and 9-cis-epoxycarotenoid dioxygenase (IbNCED) showed changes under salt stress in both genotypes, but these genes were not induced in ND98 under salt stress (Fig. 5C). The SA biosynthesis gene phenylalanine ammonia-lyase (IbPAL) was up-regulated, but other biosynthesis and signalling genes were not induced under salt stress in ND98 (Fig. 5D). The expression patterns of these genes were consistent with the results of the RNA-Seq analysis (Supplementary Table S6). These results indicated that the JA signalling pathway was involved in responses to salt stress in ND98.
Figure 5. Distribution and expression of the genes in the JA, ABA and SA biosynthesis and signalling pathways.

(A) Analysis of the distribution of genes in phytohormone signalling pathways. The P values were determined using singular enrichment analysis (http://bioinfo.cau.edu.cn/agriGO). The top three categories of phytohormones were JA, ABA and SA, accounting for 64.2% of the DEGs in the hormone class. ABA, abscisic acid; BR, brassinosteroid; CK, cytokinins; ET, ethylene; GA, gibberellin; JA, jasmonic acid; and SA, salicylic acid. (B), (C) and (D) Expression of the key genes in the JA, ABA and SA biosynthesis and signalling pathways, respectively, under salt stress. IbLOX, c154376.graph_c0; IbAOS, c110075.graph_c0; IbAOC, c136730.graph_c0; IbOPR3, c151632.graph_c0; IbCOI1, c148337.graph_c2; IbPDF1.2, c136835.graph_c0; IbMYC2, c143913.graph_c0; IbZEP, c158900.graph_c0; IbAO, c150446.graph_c0; IbNCED, c156411.graph_c0; IbSnRK2, c148195.graph_c0; IbABF1,c125138.graph_c1; IbPAL, c147773.graph_c3; IbNPR1, c151920.graph_c0; IbPR1, c160535.graph_c0.
To verify the expression patterns of the genes involved in phytohormone metabolism, the contents of endogenous JA, ABA and SA were measured in the roots and leaves of Lizixiang and ND98. Significant differences were not observed in either the ABA or the SA contents in the leaves or roots between Lizixiang and ND98 before or after salt stress treatment (Fig. 6A,B). Though the JA content was significantly higher in the leaves and roots of ND98 than in Lizixiang under normal conditions, the JA content in the ND98 roots was significantly increased after 12 h of salt stress compared to that of untreated ND98, while the JA content in the Lizixiang roots showed no significant difference before or after salt stress treatment, indicating that JA signalling pathway can be activated by salt stress in ND98 (Fig. 6A,B). These results further showed that JA plays a crucial role in the enhanced salinity tolerance of ND98 relative to that of Lizixiang plants.
Figure 6. Responses of JA, ABA and SA in the roots and leaves of Lizixiang and ND98 under salt stress.

(A) JA, ABA and SA contents in the leaves. (B) JA, ABA and SA contents in the roots. The data are presented as the means ± SD (n = 3). *P ≤ 0.05 for significant differences between Lizixiang and ND98 according to Student’s t-test.
Analysis of stomatal conductance and transpiration rate
Stomatal conductance and transpiration rate are important parameters for evaluating the degree of stomatal opening; these parameters are negatively correlated with stomatal closure43. Accordingly, the stomatal conductance and transpiration rates of pot-grown Lizixiang and ND98 leaves were measured under normal and saline conditions. The stomatal conductance and transpiration rate were lower in ND98 than in Lizixiang under normal conditions, and these parameters were considerably reduced in ND98, compared to those in Lizixiang, under salt stress (Fig. 7A,B). The contents of JA and ABA were measured in the leaves of the corresponding pot-grown Lizixiang and ND98 plants; the JA content was significantly higher in ND98 than in Lizixiang after salt stress, whereas the ABA content was similar between the two genotypes after salt stress (Fig. 7C,D). Stomatal opening- and closure-related genes in the DEGs were analyzed. Root phototropism protein 2 (IbRPT2), which is involved in stomatal opening, exhibited significantly lower expression (Fig. 7E, Supplementary Table S6), whereas transcription factor IbbHLH93, which leads to a weak stomatal phenotype, showed significantly higher expression in ND98 than in Lizixiang (Fig. 7F, Supplementary Table S6). These results indicated that the stomatal closure enhancement in response to the increased JA content and the response of stomatal closure-related genes improved water conservation, stabilized osmotic pressure and reduced membrane damage in the leaves of ND98 under salt stress.
Figure 7. Different stomatal opening and closing phenotypes and Na+ and K+ contents in Lizixiang and ND98.

(A) Stomatal conductance. (B) Transpiration rates. (C) JA content. (D) ABA content. (E) The expression levels of stomatal opening-related gene IbRPT2 validated using qRT-PCR. IbRPT2, c158200.graph_c0. (F) The expression levels of stomatal closure-related gene IbbHLH93 validated using qRT-PCR. IbbHLH93, c152736.graph_c0. (G) Na+ and K+ content and K+/Na+ ratio. The data are presented as the means ± SD (n = 3). *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001 indicate significant differences between Lizixiang and ND98 (Student’s t-test).
Analysis of Na+ content
The ion transport-related genes potassium transporter 1 (HKT1), Na+/H+ antiporter 1(NHX1) and Na+/H+ antiporter 18 (NHX18) were up-regulated in ND98 roots, but down-regulated in Lizixiang roots under salt stress (Supplementary Table S6). The maintenance of a high intracellular K+/Na+ ratio is important for avoiding Na+ poisoning44. The Na+ and K+ contents and the K+/Na+ ratios in the roots of pot-grown Lizixiang and ND98 were measured under normal and salt conditions. Similar Na+ and K+ contents were observed in Lizixiang and ND98 under conditions where salt stress was not present, while Na+ accumulation was significantly reduced and the K+ content and K+/Na+ ratio were significantly increased in ND98 compared with Lizixiang under salt stress (Fig. 7G). These results showed that ND98 exhibited a more balanced ion state than Lizixiang, contributing to the improved adaptability of ND98 to salt stress.
Discussion
The JA signalling pathway plays important roles in the response of sweet potato to salt stress
Sweet potato is one of the most widely cultivated root and tuber crops and has considerable potential for cultivation on saline land. However, the current understanding of sweet potato responses to salt stress remains limited. Plant roots alter their developmental process to rapidly adapt to salt stress. The salt tolerance of sweet potato is improved through increasing betaine, proline and ABA contents, activating ROS scavenging and enhancing myo-inositol biosynthesis, photosynthesis and ion homoestasis29,30,31,32,45,46,47,48,49,50.
JA, which is naturally synthesized by plants, plays an important role as a signal molecule that induces tolerance mechanisms under the influence of abiotic stresses4,5,6,7,8,9,10,11,51. However, the roles of the JA signalling pathway in response to abiotic stresses in sweet potato have not been reported to date. In the present study, the salt-sensitive variety Lizixiang and the salt-tolerant line ND98 were used to identify the genes and pathways involved in the salt tolerance of sweet potato via high-throughput RNA-sequencing technology. The DEGs between the plants included a number of genes involved in plant hormone synthesis and signalling (Fig. 2B). The ABA and SA biosynthesis and signalling genes showed changes, but most of these genes were not induced in both genotypes under salt stress. Significant differences were also not observed in the ABA and SA contents in the leaves and roots of Lizixiang and ND98 before and after salt treatments (Fig. 6A,B). Interestingly, the expression levels of the JA biosynthesis and signalling genes (IbLOX, IbAOS, IbAOC, IbOPR3, IbCOI1 and IbPDF1.2) and the content of JA were significantly higher in ND98 than in Lizixiang under salt stress (Fig. 5B). These results indicate that the salt tolerance in ND98 is related to JA content, suggesting that the JA signalling pathway plays important roles in the response of sweet potato to salt stress (Fig. 8).
Figure 8. Model of the regulatory networks activated in response to salt stress in ND98.

JA signalling may regulate stomatal closure in sweet potato
Plants show enhanced stomatal closure and maintain osmotic pressure in response to abiotic stresses52. JA stimulates stomatal closure and thus prevents dehydration under salt and drought stresses53. Stomatal conductance and transpiration rates are negatively correlated with stomatal closure, and transpiration rates provide an important index to measure the loss of water from leaves43. In the present study, the stomatal conductance and transpiration rates were strongly reduced in JA-sufficient ND98 plants, compared with those in Lizixiang plants, under salt stress (Fig. 7A,B,C). Therefore, a lower degree of JA-induced stomatal closure was observed in ND98 than in Lizixiang, and reduced transpiration rates led to decreased leaf water losses in ND98. In addition, the stomatal opening-related gene IbRPT2 was significantly down-regulated and the stomatal closure-related transcription factor IbbHLH93 was significantly up-regulated in ND98 (Fig. 7E,F), consistent with the results of Inada et al.54 and Nadeau et al.55. These results indicate that JA regulates stomatal closure and reduces leaf water losses, thereby improving the salt tolerance of ND98 (Fig. 8).
JA and the ion transport network may contribute to the salt tolerance of sweet potato
High soil salinity imposes ion toxicity on plants, in addition to osmotic stress, leading to cell damage and growth arrest. The treatment of salt-stressed plants with exogenous JA has been shown to improve Na+ exclusion by decreasing Na+ uptake at the root surface7,56. The ion transport-related gene HKT1 plays a key role in protecting against Na+ toxicity through the promotion of K+ accumulation44, and another gene, NHX1, plays an important function in transporting Na+ into the vacuole57. The expression of MaNHX genes is induced by MeJA58, and the signalling network involving MeJA and ion transport controls plant growth under salt stress.
In the present study, the ion transport-related genes IbHKT1, IbNHX1 and IbNHX18 were significantly up-regulated and the Na+ content was significantly reduced in JA-sufficient ND98 roots, whereas these genes were significantly down-regulated and the Na+ content was significantly increased in Lizixiang roots under salt stress (Fig. 7G, Supplementary Table S6). These results suggest that the increased JA content induces the expression of ion transport-related genes and maintains a relative ion balance, thereby improving the salt tolerance of ND98 (Fig. 8).
Decreased DNA methylation may up-regulate salt stress-responsive genes in sweet potato
Environmental stresses induce changes in global gene expression via DNA methylation or demethylation, with hypermethylation leading to gene down-regulation and hypomethylation leading to gene up-regulation59,60,61. The high expression of the cold stress-related gene ZmMI1 is associated with a reduction in methylation under cold stress in maize roots62, and the hypomethylation of genomic DNA has been reported to lead to the up-regulation of stress-responsive genes in tobacco plants under tobacco mosaic virus (TMV) treatment63. Key demethylation-related gene ROS1 initiates the erasure of 5-methylcytosine through a base excision repair process, and DML3 is required to remove DNA methylation marks from improperly methylated cytosine and repairs high methylation levels in properly targeted sites64. In the present study, the expression levels of demethylation-related genes IbROS1 and IbDML3 were significantly higher in ND98 than in Lizixiang (Fig. 4B), and ND98 exhibited reduced methylation compared with Lizixiang under salt stress (Table 2). The proportion of genes with FPKM ≥ 1 was 13.9 and 15.2% higher under the 12- and 48-h salt treatments, respectively, in ND98 than in Lizixiang (Fig. 1B). This result was consistent with the low methylation level of ND98 under salt stress. These results demonstrate that decreased DNA methylation may up-regulate salt stress-responsive genes in sweet potato, which also contributes to the salinity tolerance of ND98 (Fig. 8). The decreased DNA methylation may be caused by the increased JA level and the further study is needed65,66.
In conclusion, for the first time, the transcriptome of sweet potato under salt stress was analyzed using the salt-sensitive variety Lizixiang and the salt-tolerant line ND98. The results of the transcript profile analysis have revealed that the JA signalling pathway plays important roles in the responses of sweet potato to salt stress, and the increased JA level enhances the salt tolerance of sweet potato by regulating stomatal closure and maintaining ion homeostasis. Decreased DNA methylation may also contribute to the salt tolerance of this crop. These results provide insights into the mechanisms and genes involved in the salt tolerance of sweet potato.
Methods
Plant materials
The salt-sensitive sweet potato variety Lizixiang and the salt-tolerant sweet potato line ND98 were used in the present study. For the RNA-Seq analysis, the plants were grown in MS medium at 27 ± 1 °C for 13 h/day under cool-white fluorescent light at 54 μM/m2/s for 4 weeks and were subsequently cultivated in Hoagland solution with 200 mM NaCl for 0, 12 and 48 h according to our previous studies30,31,32,50. The roots were excised, immediately frozen in liquid nitrogen and subsequently stored at −80 °C until further use. Two independent biological replicates were performed.
RNA extraction, library construction and RNA-Seq
Total RNA was extracted from the roots of Lizixiang and ND98 plants using the RNAprep Pure Plant Kit (Tiangen Biotech, Beijing, China). The purified RNA concentrations were quantified using a spectrophotometer (UV-Vis Spectrophotometer, Quawell Q5000, San Jose, CA, USA), and the integrity of the RNA samples was examined using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
The RNA-Seq library was constructed using the Illumina TruSeq RNA Sample Preparation Kit (Illumina Inc., San Diego, CA, USA). Poly-A mRNA was isolated from the total RNA samples using poly-T oligo-attached magnetic beads. The mRNA was fragmented using an RNA fragmentation kit (Ambion, Austin, TX, USA) prior to cDNA synthesis to avoid priming bias. The cleaved RNA fragments were transcribed into first-strand cDNA using reverse transcriptase and random primers followed by second-strand cDNA synthesis using DNA polymerase I and RNase H (Invitrogen, Carlsbad, CA, USA). The fragments were ligated to sequencing adaptors and sequenced on an Illumina HiSeqTM 2500 platform.
Transcript assembly and differential expression
The Trinity method was used for the de novo assembly of clean read data67. The contigs were linked to transcripts according to the paired-end information of the sequences, and the transcripts were subsequently clustered based on their nucleotide sequence identity. The longest transcripts in the cluster units were regarded as unigenes to eliminate redundant sequences, and the unigenes were subsequently combined to produce the final assembly used for annotation. The transcripts were calculated and normalized to FPKM, representing the gene expression level68. DESeq software was used to identify DEGs in pair-wise comparisons. The sequences were deemed significantly differentially expressed at FDR < 0.01, with at least a two-fold change in FPKM.
Gene annotation
Using BLAST (e < 1e-5) and HMMER (e < 1e-10), the assembled non-redundant transcripts sequences were aligned to various databases, including the NCBI non-redundant (Nr) protein and nucleotide (Nt) databases, the Swiss-Prot protein database, the Cluster of Orthologous Groups (COG) database, the GO database, the euKaryotic Clusters of Orthologous Groups (KOG) database, the KEGG pathway database, the Translated EMBL Nucleotide Sequence Database (TrEMBL) and the protein families (Pfam) database, and Sweetpotato GARDEN33. When parsing the alignment results, we assigned the functional description of the top 1 hit with the highest homology to annotate the transcripts in sweet potato.
Analysis of DNA methylation
The total genomic DNA of the sweet potato roots was extracted using the cetyltrimethylammonium bromide (CTAB) method, and the quality and concentration of DNA were measured using agarose gel electrophoresis (1%) and spectrophotometric assays.
MSAP analysis was performed to detect methylation-sensitive restriction sites in sweet potato root samples using twenty EcoRI + HpaII/MspI primer combinations (Supplementary Table S8) according to the protocol of Gupta et al.69. The experiments were repeated twice. The polymorphic and well-resolved bands were scored on the basis of the presence (1) or absence (0) of bands of the amplified DNA fragments and were subsequently translated into a data matrix. Four different patterns of PCR amplifications reflected the methylation status and level at the CCGG sites: 1/1, unmethylated (pattern HpaII/MspI = 1/1); 0/1, fully methylated (pattern HpaII/MspI = 0/1); 1/0, hemi-methylated (pattern HpaII/MspI = 1/0); and 0/0, either full methylation or nonexistence of the site (pattern HpaII/MspI = 0/0). Specific primers are listed in Supplementary Table S8.
Expression analysis of key genes involved in JA, ABA and SA signalling pathways
Roots of plants treated for 0, 1, 6, 12, 24 or 48 h with 200 mM NaCl in Hoagland solution were used to extract total RNA using the RNAprep Pure Plant Kit (Tiangen Biotech, Beijing, China). RNA samples were reverse-transcribed using the Quantscript Reverse Transcriptase Kit (Tiangen Biotech, Beijing, China). The cDNA solution was used as the template for PCR amplification with specific primers (Supplementary Table S8). The sweet potato β-actin gene was used as an internal control (Supplementary Table S8). The amplifications were performed according to Zhai et al.50.
Measurements of the related components
The stomatal conductance and transpiration rate in the leaves and the Na+ and K+ contents in the roots of pot-grown Lizixiang and ND98 plants treated with 150 mM NaCl were measured according to the methods of Wang et al.32 and Zhai et al.50. JA and ABA were quantified using an indirect enzyme-linked immunosorbent assay (ELISA) according to Yang et al.70. SA was estimated using a Systronics single-beam UV-Visible spectrophotometer at a λ max of 300 nm according to Rao et al.71.
Additional Information
Accession codes: Transcriptome datasets supporting the conclusions of this article are available in the NCBI SRA repository under the accession number SRP092215.
How to cite this article: Zhang, H. et al. Transcript profile analysis reveals important roles of jasmonic acid signalling pathway in the response of sweet potato to salt stress. Sci. Rep. 7, 40819; doi: 10.1038/srep40819 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This research was financially supported by grants from the National Natural Science Foundation of China (Grant no. 31371680), the Beijing Food Crops Innovation Consortium Program (BAIC09–2016) and the China Agriculture Research System (CARS-11).
Footnotes
Author Contributions H.Z. performed most of the experiments and drafted the manuscript. Q.Z., H.Z. and Y.L. conducted measurements of the related components and drafted the manuscript. X.W. participated in data analysis and revised the manuscript. Q.L. and S.H. conceived and designed the experiments and drafted the manuscript. All authors have read and approved the final manuscript.
References
- Munns R. The impact of salinity stress. (Date of access: 09/08/2016) http://www.plantstress.com/articles/salinity_i/salinity_i.htm. (2002).
- Zhang Y. et al. Expression partitioning of homeologs and tandem duplications contribute to salt tolerance in wheat (Triticum aestivum L.). Sci Rep. 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal E. et al. Transcriptome profiling of the salt-stress response in Triticum aestivum cv. Kharchia Local. Sci Rep. 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z., Guo J., Zhu A., Zhang L. & Zhang M. Exogenous jasmonic acid can enhance tolerance of wheat seedlings to salt stress. Ecotoxicol Environ Saf. 104, 202–208 (2014). [DOI] [PubMed] [Google Scholar]
- Salimi F., Shekari F. & Hamzei J. Methyl jasmonate improves salinity resistance in German chamomile (Matricaria chamomilla L.) by increasing activity of antioxidant enzymes. Acta Physiol Plant. 38, 1–14 (2016). [Google Scholar]
- Du H., Liu H. & Xiong L. Endogenous auxin and jasmonic acid levels are differentially modulated by abiotic stresses in rice. Front Plant Sci. 4, 397 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walia H. et al. Large-scale expression profiling and physiological characterization of jasmonic acid-mediated adaptation of barley to salinity stress. Plant Cell Environ. 30, 410–421 (2007). [DOI] [PubMed] [Google Scholar]
- Zhao Y. et al. A wheat allene oxide cyclase gene enhances salinity tolerance via jasmonate signaling. Plant Physiol. 164, 1068–1076 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedranzani H. et al. Salt tolerant tomato plants show increased levels of jasmonic acid. Plant Growth Regul. 41, 149–158 (2003). [Google Scholar]
- Ding H. et al. Jasmonate complements the function of Arabidopsis lipoxygenase3 in salinity stress response. Plant Sci. 244, 1–7 (2016). [DOI] [PubMed] [Google Scholar]
- Fang L. et al. Expression of Vitis amurensis NAC26 in Arabidopsis enhances drought tolerance by modulating jasmonic acid synthesis. J Exp Bot. 67, 2829–2845 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhita D., Kolla V. A., Vavasseur A. & Raghavendra A. S. Different signaling pathways involved during the suppression of stomatal opening by methyl jasmonate or abscisic acid. Plant Sci. 164, 481–488 (2003). [Google Scholar]
- Munemasa S., Hossain M. A., Nakamura Y., Mori I. C. & Murata Y. The Arabidopsis calcium-dependent protein kinase, CPK6, functions as a positive regulator of methyl jasmonate signaling in guard cells. Plant Physiol. 155, 553–561 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montillet J. L. et al. An abscisic acid-independent oxylipin pathway controls stomatal closure and immune defense in Arabidopsis. PLoS Biol. 11, e1001513 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J. K. Salt and drought stress signal transduction in plants. Annu Rev Plant Biol. 53, 247 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moons A., Bauw G., Prinsen E., Van Montagu M. & Van Der Straeten D. Molecular and physiological responses to abscisic acid and salts in roots of salt-sensitive and salt-tolerant Indica rice varieties. Plant Physiol. 107, 177–186 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larosa P. C., Handa A. K., Hasegawa P. M. & Bressan R. A. Abscisic acid accelerates adaptation of cultured tobacco cells to salt. Plant Physiol. 79, 138–142 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. & Davies W. J. Abscisic acid produced in dehydrating roots may enable the plant to measure the water status of the soil. Plant Cell Environ. 12, 73–81 (1989). [Google Scholar]
- Szepesi A. et al. Role of salicylic acid pre-treatment on the acclimation of tomato plants to salt-and osmotic stress. Acta Biol. Szeged. 49, 123–125 (2005). [Google Scholar]
- Senaratna T., Touchell D., Bunn E. & Dixon K. Acetyl salicylic acid (Aspirin) and salicylic acid induce multiple stress tolerance in bean and tomato plants. Plant Growth Regul. 30, 157–161 (2000). [Google Scholar]
- Németh M., Janda T., Horváth E., Páldi E. & Szalai G. Exogenous salicylic acid increases polyamine content but may decrease drought tolerance in maize. Plant Sci. 162, 569–574 (2002). [Google Scholar]
- Su Z. et al. Flower development under drought stress: morphological and transcriptomic analyses reveal acute responses and long-term acclimation in Arabidopsis. Plant Cell. 25, 3785–3807 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar R., Bhattacharjee A. & Jain M. Transcriptome analysis in different rice cultivars provides novel insights into desiccation and salinity stress responses. Sci Rep. 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin J. A. et al. A near complete snapshot of the Zea mays seedling transcriptome revealed from ultra-deep sequencing. Sci Rep. 4 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F. et al. Genetic regulation of salt stress tolerance revealed by RNA-Seq in cotton diploid wild species, Gossypium davidsonii. Sci Rep. 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Tao X. et al. Digital gene expression analysis based on integrated de novo transcriptome assembly of sweet potato [Ipomoea batatas (L.) Lam]. PLoS One. 7, e36234 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R. et al. De novo transcriptome sequencing of the orange-fleshed sweet potato and analysis of differentially expressed genes related to carotenoid biosynthesis. Int J Genomics (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. H. et al. Expression of both CuZnSOD and APX in chloroplasts enhances tolerance to sulfur dioxide in transgenic sweet potato plants. C R Biol. 338, 307–313 (2015). [DOI] [PubMed] [Google Scholar]
- Liu D. et al. Overexpression of IbP5CR enhances salt tolerance in transgenic sweetpotato. Plant Cell Tiss Org. 117, 1–16 (2014). [Google Scholar]
- Liu D. et al. A novel α/β-hydrolase gene IbMas enhances salt tolerance in transgenic sweetpotato. PloS one. 9, e115128 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D. et al. IbSIMT1, a novel salt-induced methyltransferase gene from Ipomoea batatas, is involved in salt tolerance. Plant Cell Tiss Org. 120, 701–715 (2015). [Google Scholar]
- Wang B. et al. A vacuolar Na+/H+ antiporter gene, IbNHX2, enhances salt and drought tolerance in transgenic sweetpotato. Sci Hortic. 201, 153–166 (2016). [Google Scholar]
- Hirakawa H. et al. Survey of genome sequences in a wild sweet potato, Ipomoea trifida (H. B. K.) G. Don. DNA Res. 22, 171–179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L. et al. Analyses of the complete genome and gene expression of chloroplast of sweet potato [Ipomoea batata]. PLoS One. 10, e0124083 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. et al. De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweetpotato (Ipomoea batatas). BMC Genomics. 11, 1 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firon N. et al. Transcriptional profiling of sweetpotato (Ipomoea batatas) roots indicates down-regulation of lignin biosynthesis and up-regulation of starch biosynthesis at an early stage of storage root formation. BMC Genomics. 14, 1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie F. et al. De novo sequencing and a comprehensive analysis of purple sweetpotato (Ipomoea batatas L.) transcriptome. Planta. 236, 101–113 (2012). [DOI] [PubMed] [Google Scholar]
- Min L. et al. Sugar and auxin signaling pathways respond to high-temperature stress during anther development as revealed by transcript profiling analysis in cotton. Plant Physiol. 164, 1293–1308 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller M. J. Enzymes involved in jasmonic acid biosynthesis. Physiol Plant. 100, 653–663 (1997). [Google Scholar]
- Nambara E. & Marion-Poll A. Abscisic acid biosynthesis and catabolism. Annu Rev Plant Biol. 56, 165–185 (2005). [DOI] [PubMed] [Google Scholar]
- Lee H. I., León J. & Raskin I. Biosynthesis and metabolism of salicylic acid. Proc Natl Acad Sci. 92, 4076–4079 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Fan W., Kinkema M., Li X. & Dong X. Interaction of NPR1 with basic leucine zipper protein transcription factors that bind sequences required for salicylic acid induction of the PR-1 gene. Proc Natl Acad Sci. 96, 6523–6528 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farquhar G. D. & Sharkey T. D. Stomatal conductance and photosynthesis. Annu Rev Plant Physiol. 33, 317–345 (1982). [Google Scholar]
- Rubio F., Gassmann W. & Schroeder J. I. Sodium-driven potassium uptake by the plant potassium transporter HKT1 and mutations conferring salt tolerance. Science. 270, 1660 (1995). [DOI] [PubMed] [Google Scholar]
- Gao S. et al. Overexpression of SOS genes enhanced salt tolerance in sweetpotato. J Integr Agr. 11, 378–386 (2012). [Google Scholar]
- Fan W., Zhang M., Zhang H. & Zhang P. Improved tolerance to various abiotic stresses in transgenic sweet potato (Ipomoea batatas) expressing spinach betaine aldehyde dehydrogenase. PLoS One. 7, e37344 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. H. et al. Cloning and characterization of an Orange gene that increases carotenoid accumulation and salt stress tolerance in transgenic sweetpotato cultures. Plant Physiol Biochem. 70, 445–454 (2013). [DOI] [PubMed] [Google Scholar]
- Kim S. H. et al. Downregulation of the lycopene ε-cyclase gene increases carotenoid synthesis via the β-branch-specific pathway and enhances salt-stress tolerance in sweetpotato transgenic calli. Physiol Plant. 147, 432–442 (2013). [DOI] [PubMed] [Google Scholar]
- Liu D. et al. An Ipomoea batatas iron-sulfur cluster scaffold protein gene, IbNFU1, is involved in salt tolerance. PloS one. 9, e93935 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai H. et al. A myo-inositol-1-phosphate synthase gene, IbMIPS1, enhances salt and drought tolerance and stem nematode resistance in transgenic sweet potato. Plant Biotechnol J. 14, 592–602 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manar T. et al. The effects of JA treatment on the growth and some enzyme activities of eggplant embryos grown in vitro under salt stress conditions. Res J Biotechnol. 8, 12 (2013). [Google Scholar]
- Zhu J. K. Salt and drought stress signal transduction in plants. Annu Rev Plant Biol. 53, 247 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Mata C. & Lamattina L. Nitric oxide induces stomatal closure and enhances the adaptive plant responses against drought stress. Plant Physiol. 126, 1196–1204 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inada S., Ohgishi M., Mayama T., Okada K. & Sakai T. RPT2 is a signal transducer involved in phototropic response and stomatal opening by association with phototropin 1 in Arabidopsis thaliana. Plant Cell. 16, 887–896 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau J. A. Stomatal development: new signals and fate determinants. Curr Opin Plant Biol. 12, 29–35 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahzad A. N. et al. Maize genotypes differing in salt resistance vary in jasmonic acid accumulation during the first phase of salt stress. J Agron Crop Sci. 201, 443–451 (2015). [Google Scholar]
- Apse M. P., Aharon G. S., Snedden W. A. & Blumwald E. Salt tolerance conferred by overexpression of a vacuolar Na+/H+ antiport in Arabidopsis. Science. 285, 1256–1258 (1999). [DOI] [PubMed] [Google Scholar]
- Cao B. et al. Molecular characterization and expression analysis of the mulberry Na+/H+ exchanger gene family. Plant Physiol Biochem. 99, 49–58 (2016). [DOI] [PubMed] [Google Scholar]
- Zhu J. K. Epigenome sequencing comes of age. Cell. 133, 395–397 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui T. et al. Transcriptional silencing of secreted frizzled related protein 1 (SFRP1) by promoter hypermethylation in non-small-cell lung cancer. Oncogene. 24, 6323–6327 (2005). [DOI] [PubMed] [Google Scholar]
- Jaenisch R. & Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 33, 245–254 (2003). [DOI] [PubMed] [Google Scholar]
- Steward N., Ito M., Yamaguchi Y., Koizumi N. & Sano H. Periodic DNA methylation in maize nucleosomes and demethylation by environmental stress. J Biol Chem. 277, 37741–37746 (2002). [DOI] [PubMed] [Google Scholar]
- Wada Y., Miyamoto K., Kusano T. & Sano H. Association between up-regulation of stress-responsive genes and hypomethylation of genomic DNA in tobacco plants. Mol Genet Genomics. 271, 658–666 (2004). [DOI] [PubMed] [Google Scholar]
- Ortega-Galisteo A. P., Morales-Ruiz T., Ariza R. R. & Roldán-Arjona T. Arabidopsis DEMETER-LIKE proteins DML2 and DML3 are required for appropriate distribution of DNA methylation marks. Plant Mol Biol. 67, 671–681 (2008). [DOI] [PubMed] [Google Scholar]
- Ahmad F. et al. Comprehensive gene expression analysis of the DNA (cytosine-5) methyltransferase family in rice (Oryza sativa L.). Genet. Mol. Res. 13, 5159–5172 (2014). [DOI] [PubMed] [Google Scholar]
- Kissoudis C., van de Wiel C., Visser R. G. & van der Linden G. Enhancing crop resilience to combined abiotic and biotic stress through the dissection of physiological and molecular crosstalk. Front Plant Sci. 5, 207 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 29, 644–652 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 28, 511–515 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta V., Bijo A., Kumar M., Reddy C. & Jha B. Detection of epigenetic variations in the protoplast-derived germlings of Ulva reticulata using methylation sensitive amplification polymorphism (MSAP). Mar Biotechnol. 14, 692–700 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Zhang J., Wang Z., Zhu Q. & Wang W. Hormonal changes in the grains of rice subjected to water stress during grain filling. Plant Physiol. 127, 315–323 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A. N., Sivasankar B. & Sadasivam V. Kinetic study on the photocatalytic degradation of salicylic acid using ZnO catalyst. J Hazard Mater. 166, 1357–1361 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.