

Summary
Cholinergic interneurons (CHIs) play a major role in motor and learning functions of the striatum. As acetylcholine does not directly evoke postsynaptic events at most striatal synapses, it remains unclear how postsynaptic cholinergic receptors encode the firing patterns of CHIs in the striatum. To examine the dynamics of acetylcholine release, we used optogenetics and paired recordings from CHIs and medium spiny neurons (MSNs) virally overexpressing G-protein activated inwardly rectifying potassium (GIRK) channels. Due to the efficient coupling between endogenous muscarinic receptors and GIRK channels, we found that firing of individual CHIs resulted in monosynaptic spontaneous inhibitory post-synaptic currents (IPSCs) in MSNs. Paired CHI-MSN recordings revealed that the high probability of acetylcholine release at these synapses allowed muscarinic receptors to faithfully encode physiological activity patterns from individual CHIs without failure. These results indicate that muscarinic receptors in striatal output neurons reliably decode CHI firing.
eTOC blurb
Mamaligas and Ford examine the activation of muscarinic M4-receptors on direct-pathway medium spiny neurons in the striatum. By measuring synaptic currents through overexpressed GIRK channels, they find that cholinergic interneuron firing evokes transient spontaneous events through these metabotropic receptors.
Introduction
Cholinergic interneurons (CHIs) are the major source of ACh in the striatum (Bolam et al., 1984; Kawaguchi, 1993; Lim et al., 2014; Wilson et al., 1990). While these cells represent a small population of striatal neurons (1 – 2%), their broad arborizations and tiled distribution provide dense ACh innervation throughout the striatum. CHI firing in vivo switches from tonic mode to a transient pause followed by a rebound increase that can be tightly locked to rewarding cues and related stimuli that occur during associative motor learning tasks (Aosaki et al., 1994; Atallah et al., 2014; Morris et al., 2004). The timing of firing patterns in cholinergic interneurons (CHIs) is associated with behaviors in response to expected rewards as well as aversive stimuli (Shimo and Hikosaka, 2001).
In the striatum, ACh signals through both ligand-gated ion channel nicotinic receptors and metabotropic G-protein coupled muscarinic receptors. Through activation of these receptors, cholinergic transmission modulates the activity of multiple striatal circuits to regulate output activity from the striatum (Goldberg et al., 2012; Koos and Tepper, 2002; Kreitzer, 2009; Lim et al., 2014). Although many studies have examined the role of CHIs in striatal microcircuitry, the mechanisms by which ACh mediates synaptic transmission in the striatum still remain unclear. Striatal cholinergic transmission has often been studied by measuring the di-synaptic modulation of dopamine and GABA release. ACh mediated release of these transmitters involves activation of presynaptic nicotinic receptors on dopamine terminals or GABAergic interneurons and requires synchronous activation of multiple CHIs (Cachope et al., 2012; Nelson et al., 2014b; Threlfell et al., 2012). As a result of these indirect synaptic connections, it has been difficult to directly determine how the release of ACh as a result of CHI firing drives this modulation.
Muscarinic G-protein coupled receptors regulate the integration of synaptic inputs, plasticity, firing patterns, collateral connections and the transition to up-states of medium spiny neurons (MSNs), the output neuron of the striatum (Ding et al., 2006; Goldberg et al., 2012; Higley et al., 2009; Perez-Rosello et al., 2005; Yamamoto et al., 2013). In the striatum, muscarinic receptors do not directly couple to endogenous ion channels but instead indirectly alter excitability through 2nd messenger signaling cascades (Bernard et al., 1992; Calabresi et al., 1998; Goldberg et al., 2012; Pakhotin and Bracci, 2007; Pisani et al., 2002; Shen et al., 2007; Sullivan et al., 2008). Due to the multistep processes underlying muscarinic signal transduction, it has been difficult to separate receptor activation from signaling, limiting the resolution of cholinergic signaling events onto MSNs. The pacemaking of cholinergic interneurons (Bennett and Wilson, 1999) and broad axonal arborizations (Bolam et al., 1984; Perez-Rosello et al., 2005) have been thought to create a background tone of ACh that tonically drives muscarinic receptor activation. CHIs exhibit a range of firing patterns including regular and irregular tonic activity as well as bursts and pauses (Bennett et al., 2000; Bennett and Wilson, 1999; Goldberg and Reynolds, 2011). However, it remains unclear how the patterns of CHI firing and time course of ACh release activate cholinergic receptors on MSNs.
In the present study, G-protein activated potassium channels (GIRK2; Kir3.2) were virally overexpressed in MSNs to provide a rapid electrophysiological readout of muscarinic receptor activation. We find that cholinergic interneurons make robust monosynaptic muscarinic connections with MSNs. Using paired CHI-MSN recordings, we show that the firing of an individual CHI is sufficient to drive a muscarinic receptor mediated synaptic event. Despite the intrinsically slow kinetics of GPCR signaling, the rapid dynamics of these synaptic events allows the firing of individual cholinergic interneurons to be discretely encoded within MSNs as spontaneous events as opposed to tonic muscarinic receptor activation. These properties allow for reliable cholinergic transmission in response to different patterns of CHI firing.
Results
Spontaneous muscarinic IPSCs in MSNs overexpressing GIRK2
To examine how muscarinic receptors encode cholinergic interneuron firing patterns in striatal output neurons, we virally overexpressed G-protein coupled inwardly rectifying K+ channels (GIRK2; Kir3.2) in MSNs (Marcott et al., 2014). Injection of an adeno-associated virus (AAV) encoding tdTomato and GIRK2 under a synapsin promoter into the dorsal striatum led to the co-expression of both GIRK2 and the soluble fluorophore tdTomato (Figure 1A) (Marcott et al., 2014). Out of 409 tdTomato+ neurons, 366 co-expressed GIRK2 (Figure 1A and 1B). Expression was largely restricted from cholinergic neurons as limited GIRK2 immunoreactivity or tdTomato fluorescence could be detected in choline acetyltransferase (ChAT)-positive neurons (4 of 71 ChAT+ co-expressed GIRK2) (Figure 1A and 1B). Weak expression of GIRK2 in CHIs was likely due to the use of a synapsin promoter as we found that other AAVs using this promoter also failed to effectively transfect CHIs (Figure S1A). As seen previously (Marcott et al., 2014), no GIRK2 immunoreactivity could be detected in control uninjected striatal slices (Figure S1B).
Figure 1. Muscarinic receptors evoke an outward current in MSNs following viral mediated expression of GIRK2.

(A) Immunohistochemical image illustrating AAV induced expression of GIRK2 and tdTomato in striatal sections two weeks following injection of AAV2/9.hSyn.tdTomato.T2A.GIRK2.WPRE into the dorsal striatum. Shown are tdTomato, GIRK2 and choline acetyl transferase (ChAT) immunoreactivity in striatal cells indicating that AAV drove the expression of GIRK2 in non-cholinergic striatal neurons. 20 μm scale bar.
(B) Quantification tdTomato+ and ChAT+ neurons expressing GIRK2 immunoreactivity.
(C) Example voltage-clamp recording from a tdTomato+ MSN. Bath application of the muscarinic agonist oxotremorine-M (1 μM) evoked an outward current.
(D) Concentration-response curves for the outward current produced from GIRK2+ MSN by bath application of Oxo-M > 21-days (purple) or 4-days (magenta) following AAV.GIRK2 injection into the striatum. There is an increase in the maximum effect produced by Oxo-M, but no change in the EC50. Error bars indicate ± SEM. n = 4 – 11 ** = p < 0.01, Mann-Whitney.
(E) Example trace from a tdTomato+ MSN in an AAV-injected hemisphere (top) and a control non-fluorescent MSN in a control uninjected slice (bottom). Iontophoretic application of ACh (100 μM, 100 ms) evoked an outward current that was blocked by the muscarinic antagonist scopolamine (1 μM) (top). No current could be evoked in control MSNs from uninjected hemispheres (bottom).
(F) ACh-mediated current voltage relationship. 10mV voltage steps were given from −50 to −130 mV. Subtraction of the control traces from those recorded in ACh reveals a current that shows inward rectification and reverses near the predicted K+ reversal potential.
To examine if exogenous GIRK2 channels could couple to muscarinic receptors (mAChRs) in MSNs, we performed whole-cell voltage clamp recordings of tdTomato+ neurons in the dorsal striatum. MSNs were identified by their low input resistance, hyperpolarized resting membrane potential, and lack of hyperpolarization-activated current (Kreitzer, 2009; Marcott et al., 2014). After allowing 21 days for expression, application of the muscarinic agonist oxotremorine M (Oxo-M) (1 μm) evoked an outward current in tdTomato+ MSNs (Figure 1C). Overexpressed GIRK2 channels couple efficiently to endogenous Gαi/o coupled GPCRs without altering the affinity of transmitter for the receptor (Marcott et al., 2014). To confirm that overexpression of GIRK2 did not change the affinity of muscarinic receptors, we varied the extent of GIRK2 expression by recording from MSNs taken 3–5 days post AAV injection. In brain slices taken from mice > 21 days post-AAV injection, Oxo-M evoked larger outward currents in tdTomato+ neurons than in animals expressing GIRK for only 3–5 days (Figure 1D). Although the amplitude of Oxo-M induced outward currents were decreased under conditions of reduced expression, the EC50 of Oxo-M was unchanged (4 day expression: 431 nM, 95% confidence interval = 121 – 741 nM; > 21 day expression 234 nM, 95 % confidence interval = 118 – 350 nM) (Figure 1D). Iontophoresis of exogenous ACh (100 μM, 100 ms) also evoked an outward current of 285 ± 84 pA in tdTomato+ MSNs (n = 8) (Figure 1E). The current was eliminated by the mAChR antagonist scopolamine (1 μM) (2 ± 1 pA, n = 5, p < 0.05, Wilcoxon) (Figure 1E). The current-voltage relationship in response to ACh showed inward rectification and a reversal potential near the predicted K+ equilibrium potential, consistent with activation of a GIRK conductance (Figure 1F). Iontophoresis of ACh failed to evoke outward currents in control MSNs from the uninjected hemisphere (1 ± 1 pA, n = 27) (Figure 1E). Together the results show that overexpressed GIRK2 channels couple to endogenous muscarinic receptors in MSNs without a change in the apparent affinity of receptors.
To isolate cholinergic transmission through muscarinic receptors, we performed whole-cell recordings (Vh = − 60 mV) from GIRK2+ MSNs in the presence of glutamate, GABA and dopamine receptor antagonists. In the absence of stimulation, recordings revealed spontaneous IPSCs (sIPSCs) that were abolished by the mAChR antagonists scopolamine (1 μM, n = 11, p < 0.001, Wilcoxon) or tropicamide (1 μM, n = 5, p < 0.05, Wilcoxon) (Figure 2A and 2F). The frequency of sIPSCs was similar to the range of frequencies reported for CHI pacemaker firing in slices (Bennett and Wilson, 1999; Wilson et al., 1990). While Gαq-coupled M1 mAChRs are expressed in all MSNs, Gαi/o-coupled M4 mAChRs are expressed predominantly in direct pathway MSNs (Bernard et al., 1992; Goldberg et al., 2012; Lim et al., 2014; Yan et al., 2001). Spontaneous IPSCs occurred in roughly half of all MSNs (96 out of 202 GIRK2+ MSNs, Figure 2B). In a subset of experiments, we expressed GIRK2 in Drd1-eYFP mice that express YFP in direct pathway, D1-receptor-containing MSNs (dMSN). We found that sIPSCs were present in all D1-eYFP neurons tested (10 out of 10 YFP+ neurons, Figure 2B). As GIRK channels preferentially couple to Gαi/o-coupled GPCRs (Luscher and Slesinger, 2010), scopolamine-sensitive sIPSCs in dMSNs likely result from activation of muscarinic M4-receptors.
Figure 2. CHI firing evokes spontaneous muscarinic-IPSCs in GIRK2+ MSNs.

(A) Representative traces of sIPSCs in the absence of stimulation from a GIRK2+ MSN (black) that were blocked by the muscarinic antagonist scopolamine (1 μM) (grey).
(B) Percentage of neurons displaying sIPSCs in GIRK2+ WT MSNs and D1-eGFP+ (dMSN) MSNs.
(C) Amplitude distribution of sIPSCs from the MSN shown in (A) (10 pA bin size).
(D) Rise time distribution of sIPSCs from the same neuron illustrating that rise time does not correlate with amplitude of event (n = 51 events, amplitude distribution: p > 0.05, Chi-squared test; amplitude vs rise: r2 = 0.03, p = 0.21, Pearson correlation).
(E) Representative trace illustrating the sIPSCs were blocked by TTX (200 nM).
(F) Summary of pharmacological effects on sIPSCs frequency. sIPSCs were eliminated in the presence of scopolamine (1 μM), tropicamide (1 μM), Ba2+ (200 μM), vesamicol (2 μM), low Ca2+ (none added) and TTX (200 μM) (* = p < 0.05, *** = p < 0.001, Wilcoxon matched-pairs signed rank).
(G) Representative immunohistochemical images showing selective expression of ChR2-eYFP in a ChAT+ neuron.
(H) Optogenetic activation of ChR2-expressing CHIs in the striatum evokes a muscarinic-IPSC in GIRK2+ MSNs. Representative trace illustrates that the light-evoked IPSC (2 ms flash, 470 nm light) was blocked by the muscarinic antagonist scopolamine (1μM).
(I) Summary data illustrating that optogenetic activation of multiple CHIs evokes muscarinic-IPSCs larger and longer in duration than muscarinic sIPSCs (n = 8 neurons, p < 0.01 for amplitude and p < 0.05 for duration, Wilcoxon matched-pairs signed rank).
(J) Optogenetic silencing of eNpHR-expressing CHIs in the striatum eliminates sIPSCs in GIRK2+ MSNs. Representative trace illustrates the effect of green light (5 s, 530 nm).
(K) Summary data illustrating that optogenetic silencing of CHIs eliminates sIPSCs yet does not alter the baseline holding current (frequency: p < 0.001, holding current: p > 0.05, Wilcoxon matched-pairs signed rank).
Spontaneous IPSCs occurred with a mean frequency of 1.4 ± 0.1 Hz (2489 events, 32 cells) (Figures 2A and 2F) and displayed an average amplitude of 40 ± 4 pA (2865 events, 40 cells) that varied between 6 and 456 pA. As the level of AAV-driven GIRK2 expression varies among MSNs (Marcott et al., 2014), the amplitude of sIPSCs was not compared across neurons. However, the amplitude of events within each MSN was normally distributed (56 ± 4 pA, n = 51 events, single cell, p > 0.05, Chi-squared test) (Figure 2C) and did not correlate with 10 – 90% rise time (70 ± 1 ms, n = 51 events, single cell, r2 = 0.03, p = 0.21, Pearson’s correlation) (Figure 2D). These results suggest that the rate of mAChR activation underlying sIPSCs is independent of the amount of ACh released.
To examine the mechanism underlying sIPSCs, we recorded events in tetrodotoxin (TTX, 200 nM). Application of TTX abolished sIPSCs (n = 5, p < 0.05, Wilcoxon), indicating that events were action potential dependent (Figure 2E and 2F). In the presence of TTX, it was not possible to resolve individual miniature IPSCs above the baseline noise. Limiting Ca2+ entry by using ACSF without added Ca2+ (n = 5, p < 0.05, Wilcoxon), disrupting the vesicular ACh transporter with vesamicol (2 μM, n = 5, p < 0.05, Wilcoxon), or blocking GIRK channels with Ba2+ (200 μM, n = 5, p < 0.05, Wilcoxon) eliminated sIPSCs (Figure 2F). These results indicate that muscarinic sIPSCs resulted from the activity dependent vesicular release of ACh. To confirm that ACh underlying sIPSCs originated from local CHIs, we selectively expressed the light activated cation channel, channelrhodopsin-2 (ChR2), in CHIs by co-injecting a Cre-dependent AAV (AAV.DIO.ChR2.eYFP) into the striatum of choline acetyltransferase-IRES-Cre transgenic mice (ChAT-Cre) (Figure 2G). Photoactivation of ChR2-expressing CHIs (2 ms widefield light, 470 nm, ~1 mW, Figure S2A) resulted in scopolamine-sensitive muscarinic IPSCs in roughly half of all GIRK2+ MSNs examined (n = 8 of 19 neurons) (Figure 2H). Optogenetically evoked IPSCs (oIPSCs) were larger in amplitude and longer in duration than sIPSCs (sIPSCs: 38 ± 6 pA, 197 ± 11 ms half width n = 8; oIPSCs: 517 ± 112 pA, 237 ± 14 ms half width, n = 8 neurons, p < 0.01 for amplitude and p < 0.05 for half width, Wilcoxon) (Figure 2I). Electrical stimulation (0.7 ms, 20 – 40 μA) also evoked large IPSCs that were longer than sIPSCs (302.1 ± 18.8 ms, n = 16, p < 0.05 vs sIPSC 20% width, Mann-Whitney). Thus, the synchronous activation of multiple CHI terminals led to a large increase in mAChR activation.
We confirmed that CHI firing was necessary to drive muscarinic sIPSCs using the light-driven Cl− pump, halorhodopsin, expressed in cholinergic interneurons. We co-injected a Cre-dependent AAV (AAV.DiO.eNpHR3.0-eYFP) into the dorsal striatum of ChAT-Cre mice along with AAV.GIRK2 (Figure S2B). Wide-field illumination of the striatum with long pulses of green light (5 s, 530 nM) led to a silencing of CHI firing during current-clamp recordings (Figure S2C). This resulted in a pause in sIPSCs in GIRK2+ MSNs (7.6 ± 2% of control, n = 5 neurons, p < 0.01, Wilcoxon) (Figure 2K). We found that while inhibiting CHI firing with halorhodopsin eliminated sIPSCs, there was no change in the holding current (99.4 ± 0.7% of control, n = 5 neurons, p > 0.05, Wilcoxon) (Figure 2K). The lack of a standing outward current upon silencing CHI activity suggests that muscarinic receptors on MSNs encode tonic firing of CHIs as a series of phasic events rather than a sustained level of receptor activation.
Cholinergic interneuron firing evokes unitary IPSCs
Synchronous activation of cholinergic interneurons drives the release of dopamine and GABA in the striatum from SNc terminals via presynaptic nicotinic receptors (Cachope et al., 2012; Nelson et al., 2014b; Threlfell et al., 2012). As synchronous optogenetic activation of multiple CHIs evoked oIPSCs that were prolonged relative to sIPSCs (Figure 2I), we next performed paired recordings between cholinergic interneurons and GIRK2+ MSNs to determine if the firing of an individual CHI was sufficient to evoke a muscarinic receptor-mediated IPSC. MSNs were voltage clamped at −60 mV, and CHIs were recorded in current clamp configuration. CHIs were identified by their large size, lack of tdTomato fluorescence (Figure 1A), and presence of hyperpolarization activated inward current (Figure S2B). CHI-MSN pairs were separated by less than 200 μm. Approximately 70% of recorded CHIs were synaptically connected to GIRK2+ MSNs that exhibited sIPSCs (62 of 89 paired attempts). Cholinergic interneurons were hyperpolarized to just below threshold to prevent pacemaker firing. In synaptically coupled pairs, current injection (4 ms, 150 – 500 pA) triggered a single action potential (AP) in a CHI that was time locked to a unitary muscarinic IPSC (uIPSCs) in the postsynaptic MSN (Figure 3A and 3B). Action potentials reliably evoked uIPSCs, as no failures were apparent in 560 events from 24 pairs (Figure 3C). CHI-triggered uIPSCs activated following a lag of 39 ± 1 ms (time to 10%, n = 532 events, 24 pairs; 10 – 90% rise time: 76 ± 1 ms, n = 537 events, 24 pairs) (Figure 3B). The latency to activation is similar to that seen in isolated systems where saturating concentrations of agonist have been applied to evoke GPCR mediated GIRK currents (Courtney and Ford, 2014; Ford et al., 2009; Sodickson and Bean, 1996) and likely results from the intrinsic kinetics of the receptor/G-protein-GIRK signaling complex (Ford et al., 2009). Paired uIPSCs were abolished in the presence of scopolamine (1 μM) (control: 42.4 ± 19 pA, scopolamine: 3.4 ± 1 pA, n = 6 pairs, p < 0.05, Wilcoxon) (Figure 3D). Subthreshold depolarizations of CHIs did not evoke IPSCs. Together the results indicate that individual CHIs make monosynaptic connections with MSNs and that following release, muscarinic receptors are exposed to a high concentration of ACh.
Figure 3. Muscarinic receptors mediate unitary IPSCs at CHI-MSN synapses.

(A) In paired recordings between CHIs and GIRK2+ MSNs, an action potential in a current-clamped CHI elicits a unitary IPSC in a voltage-clamped MSN. MSNs were held at −60 mV. CHIs were hyperpolarized by negative DC current injection and depolarized to threshold with 4 ms 150 pA steps (bottom trace). Two uIPSCs (u) are shown that were time locked to the firing of the paired CHI and two sIPSCs (s) occurred that were unpaired with the firing of the recorded presynaptic CHI.
(B) Averaged MSN responses following single action potentials in the synaptically coupled CHI pair illustrated in (A).
(C) uIPSC amplitude histogram (mean = 113 ± 0.3, n = 40 uIPSCs, 10 pA bin size) and latency distribution histogram (time to 10% of peak, mean = 32.9 ± 0.09, n = 39 uIPSCs, 2 ms bin size) for the synaptically coupled CHI pair illustrated in (A). All action potentials resulted in a resolvable IPSC.
(D) Summary of average uIPSC amplitude that was blocked the muscarinic receptor antagonist scopolamine (1 μM) (n = 6, p < 0.05, Wilcoxon matched-pairs signed rank).
(E) No difference was found in the amplitude or duration between sIPSCs and uIPSCs in all CHI-GIRK2+ MSN paired recordings (Mann-Whitney).
(F) For each paired CHI-MSN recording, the coefficient of variation (CV) for uIPSCs was less than that of sIPSCs (n = 23 pairs, p < 0.001, Wilcoxon matched-pairs signed rank).
In a subset of experiments, we made paired recordings in the absence of GABA, and glutamate receptor antagonists. Unitary IPSCs were unaffected by blocking GABA, or glutamate receptors (uIPSC amplitude with antagonists: 111 ± 6% of control, n = 4 pairs, p > 0.05, Wilcoxon) (Figure S3A and S3B). In addition, in the absence of antagonists, no paired GABA or glutamate synaptic events were observed from CHIs to MSNs (Figure S3A). This was in contrast to the fast inward synaptic current that was seen in MSNs following synchronous photo-activation of multiple CHIs via ChR2 (Figure S3C). This confirms that synchronous CHI activation is required to evoke the disynaptic release of GABA from dopamine terminals and GABAergic interneurons (English et al., 2012; Nelson et al., 2014b; Tritsch et al., 2012) and the co-release of glutamate from cholinergic interneurons (Higley et al., 2011; Nelson et al., 2014a) onto MSNs.
During paired recordings, we observed sIPSCs unpaired with firing of the recorded CHI (Figure 3A). Like sIPSCs, unitary IPSCs exhibited a range of amplitudes across MSNs. The average amplitude and duration of sIPSCs were similar to that of uIPSCs when compared across cells (p > 0.05 for all sIPSC vs uIPSC, Mann-Whitney) (Figure 3E). Within pairs of CHIs and MSNs uIPSCs exhibited little variability across events (coefficient of variation, CV = 0.27 ± 0.01, n = 23 pairs) (Figure 3A and 3F). The stable amplitude of uIPSCs is consistent with the idea that ACh may be released from multiple CHI terminals after each AP as a result of their highly branched and varicose axonal arborizations (Bolam et al., 1984; Contant et al., 1996; Descarries and Mechawar, 2000). Within individual MSNs, the amplitude of sIPSCs was much more variable when compared to paired uIPSCs (CV sIPSCs: 0.52 ± 0.01 CV: uIPSCs 0.27 ± 0.01, p = 0.0005, Wilcoxon) (Figure 3F). As each CHI-MSN paired recording showed a low variability of uIPSCs, the increased CV of sIPSCs likely results from the fact that multiple CHIs with a different number of intact synaptic connections in the slice synapse onto a given MSN. This idea is supported by the fact that in recordings made near the surface of the brain slice where axonal inputs from most CHIs would likely have been severed, MSNs exhibited low frequency consistent amplitude sIPSCs (CV = 0.29 ± 0.01, n = 6) suggestive of only a single CHI input (Figure S3D). In these cases sIPSCs were highly regular and the CV among events was similar to that of uIPSCs from paired recordings (p > 0.05, Mann-Whitney) (Figure S3D). Analysis of the CV from uIPSCs (0.27 ± 0.01) predicted that several CHIs may synapse onto each MSN. The predicted CV of spontaneous events when 3 CHIs were estimated to connect to each MSN (0.52) was in close agreement with that observed during recordings (0.52 ± 0.01). Together these results suggest that multiple CHIs are connected to a single MSN.
In paired recordings between CHIs and GIRK2+ MSNs we also found that the amplitude of uIPSCs was unaffected whether or not they were preceded by an unpaired sIPSC (isolated: 47 ± 7 pA, preceded: 52 ± 8 pA, n = 15 pairs of cells, p > 0.05, Wilcoxon, Figure 4A and 4B). This indicates that inputs from different CHIs are likely independent. To further examine the connectivity between CHIs and MSNs, we next made loose, cell-attached recordings of CHIs while making whole cell voltage clamp recordings from GIRK2+ MSNs. Under these conditions, roughly 1/3rd of sIPSCs recorded could be attributed to the paired CHI (Figure 4C). In the cell illustrated in Figure 3C, 31% of IPSCs were paired and 69% were unpaired (Figure 4D). This suggests that possibly three CHIs were synaptically coupled to that MSN. This estimation is likely an underestimate as some CHIs more superficial to the MSN recorded may have had axons severed during slicing. Across paired recordings, 38 ± 2% of all events were paired uIPSCs (n = 18 pairs) (Figure 4E). Among cell-attached paired recordings, there was a wide distribution of the ratio between paired and total IPSCs. For individual pairs, between 23% and 55% of all sIPSCs recorded were contributed by a paired CHI (n = 18 pairs, Figure 4F). This range indicates that between 1/5 and 1/2 of events were paired and suggests that in our slice recordings, a given MSN may receive input from between 2 and 5 CHIs (mean: 2.6 ± 0.2 CHIs per MSN) (Figure 4G). This connectivity is similar to that predicted by analyzing the CV of events (Figure 3F, above). While CHIs with faster firing rates would contribute more to the apparent connectivity, these recordings suggest that in slices several CHIs independently converge onto each MSN.
Figure 4. Convergence and functional independence of CHI inputs onto MSNs.

(A) Two traces from a paired CHI-MSN recording separated by 5 s illustrating that the amplitude of uIPSCs were unaffected if immediately preceded by an unpaired sIPSC (grey box). CHIs were hyperpolarized by negative DC current injection and depolarized to threshold with 4 ms 150 pA steps to trigger uIPSCs (bottom trace).
(B) Quantification of uIPSC amplitudes of isolated uIPSCs (not preceeded by an action potential for > 5s) and uIPSCs where a sIPSC occurred in < 1 s prior (n = 15 paired recordings, p > 0.05, Wilcoxon matched-pairs signed rank).
(C) Representative trace of cell-attached paired recordings showing paired and unpaired IPSCs. Dashed line indicate uIPSCs that are paired with the recorded CHI.
(D) Quantified percentage of paired and unpaired IPSCs for the paired recording represented in (A).
(E) Group ratio of paired or unpaired IPSCs to the total number of IPSCs (n = 18 pairs, p < 0.0001, chi-square = 16.067, 1 DOF).
(F) Distribution histogram of paired IPSC ratios (0.1 bin size, n = 18 pairs).
(G) Number of CHIs connected to an MSN in each paired recording. Mean value represented by horizontal line (2.9 ± 0.2 CHIs, n = 18).
Acetylcholinesterase limits the duration of mAChR activation
The striatum contains the highest levels of acetylcholinesterase (AChE) in the brain (Hebb and Silver, 1961; Macintosh, 1941). As the activation kinetics of sIPSCs (39 ± 1 ms lag till onset, n = 532 events) were greater than 2-fold faster than GIRK currents evoked when saturating concentrations (100 μM) of agonists were applied to other Gαi/o-coupled GPCRs on membrane patches (Courtney and Ford, 2014; Ford et al., 2009; Sodickson and Bean, 1996, 1998), the activation of muscarinic receptors underlying sIPSCs likely resulted from a local high concentration of ACh. To determine the role of AChE in regulating the extent of muscarinic receptor activation, we applied a non-saturating concentration of the AChE inhibitor ambenonium (10 nM). The amplitude (control: 48 ± 11, n = 274 events, n = 6 cells; ambenonium: 85 ± 14 pA, n= 278 events, n = 6 cells; p<0.0001, Kolmogorov-Smirnov test) and half width (146 ± 8; ambenonium: 371 ± 23 ms n = 6 cells, p < 0.05, Wilcoxon) of sIPSCs increased in the presence of ambenonium (Figure 5A – 5D). This suggests that due to efficient enzymatic degradation, AChE limits the duration of muscarinic receptor activation.
Figure 5. The extent of muscarinic receptor activation is regulated by the enzymatic degradation of ACh.

(A) Representative sIPSCs recorded under control conditions and in the presence of the AChE inhibitor ambenonium (10 nM).
(B) Averaged superimposed sIPSCs from the above cell under control conditions and in the presence of ambenonium (10 nM) illustrating that inhibiting AChE increases the amplitude and duration of sIPSCs.
(C) Summary of the effect of ambenonium (10 nM) on 10 – 90% rise time and duration (260 ± 24% control width) on sIPSCs (n = 6, p < 0.05, Wilcoxon matched-pairs signed rank).
(D) Amplitude distribution histograms (20 pA bin size) of sIPSCs in control and in the presence of ambenonium (10 nM) (p < 0.001, Kolmogorov-Smirnov test).
(E) Representative trace illustrating the effect of optogenetic silencing of eNpHR-expressing CHIs on muscarinic IPSCs in the presence of ambenonium (80 nM).
(F) Summary of the average baseline current amplitude measured before and after optogenetic silencing of eNpHR-expressing CHIs under control conditions and in the presence of ambenonium (80 nM) (p < 0.05, Mann-Whitney).
Since silencing CHI firing with halorhodopsin did not lead to a change in holding current under control conditions (Figure 2J and 2K), we next used halorhodopsin to inhibit CHI firing while blocking AChE. In the presence of ambenonium (80 nM), photoactivation of halorhodopsin (5 s) led to a decrease in the baseline holding current (control: 2 ± 3 pA change, ambenonium 70 ± 13 pA change, n = 5 cells, p < 0.05, Wilcoxon, Figure 5F). A parallel depolarization of the membrane potential during recordings in current-clamp mode was also observed in the presence of ambenonium, but not in control conditions (Figure S5). This indicates that in brain slices, tonic receptor activation only occurs when AChE is blocked. Thus despite the background tonic firing of CHIs, AChE functions to limit the extracellular tone of ACh to prevent the tonic activation of muscarinic receptors.
Cholinergic interneuron regulation of MSN output through muscarinic receptors
We found that muscarinic receptor activation led to the phasic activation of overexpressed GIRK channels, so we next wanted to test if muscarinic transmission could functionally regulate endogenous signaling cascades in dMSNs over similar timescales. The next experiments were performed without overexpressing GIRK. MSNs make recurrent synapses on neighboring MSNs (Koos et al., 2004; Taverna et al., 2008; Tecuapetla et al., 2007). As muscarinic agonists inhibit GABA transmission between MSNs (Yamamoto et al., 2013), we next sought to examine whether the release of ACh from CHI could also transiently regulate GABA transmission through these collaterals. D1-receptor expressing dMSNs have a higher connectivity with other dMSNs than indirect pathway MSNs (iMSNs) (Taverna et al., 2008). We crossed D1-eYFP mice with ChAT-Cre mice in order to both visualize dMSNs and express halorhodopsin in CHIs using AAV.DiO.eNpHR3.0-eYFP. CHIs exhibit a rebound burst of APs following strong hyperpolarizations due to activation of an HCN conductance (Straub et al., 2014; Wilson, 2005). In recordings from halorhodopsin expressing CHIs, we found that following wide-field illumination (0.5 – 4 s; 530 nm; ~1 mW), termination of photostimulation led to a transient burst of 2–3 APs in CHIs that was time-locked to the end of the light pulse (latency from end of light pulse to first AP: 71 ± 8 ms; burst interspike interval: 64 ± 7 ms) (Figure 6A and 6B). We recorded dMSN-dMSN pairs in the presence of the nicotinic receptor antagonist mecamylamine (5 μM) to isolate muscarinic transmission from CHIs. In successfully coupled dMSN-dMSN pairs, we found that the amplitude of GABAA IPSCs was decreased by triggering bursts of APs in CHIs 500ms prior to evoking GABAA IPSCs (16 ± 2 % decrease, n = 7 pairs, p < 0.05, Wilcoxon) (Figure 6C and 6D). No inhibition was observed when repeating these experiments in the presence of the muscarinic antagonist scopolamine (1 μM) (10 ± 9 % increase, n = 5 pairs, p > 0.05, Wilcoxon) (Figure 6E). Thus while overexpressed GIRK channels allow for rapid sensing of muscarinic receptor activation at cholinergic synapses, these results show that CHI firing also endogenously drives transient inhibition of GABA release from dMSN collateral terminals though these receptors on similar timescales.
Figure 6. Muscarinic inhibition of dMSN GABA collaterals.
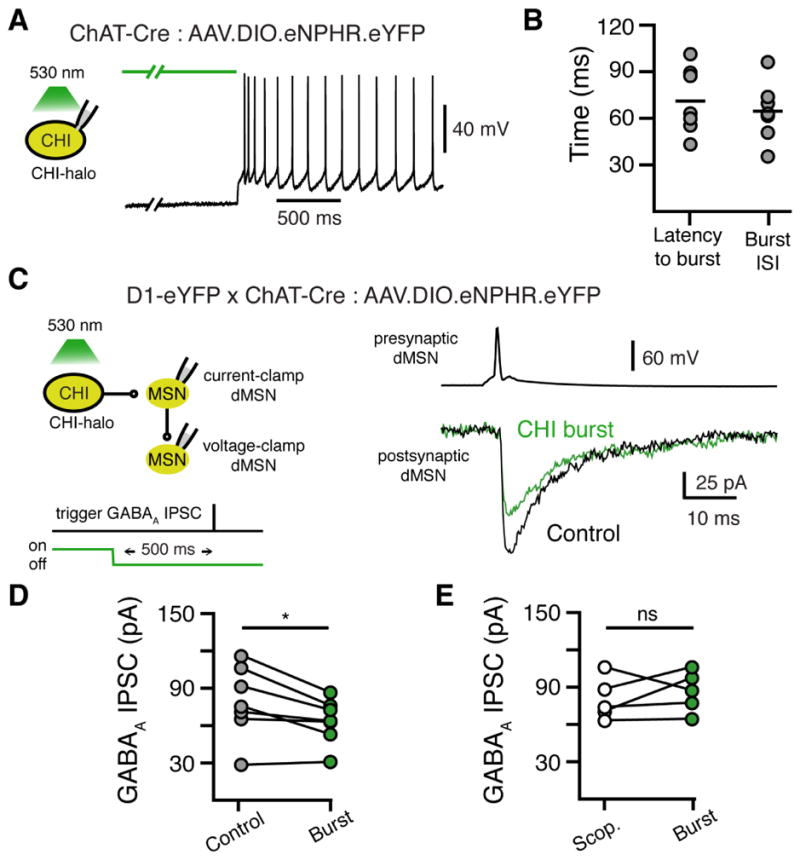
(A) Optogenetic silencing of eNpHR-expressing CHIs leads to a rebound burst of APs upon termination of wide-field (530 nm) illumination.
(B) Quantification of the latency to 1st AP following termination of 530 nm light and quantification of the ISI of the first 2 APs in the initial rebound burst.
(C) Representative recording from a dMSN-dMSN paired recording in slices from a D1-eYFP x ChAT-Cre mouse expressing eNpHR in CHIs. An AP in the presynaptic dMSN triggered a monosynaptic GABAA IPSC in the postsynaptic dMSN. The amplitude of the GABAA IPSC was reduced by terminating wide-field (530 nm) illumination 500 ms before triggering the AP in the presynaptic MSN. Traces are the average of 6 sweeps for each condition. GABAA IPSCs were recorded in the presence of DNQX (10 μM) and SKF 38393 (10 nM).
(D) Quantification of the average amplitude of GABAA IPSCs under control conditions versus 500 ms following termination of 530 nm illumination (n = 7 pairs, p <0.05, Wilcoxon matched-pairs signed rank).
(E) Quantification of the average amplitude of GABAA IPSCs under control conditions versus 500 ms following termination of 530 nm illumination in the presence of scopolamine (1 μM) (n = 5 pairs, p >0.05, Wilcoxon matched-pairs signed rank).
Regulation of ACh release transmission at muscarinic synapses
As the probability of transmitter release (Pr) decreases at many synapses with repeated stimulations, we next examined whether CHI firing resulted in a short-term depression of muscarinic transmission. To assess the probability of ACh release from CHIs onto GIRK2+ MSNs, we measured the paired pulse ratio (PPR) of uIPSCs elicited by two action potentials. The amplitude of a second uIPSC was depressed relative to the first with short interpulse intervals (PPR 250 ms: 0.48 ± 0.04, n = 5 pairs, p < 0.01; PPR 500 ms: 0.6 ± 0.1, n = 5 pairs, p < 0.01, Mann-Whitney) and required several seconds for full recovery (PPR 7 s: 1 ± 0.1, n = 5 pairs, p > 0.05, Mann-Whitney) (Figure 7A and 7D). Short-term depression is often indicative of a change in the pre-synaptic release of transmitter that occurs at synapses with a high probability of release. However as both pre- and post-synaptic mechanisms can contribute to short-term depression, we also applied ACh using paired iontophoretic applications. The amplitude of the two resulting currents when ACh was applied with a 500 ms interpulse interval were the same (PPR: 1 ± 0.1, n = 3, p > 0.05, Wilcoxon), suggesting that the depression of uIPSCs was pre-synaptic in origin.
Figure 7. Release properties of ACh at CHI-MSN muscarinic synapses.
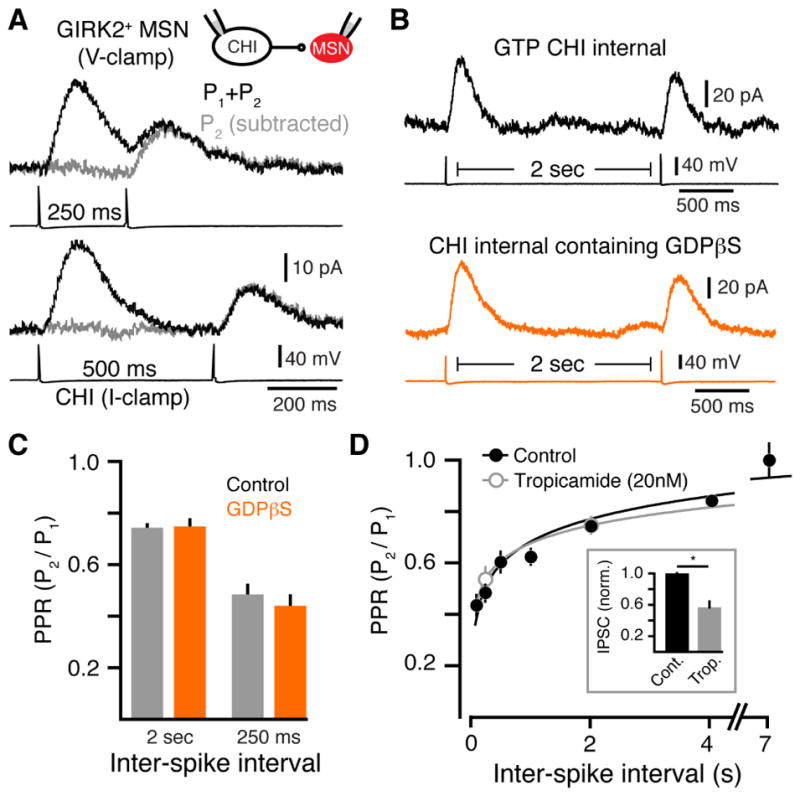
(A) Representative traces of uIPSCs evoked by paired pulse stimulation with 250 ms (above) and 500 ms (below) intervals. IPSCs evoked by the second action potential (AP) (grey) were calculated by subtracting the single pulse IPSC for intervals 100 and 250 ms apart.
(B) Representative traces illustrating that the PPR of uIPSCs was not altered when GTP was replaced with GDPβs (0.6 mM) in the intracellular solution.
(C) Summary data of PPRs of uIPSCs evoked at 250 or 2000 ms ISI with either GTP or GDBβs in the intracellular solution.
(D) PPR (P2/P1) for uIPSCs plotted against ISIs of 100, 250, 500, 1000, 2000, 4000 and 7000 ms. PPR is illustrated for uIPSCs under control conditions (black) and in the presence of an IC50 concentration of tropicamide (20 nM) (grey). Inset: inhibition of evoked IPSCs by the IC50 concentration tropicamide (20 nM) (p < 0.05, n = 5, Wilcoxon matched-pairs signed rank).
Cholinergic interneurons express muscarinic autoreceptors, which inhibit their activity. To determine if activation of presynaptic autoreceptors contributed to the reduction in ACh release, we replaced the GTP in the presynaptic intracellular solution with the GDP analog guanosine 5′-2-thiodiphosphate (GDPβs) (0.6mM) to selectively block G-protein activation in CHIs without altering muscarinic signaling in postsynaptic MSNs. The GDP analog functions as a competitive antagonist of GTP binding to G-proteins. Dialysis of CHIs with (GDPβs) (0.6 mM) for 6 minutes eliminated dopamine D2-receptor mediated IPSCs (Figure S7) (Chuhma et al., 2014; Straub et al., 2014), confirming that GDPβs blocked GPCR signaling in CHIs. We next repeated the PPR experiment after allowing for 8 – 10 minutes for GDPβs to fully dialyze CHIs. GDPβs dialysis of CHIs however did not alter the PPR of muscarinic uIPSCs at interspike intervals of 250 ms and 2 seconds when compared with control (intracellular GTP) recordings (PPR 250 ms: 0.44 ± 0.04; PPR 2 seconds: 0.75 ± 0.03, n = 7 pairs, p > 0.05 relative to control, Mann-Whitney) (Figure 7B and 7C). While the D2-receptors underlying D2-IPSCs likely reside at somatodendritic sites as opposed to muscarinic autoreceptors at CHI terminals, the lack of effect of GDPβs on muscarinic uIPSCs suggests that over the timescales examined, autoreceptors do not alter ACh release probability at muscarinic synapses.
To test whether any of the depression resulted from postsynaptic saturation of receptor signaling to G-proteins and GIRK channels, we repeated PPR analysis in the presence of a low (~IC50) concentration of the muscarinic antagonist tropicamide (20 nM). Tropicamide reduced the amplitude of uIPSCs by approximately half (43 ± 14 %, n = 5, p < 0.05 Wilcoxon) (Figure 7D; inset). Despite the reduction in amplitude, the PPR was unchanged (PPR 250 ms: control 0.48 ± 0.04, tropicamide 0.53 ± 0.05, n = 5 pairs each, p > 0.05, Mann-Whitney; PPR 2 seconds: control 0.74 ± 0.01, n = 4, tropicamide 0.75 ± 0.03, n = 5 pairs, p > 0.05, Mann Whitney). Using GDPβS internal in conjunction with bath application of tropicamide (20 nM) produced no further reduction in uIPSC amplitude or the PPR (PPR 250 ms: 0.58 ± 0.04; PPR 2 seconds: 0.69 ± 0.05, n = 4 pairs, p > 0.05 relative to control, tropicamide, and GDPβS groups, Mann-Whitney). These results suggest that the observed depression does not result from activation of presynaptic autoreceptors or saturation of postsynaptic signaling cascades but instead may be due to intrinsic short-term depression of ACh vesicle release.
CHIs are autonomous pacemakers that fire at low frequencies (Bennett and Wilson, 1999). To determine the implications of tonic CHI firing on muscarinic transmission, we introduced trains of action potentials to CHIs at 2 Hz for 5 seconds (Figure 8A). Trains of action potentials evoked a marked depression of uIPSCs (Figure 8A). Despite the depression in amplitude, all action potentials in the train led to an IPSC such that no failures were observed (Figure 8A, 2440 events, 6 pairs). At 2 Hz, the majority of the depression occurred after the first action potential of the train such that the amplitude of the IPSC did not depress in subsequent events (p < 0.05, n = 6 pairs, Tukey-Kramer test) (Figure 8B). Coinciding with the decrease in amplitude, there was a decrease in variance of the amplitude of events across multiple trials (first pulse variance: 122 ± 44 pA2, tenth pulse: 33 ± 9 pA2, n = 6 pairs, p < 0.05, Wilcoxon) (Figure 8C). The decrease in amplitude of events and the associated decrease in variance suggest that resting CHIs display a moderately high probability release profile, which lowers upon tonic activity. Recordings in which presynaptic GTP was replaced with GDPβS showed no differences in uIPSC amplitude relative to the first pulse in the train (n = 5 pairs, p > 0.05 for all events in the train, Mann-Whitney), indicating that autoreceptors do not underlie depression due to tonic firing. Directly testing the change in Pr with low extracellular Ca2+ (0.5 mM) was difficult because of the strong regulation of CHI pacemaker firing by Ca2+-activated potassium (SK) channels (Bennett and Wilson, 1999). As a result, we found that in low extracellular Ca2+ (0.5 mM) there was dramatic increase in the frequency of sIPSCs that often summated, occluding resolvable uIPSCs.
Figure 8. Burst firing relieves depression at muscarinic synapses as a result of tonic firing of cholinergic interneurons.

(A) Representative trace of paired uIPSCs evoked by a 2 Hz train of 10 APs in a CHI.
(B) Summary data of normalized uIPSCs amplitudes as a function of pulse number.
(C) Paired uIPSC amplitude variance as a function of pulse number for the 1st and 10th pulse in a train (2 Hz) (n = 6 p < 0.05, Wilcoxon matched-pairs signed rank).
(D) Bursts of APs (5 APs at 20 Hz) generate similar amplitude uIPSCs as a single APs when CHIs when tonic firing of CHIs is prevented.
(E) Quantification of uIPSC amplitudes when evoked by a single AP or bursts of APs.
(F) Bursts of APs (5 APs at 20 Hz) generate larger amplitude uIPSCs than single APs when transmission is depressed by tonic CHI firing (2 Hz). Tonic firing of CHI was simulated by a inducing a train of 20 APs (2 Hz) in CHIs. A 1 s pause in firing was followed either by a single AP or a burst (5 at 20 Hz) of APs.
(G) Quantification of the amplitude of uIPSCs when evoked by a single AP or trains of APs following tonic firing (n = 5, p < 0.05, Wilcoxon matched-pairs signed rank).
(H) Quantification of the change in uIPSC amplitude evoked by single or bursts of APs when compared to the amplitude of the last uIPSC in a train (n = 5, p < 0.05, Wilcoxon matched-pairs signed rank).
Lastly we set out to examine how muscarinic transmission in MSNs is regulated during tonic cholinergic patterns of activity. CHIs can fire in both single spikes and burst patterns in response to salient stimuli (Goldberg and Reynolds, 2011; Morris et al., 2004; Schulz and Reynolds, 2013). While hyperpolarizing CHIs to limit pacemaker firing, we found that a burst of action potentials (5 APs at 20 Hz) produced strong depression such that the burst-evoked uIPSCs were of similar amplitude to uIPSCs evoked with a single action potential (111 ± 6 % of single uIPSC, n = 9, p > 0.05, Wilcoxon, Figure 8D and 8E). In contrast, when IPSCs were depressed by evoking a train of 20 action potentials at 2 Hz, bursts of APs following a 1 sec pause led to an alleviation of depression (230 ± 14 % of last pulse, n = 6 pairs, p < 0.05, Wilcoxon) that was not observed with only a single action potential (120 ± 9 % of last pulse, p > 0.05, n = 6 pairs, Wilcoxon) (Figure 8F – 8H). Rebound burst firing of CHIs in vivo is tightly locked to rewarding cues and associated stimuli that occur during reward-based learning tasks (Aosaki et al., 1994; Atallah et al., 2014; Morris et al., 2004). Together these findings suggest that as a result of short-term depression, the extent of ACh release can be modulated in response to these patterns of firing.
Discussion
M4-receptors are the most highly expressed class of muscarinic receptors in the striatum (Bernard et al., 1992; Yan et al., 2001). These inhibitory receptors are localized primarily in dMSNs (Bernard et al., 1992; Yan et al., 2001), where they decrease excitability (Howe and Surmeier, 1995) and oppose dopamine D1-receptors induction of glutamate long-term plasticity (Shen et al., 2015). Increasing M4-receptor activity in vivo rescues L-DOPA induced impairments in synaptic plasticity and L-DOPA induced dyskinesia in animal models (Shen et al., 2015). As animals lacking M4-receptors selectively in dMSNs exhibit increased locomotor activity and behavioral sensitization to psychostimulants (Jeon et al., 2010) muscarinic GPCR signaling through these receptors is an important regulator of the striatonigral pathway. Despite the importance of these receptors in regulating striatal function, it has remained unclear how the release of ACh drives the activation of muscarinic receptors on MSNs. Using GIRK2 channels as a readout of muscarinic activation, our results show that muscarinic receptors on dMSNs receive independent, phasic synaptic inputs from CHIs. The firing of a single action potential in CHIs evoked unitary GIRK-mediated IPSCs in post-synaptic dMSNs. Our results also indicate that the physiological and anatomical characteristics of CHIs allow for consistently depressed ACh release during CHI tonic firing. Despite this depression, muscarinic M4-receptors reliably encode ACh release evoked by physiological CHI firing patterns without failure. As a result of the depression of ACh release during physiological firing of CHIs, MSNs increase their dynamic range of receptor activation, potentially allowing for differential behavioral responses in response to influential stimuli. We found that in the absence of GIRK, muscarinic receptors also could rapidly regulate dMSN output locally in the striatum through an inhibition of axon collateral synapses. Thus, the firing of CHIs may be endogenously encoded in striatal circuits through the transient inhibition of local circuits.
Although CHIs only comprise a small percentage of neurons in the striatum, their tiled distribution and extensive arborizations position them to modulate a large MSN population. Cholinergic terminals have been found to make sparse synaptic connections (~3% synaptic incidence) with MSN dendrites, occurring primarily at symmetrical synapses (Bolam et al., 1984; Contant et al., 1996; Descarries and Mechawar, 2000). Despite this low connectivity, monosynaptic rabies-tracing studies that have mapped CHI inputs to MSNs have found CHIs are extensively connected with multiple dMSNs (Salin et al., 2009). Interestingly, the connections formed between CHIs and MSNs using rabies stands in contrast to similar studies that have examined SNc dopamine inputs onto MSNs (Wall et al., 2013). While SNc terminals provide abundant innervation to the striatum, monosynaptic rabies tracing approaches have found that only a small proportion of the total dopaminergic inputs are labeled (Wall et al., 2013). One possibility for the increased monosynaptic rabies labeling of cholinergic synapses may be that CHI terminals span shorter distances to MSN dendrites than DA synapses. The symmetrical synapses formed between CHIs and MSNs where M4-receptors have been found (Hersch et al., 1994), may therefore be the cholinergic synapses labeled with trans-synaptic rabies mapping.
We found that CHI firing resulted in consistent amplitude muscarinic IPSCs. The low failure rate, low coefficient of variation and high probability of release of IPSCs suggest that each CHI action potential drives release of transmitter from many active release sites. The large number of active terminals likely allows for the overall stable amplitude of muscarinic events. This may be similar to other synapses at which multiple release sites and a high probability of release contribute to high fidelity post-synaptic receptor activation (Buhl et al., 1994; Kraushaar and Jonas, 2000; Marty et al., 2011 ; Silver et al., 1998). During the course of these experiments we were unable to resolve miniature events resulting from spontaneous vesicle release. One possibility may be that the large number of active zones, in conjunction with the slow kinetics of relatively small events may have been below the level of detection. Future studies using mean-variance quantal analysis or noise deconvolution may elucidate the quantal characteristics of ACh release at this synapse.
ACh release exhibited robust paired pulse depression at these synapses. This depression was presynaptic in nature, as decreasing postsynaptic receptor activity did not alter the paired pulse ratio. As a result of this depression, during pacemaker firing cholinergic transmission likely exists under a basally depressed state. A decrease in uIPSC variance coincided with the depression of ACh release, indicating that not all release sites are active during a given action potential (Clements and Silver, 2000; Scheuss and Neher, 2001). The decreased probability of release as a result of short-term plasticity could allow for a larger dynamic range of muscarinic receptor activation from a single CHI. During reward based learning tasks, pauses in CHI firing are followed by a rebound burst (Aosaki et al., 1994; Atallah et al., 2014; Chuhma et al., 2014; Morris et al., 2004; Straub et al., 2014). Thus, tonic depression of ACh release at basal CHI firing rates results in differential muscarinic receptor activation in response to burst firing, effectively allowing for bi-directional dynamics in this system and amplifying the range of receptor activation in response to salient cues.
The autonomous pacemaking of cholinergic interneurons (Bennett and Wilson, 1999) has been thought to create a background tone of ACh. The present results however show that the firing of individual CHIs evokes the local release of ACh in the striatum sufficient to evoke phasic receptor activation. We found CHI firing could transiently activate muscarinic receptor signaling either through GIRK channels or the endogenous inhibition of GABAergic collaterals. Thus, although cholinergic firing does not correlate with MSN firing at the cell body level (Adler et al., 2013), time-locked CHI activity can rapidly alter terminal transmitter release from dMSNs. This is similar to the rapid muscarinic receptor-mediated inhibition that has been observed at corticostriatal synapses onto MSNs (Pakhotin and Bracci, 2007). Rather than directly modifying MSN excitability, muscarinic receptors may therefore evoke subtle changes in MSN activity. Similar to muscarinic receptors, dopamine receptors do not acutely regulate MSN excitability through direct interactions with ion channels (Gerfen and Surmeier, 2011; Kreitzer, 2009). Instead, recent studies have found that dopamine receptors can drive rapid effects through 2nd messenger signaling cascades to regulate integration and plasticity in MSNs on a short-latency millisecond timescale (Swapna et al., 2016; Yagishita et al., 2014). Dopamine D1 receptors on dMSNs have been shown to promote spine growth in response to brief stimulation of dopamine release (Yagishita et al., 2014). Dopamine D1 receptors and muscarinic M4 receptors in dMSNs oppose one another’s downstream signaling activity (Shen et al., 2015). As dopamine receptors and muscarinic receptors can both signal transiently, the opposing firing patterns of dopamine neurons and CHIs likely shape the properties of transmitter release from dMSNs. Because transient muscarinic receptor activation on dMSNs is involved in disconnecting the microcircuit through collateral inhibition in the striatum, CHIs could play an integral role in temporally segregated modulation of local striatal connectivity. Recent work studying the timing of striatal GPCR activation has begun to elucidate the rapid biochemical effects of receptor signaling. Our results show that firing of individual CHIs leads to temporally and spatially localized muscarinic receptor activation, providing insights into the initial stages activating these pathways. The faithful time locked response of muscarinic receptors on MSNs could thus prove critical in controlling CHI-regulated behaviors.
Experimental Procedures
Stereotaxic injections
All procedures were performed in compliance with guidelines of the Institutional Animal Care and Use Committee at Case Western Reserve University. Both male and female wild-type C57BL6, ChAT-internal ribosome entry site-Cre heterozygote and D1-eYFP mice were injected at postnatal day 21. D1-eYFP mice were kindly provided by Dr. Veronica Alvarez at NIAAA (NIH). All other animals were from The Jackson Laboratory, Bar Harbor, ME. The injection coordinates were (relative to bregma): AP +1.15 mm, ML −1.85 mm, DV −3.35 mm. A volume of 300 nL of AAV9.hSyn.tdTomato.T2A.mGIRK2-1-A22A.WPRE.bGH and/or 300 nL of AAV5.EF1a.DIO.hChR2(H134R)-EYFP.WPRE.hGH or 300 nL of AAV1.EF1a.DIO.eNPHR3.0-EYFP.WPRE.hGH were injected into one hemisphere of the striatum. All AAVs were from the University of Pennsylvania Viral Core unless otherwise specified. Animals were allowed to recover for ~1–4 weeks to allow for expression.
Immunohistochemistry
AAV9 injected mice were anesthetized and transcardially perfused with ice-cold PBS containing (in mM) 137 NaCl, 1.5 KH2PO4, 8 NaH2PO4, and 2.7 KCl (pH=7.4) followed by 4% paraformaldehyde in PBS. Brains were rehydrated overnight in 30% sucrose. Slide-mounted sections were permeabilized with 0.1% Triton X-100 in PBS (PBS-T) and blocked in 5% normal donkey serum in PBS-T (2 hours, room temperature). Slides were washed and incubated with 1:200 goat anti-ChAT (Millipore) and/or rabbit anti-Kir3.2 (Alomone Labs) antibodies, diluted in blocking buffer, for 72 hours (anti-ChAT) at 4°C. Slides were then w ashed and incubated for 6 hours in 1:1000 donkey anti-goat 647 (ChAT staining) and washed. For GIRK staining, slides were then incubated for 2 hours in 1:500 donkey anti-rabbit 488 (Abcam). Fluorescent confocal images were obtained using a Zeiss LSM 510 META laser scanning confocal microscope (Carl Zeiss) with a 403 Plan-Neofluar, NA 1.3, oil-immersion lens. All images were processed using ImageJ, n = 5 animals examined.
Slice preparation
Striatal coronal slices (240 μm) were made in ice-cold sucrose cutting solution containing (in mM): 75 NaCl, 2.5 KCl, 6 MgCl2, 0.1 CaCl2, 1.2 NaH2PO4, 25 NaHCO3, 2.5 D-glucose, and 50 sucrose; bubbled with 95% O2 and 5% CO2. Slices were incubated at 35°C for at least 45 minut es in ACSF containing (in mM): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.4 CaCl2, 1.2 NaH2PO4, 21.4 NaHCO3, and 11.1 D-glucose; and 10 μM MK-801 bubbled with 95% O2 and 5% CO2. Slices were transferred to the recording chamber and perfused with ACSF (34±2°C) a t 2 mL/minute. Solutions contained DNQX (10 μM), picrotoxin (100 μM), CGP55845 (300 nM), SCH 23390 hydrochloride (1 μM) and sulpiride (200 nM). MSNs were visualized using a BXWI51 scope (Olympus) with IR gradient contrast optics and custom-made LEDs.
Electrophysiology
Recordings were performed using Axopatch 200A and Axopatch 200B amplifiers (Molecular Devices). Membrane potentials were not corrected for liquid junction potentials. CHIs were identified by the presence of large h-current when stepped to −110 mV in voltage-clamp. Patch pipettes (1.5 – 2MΩ) for MSNs contained 115 mM K-methylsulphate, 20 mM NaCl, 1.5 mM MgCl2, 10 mM HEPES(K), 10 mM BAPTA-tetrapotassium, 1 mg/mL ATP, 0.1 mg/mL GTP, and 1.5 mg/mL phosphocreatine, pH 7.4, 275 mOsm. Pipettes for recordings of CHIs contained 135 mM D-gluconate(K), 10 mM HEPES(K), 0.1 mM CaCl2, 2 mM MgCl2, 0.1 mM EGTA, 1 mg/mL ATP, 0.1 mg/mL GTP, and 1.5 mg/mL phosphocreatine, pH 7.4, 275 mOsm. All recordings were acquired with Axograph X (Axograph Scientific) at 10 kHz. Voltage clamp and cell-attached recordings were filtered to 2 kHz. MSNs were held at a voltage of −60 mV. No series resistance compensation was used, and cells were discarded if their series resistance exceeded 15 MΩ. Light evoked ACh release was stimulated by wide-field illumination with 488 nm blue light (2 ms pulse, ~1.0 mW/mm2). Halorhodopsin-mediated inhibition of CHI firing was induced by wide-field illumination with 570 nm green light (2 – 5 seconds, ~1.0 mW/mm2). All drugs were perfused via bath application unless otherwise noted. ACh was applied via iontophoresis (100 mM, 160 nA ejection, 25 – 50 ms; 1 pulse/min).
For paired dMSN-dMSN recordings, the presynaptic MSN internal solution contained: 135 mM D-gluconate(K), 10 mM HEPES(K), 0.1 mM CaCl2, 2 mM MgCl2, 0.1 mM EGTA, 1 mg/mL ATP, 0.1 mg/mL GTP, and 1.5 mg/mL phosphocreatine, pH 7.4, 275 mOsm. Presynaptic MSNs were held near −60 mV and APs were evoked by a 4ms injection of 800 pA. For recording the postsynaptic MSN, an internal solution containing 135 mM CsCl, 0.1 mM CaCl2, 2 mM MgCl2, 10 mM HEPES(K), and 0.1 mM EGTA was used. Postsynaptic MSNs were held at −70 mV. GABAA IPSCs were recorded as inward currents (ECl− = ~ 0 mV). Recordings were performed in the presence of SKF 38393 (10 nM).
In the subset of experiments in which muscarinic, GABAergic, and gluatamatergic transmission was assessed, postsynaptic MSNs were recorded with an internal solution containing: 57.5 mM K-methylsulphate, 67.5 KCl, 10 mM NaCl, 50 μM CaCl2, 1.75 mM MgCl2, 10 mM HEPES(K), 5 mM BAPTA-tetrapotassium, 50 μM EGTA, 1 mg/mL ATP, 0.1 mg/mL GTP, and 1.5 mg/mL phosphocreatine, pH 7.4, 275 mOsm (final Cl− concentration = 78.4 mM).
Materials
Picrotoxin, TTX, DNQX, and MK-801 were obtained from Ascent Scientific. ACh, Vesamicol, Tropicamide, Ambenonium, CGP 55845, scopolamine hydrobromide, SKF 38393, and SCH 23390 were from Tocris Bioscience. K-methylsulphate was from Acros Organic and BAPTA was from Invitrogen. All other chemicals were from Sigma-Aldrich.
Statistics and Analysis
Data are shown as mean ± SEM. Statistical significance was determined using Pearson correlation, Wilcoxon match-pairs signed rank test, Mann-Whitney test, Kolmogorov-Smirnov test, or Tukey-Kramer test, where appropriate (InStat 3.0, Graphpad; Axograph Scientific). Statistical differences in EC50 were determined by 95% confidence interval.
The predicted CV for sIPSCs was calculated using the following formula:
where CVsIPSC and CVuIPSC represent the coefficients of variation for sIPSCs and uIPSCs, respectively, n represents the number of CHIs connected, and μuIPSC and μtotalISPC represent the mean amplitudes of uIPSCs and the total population of uIPSCs and sIPSCs, respectively.
Supplementary Material
Highlights.
Striatal cholinergic firing drives spontaneous muscarinic activation in MSNs.
Paired recordings reveal unitary muscarinic events that occur without failure.
Muscarinic receptors reliably encode physiological patterns of cholinergic firing.
Cholinergic inputs onto MSNs are independent and exhibit high release probability.
Acknowledgments
This work was funded by NIH grants R01-NS95809 and R01-DA35821.
We thank Lauren Donovan, Jared Cregg and Katherine Lobur for assistance with immunohistochemistry and genotyping, and Drs. John Williams, Stephanie Gantz and Kevin Bender for helpful comments on the manuscript.
Footnotes
Author Contributions:
AAM and CPF designed and performed the research, analyzed the data, prepared the figures and wrote the manuscript.
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adler A, Katabi S, Finkes I, Prut Y, Bergman H. Different correlation patterns of cholinergic and GABAergic interneurons with striatal projection neurons. Frontiers in systems neuroscience. 2013;7:47. doi: 10.3389/fnsys.2013.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aosaki T, Graybiel AM, Kimura M. Effect of the nigrostriatal dopamine system on acquired neural responses in the striatum of behaving monkeys. Science. 1994;265:412–415. doi: 10.1126/science.8023166. [DOI] [PubMed] [Google Scholar]
- Atallah HE, McCool AD, Howe MW, Graybiel AM. Neurons in the ventral striatum exhibit cell-type-specific representations of outcome during learning. Neuron. 2014;82:1145–1156. doi: 10.1016/j.neuron.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BD, Callaway JC, Wilson CJ. Intrinsic membrane properties underlying spontaneous tonic firing in neostriatal cholinergic interneurons. J Neurosci. 2000;20:8493–8503. doi: 10.1523/JNEUROSCI.20-22-08493.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BD, Wilson CJ. Spontaneous activity of neostriatal cholinergic interneurons in vitro. J Neurosci. 1999;19:5586–5596. doi: 10.1523/JNEUROSCI.19-13-05586.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard V, Normand E, Bloch B. Phenotypical characterization of the rat striatal neurons expressing muscarinic receptor genes. J Neurosci. 1992;12:3591–3600. doi: 10.1523/JNEUROSCI.12-09-03591.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolam JP, Wainer BH, Smith AD. Characterization of cholinergic neurons in the rat neostriatum. A combination of choline acetyltransferase immunocytochemistry, Golgi-impregnation and electron microscopy. Neuroscience. 1984;12:711–718. doi: 10.1016/0306-4522(84)90165-9. [DOI] [PubMed] [Google Scholar]
- Buhl EH, Halasy K, Somogyi P. Diverse sources of hippocampal unitary inhibitory postsynaptic potentials and the number of synaptic release sites. Nature. 1994;368:823–828. doi: 10.1038/368823a0. [DOI] [PubMed] [Google Scholar]
- Cachope R, Mateo Y, Mathur BN, Irving J, Wang HL, Morales M, Lovinger DM, Cheer JF. Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: setting the tone for reward processing. Cell reports. 2012;2:33–41. doi: 10.1016/j.celrep.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Pisani A, Sancesario G, North RA, Bernardi G. Muscarinic IPSPs in rat striatal cholinergic interneurones. J Physiol. 1998;510(Pt 2):421–427. doi: 10.1111/j.1469-7793.1998.421bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuhma N, Mingote S, Moore H, Rayport S. Dopamine neurons control striatal cholinergic neurons via regionally heterogeneous dopamine and glutamate signaling. Neuron. 2014;81:901–912. doi: 10.1016/j.neuron.2013.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD, Silver RA. Unveiling synaptic plasticity: a new graphical and analytical approach. Trends Neurosci. 2000;23:105–113. doi: 10.1016/s0166-2236(99)01520-9. [DOI] [PubMed] [Google Scholar]
- Contant C, Umbriaco D, Garcia S, Watkins KC, Descarries L. Ultrastructural characterization of the acetylcholine innervation in adult rat neostriatum. Neuroscience. 1996;71:937–947. doi: 10.1016/0306-4522(95)00507-2. [DOI] [PubMed] [Google Scholar]
- Courtney NA, Ford CP. The timing of dopamine- and noradrenaline-mediated transmission reflects underlying differences in the extent of spillover and pooling. J Neurosci. 2014;34:7645–7656. doi: 10.1523/JNEUROSCI.0166-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descarries L, Mechawar N. Ultrastructural evidence for diffuse transmission by monoamine and acetylcholine neurons of the central nervous system. Prog Brain Res. 2000;125:27–47. doi: 10.1016/S0079-6123(00)25005-X. [DOI] [PubMed] [Google Scholar]
- Ding J, Guzman JN, Tkatch T, Chen S, Goldberg JA, Ebert PJ, Levitt P, Wilson CJ, Hamm HE, Surmeier DJ. RGS4-dependent attenuation of M4 autoreceptor function in striatal cholinergic interneurons following dopamine depletion. Nat Neurosci. 2006;9:832–842. doi: 10.1038/nn1700. [DOI] [PubMed] [Google Scholar]
- English DF, Ibanez-Sandoval O, Stark E, Tecuapetla F, Buzsaki G, Deisseroth K, Tepper JM, Koos T. GABAergic circuits mediate the reinforcement-related signals of striatal cholinergic interneurons. Nat Neurosci. 2012;15:123–130. doi: 10.1038/nn.2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Phillips PE, Williams JT. The time course of dopamine transmission in the ventral tegmental area. J Neurosci. 2009;29:13344–13352. doi: 10.1523/JNEUROSCI.3546-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annual review of neuroscience. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JA, Ding JB, Surmeier DJ. Muscarinic modulation of striatal function and circuitry. Handb Exp Pharmacol. 2012:223–241. doi: 10.1007/978-3-642-23274-9_10. [DOI] [PubMed] [Google Scholar]
- Goldberg JA, Reynolds JN. Spontaneous firing and evoked pauses in the tonically active cholinergic interneurons of the striatum. Neuroscience. 2011;198:27–43. doi: 10.1016/j.neuroscience.2011.08.067. [DOI] [PubMed] [Google Scholar]
- Hebb CO, Silver A. Gradient of choline acetylase activity. Nature. 1961;189:123–125. doi: 10.1038/189123a0. [DOI] [PubMed] [Google Scholar]
- Hersch SM, Gutekunst CA, Rees HD, Heilman CJ, Levey AI. Distribution of m1–m4 muscarinic receptor proteins in the rat striatum: light and electron microscopic immunocytochemistry using subtype-specific antibodies. J Neurosci. 1994;14:3351–3363. doi: 10.1523/JNEUROSCI.14-05-03351.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley MJ, Gittis AH, Oldenburg IA, Balthasar N, Seal RP, Edwards RH, Lowell BB, Kreitzer AC, Sabatini BL. Cholinergic interneurons mediate fast VGluT3-dependent glutamatergic transmission in the striatum. PloS one. 2011;6:e19155. doi: 10.1371/journal.pone.0019155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley MJ, Soler-Llavina GJ, Sabatini BL. Cholinergic modulation of multivesicular release regulates striatal synaptic potency and integration. Nat Neurosci. 2009;12:1121–1128. doi: 10.1038/nn.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe AR, Surmeier DJ. Muscarinic receptors modulate N-, P-, and L-type Ca2+ currents in rat striatal neurons through parallel pathways. J Neurosci. 1995;15:458–469. doi: 10.1523/JNEUROSCI.15-01-00458.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon J, Dencker D, Wortwein G, Woldbye DP, Cui Y, Davis AA, Levey AI, Schutz G, Sager TN, Mork A, et al. A subpopulation of neuronal M4 muscarinic acetylcholine receptors plays a critical role in modulating dopamine-dependent behaviors. J Neurosci. 2010;30:2396–2405. doi: 10.1523/JNEUROSCI.3843-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y. Physiological, morphological, and histochemical characterization of three classes of interneurons in rat neostriatum. J Neurosci. 1993;13:4908–4923. doi: 10.1523/JNEUROSCI.13-11-04908.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koos T, Tepper JM. Dual cholinergic control of fast-spiking interneurons in the neostriatum. J Neurosci. 2002;22:529–535. doi: 10.1523/JNEUROSCI.22-02-00529.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koos T, Tepper JM, Wilson CJ. Comparison of IPSCs evoked by spiny and fast-spiking neurons in the neostriatum. J Neurosci. 2004;24:7916–7922. doi: 10.1523/JNEUROSCI.2163-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraushaar U, Jonas P. Efficacy and stability of quantal GABA release at a hippocampal interneuron-principal neuron synapse. J Neurosci. 2000;20:5594–5607. doi: 10.1523/JNEUROSCI.20-15-05594.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC. Physiology and pharmacology of striatal neurons. Annual review of neuroscience. 2009;32:127–147. doi: 10.1146/annurev.neuro.051508.135422. [DOI] [PubMed] [Google Scholar]
- Lim SA, Kang UJ, McGehee DS. Striatal cholinergic interneuron regulation and circuit effects. Frontiers in synaptic neuroscience. 2014;6:22. doi: 10.3389/fnsyn.2014.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C, Slesinger PA. Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat Rev Neurosci. 2010;11:301–315. doi: 10.1038/nrn2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macintosh FC. The distribution of acetylcholine in the peripheral and the central nervous system. J Physiol. 1941;99:436–442. doi: 10.1113/jphysiol.1941.sp003913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcott PF, Mamaligas AA, Ford CP. Phasic dopamine release drives rapid activation of striatal D2-receptors. Neuron. 2014;84:164–176. doi: 10.1016/j.neuron.2014.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty V, Kuzmiski JB, Baimoukhametova DV, Bains JS. Short-term plasticity impacts information transfer at glutamate synapses onto parvocellular neuroendocrine cells in the paraventricular nucleus of the hypothalamus. J Physiol. 2011;589:4259–4270. doi: 10.1113/jphysiol.2011.208082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G, Arkadir D, Nevet A, Vaadia E, Bergman H. Coincident but distinct messages of midbrain dopamine and striatal tonically active neurons. Neuron. 2004;43:133–143. doi: 10.1016/j.neuron.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Nelson AB, Bussert TG, Kreitzer AC, Seal RP. Striatal cholinergic neurotransmission requires VGLUT3. J Neurosci. 2014a;34:8772–8777. doi: 10.1523/JNEUROSCI.0901-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson AB, Hammack N, Yang CF, Shah NM, Seal RP, Kreitzer AC. Striatal cholinergic interneurons Drive GABA release from dopamine terminals. Neuron. 2014b;82:63–70. doi: 10.1016/j.neuron.2014.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakhotin P, Bracci E. Cholinergic interneurons control the excitatory input to the striatum. J Neurosci. 2007;27:391–400. doi: 10.1523/JNEUROSCI.3709-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Rosello T, Figueroa A, Salgado H, Vilchis C, Tecuapetla F, Guzman JN, Galarraga E, Bargas J. Cholinergic control of firing pattern and neurotransmission in rat neostriatal projection neurons: role of CaV2.1 and CaV2.2 Ca2+ channels. J Neurophysiol. 2005;93:2507–2519. doi: 10.1152/jn.00853.2004. [DOI] [PubMed] [Google Scholar]
- Pisani A, Bonsi P, Catania MV, Giuffrida R, Morari M, Marti M, Centonze D, Bernardi G, Kingston AE, Calabresi P. Metabotropic glutamate 2 receptors modulate synaptic inputs and calcium signals in striatal cholinergic interneurons. J Neurosci. 2002;22:6176–6185. doi: 10.1523/JNEUROSCI.22-14-06176.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salin P, Lopez IP, Kachidian P, Barroso-Chinea P, Rico AJ, Gomez-Bautista V, Coulon P, Kerkerian-Le Goff L, Lanciego JL. Changes to interneuron-driven striatal microcircuits in a rat model of Parkinson’s disease. Neurobiology of disease. 2009;34:545–552. doi: 10.1016/j.nbd.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Scheuss V, Neher E. Estimating synaptic parameters from mean, variance, and covariance in trains of synaptic responses. Biophysical journal. 2001;81:1970–1989. doi: 10.1016/S0006-3495(01)75848-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JM, Reynolds JN. Pause and rebound: sensory control of cholinergic signaling in the striatum. Trends Neurosci. 2013;36:41–50. doi: 10.1016/j.tins.2012.09.006. [DOI] [PubMed] [Google Scholar]
- Shen W, Plotkin JL, Francardo V, Ko WK, Xie Z, Li Q, Fieblinger T, Wess J, Neubig RR, Lindsley CW, et al. M4 Muscarinic Receptor Signaling Ameliorates Striatal Plasticity Deficits in Models of L-DOPA-Induced Dyskinesia. Neuron. 2015;88:762–773. doi: 10.1016/j.neuron.2015.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Tian X, Day M, Ulrich S, Tkatch T, Nathanson NM, Surmeier DJ. Cholinergic modulation of Kir2 channels selectively elevates dendritic excitability in striatopallidal neurons. Nat Neurosci. 2007;10:1458–1466. doi: 10.1038/nn1972. [DOI] [PubMed] [Google Scholar]
- Shimo Y, Hikosaka O. Role of tonically active neurons in primate caudate in reward-oriented saccadic eye movement. J Neurosci. 2001;21:7804–7814. doi: 10.1523/JNEUROSCI.21-19-07804.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver RA, Momiyama A, Cull-Candy SG. Locus of frequency-dependent depression identified with multiple-probability fluctuation analysis at rat climbing fibre-Purkinje cell synapses. J Physiol. 1998;510(Pt 3):881–902. doi: 10.1111/j.1469-7793.1998.881bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodickson DL, Bean BP. GABAB receptor-activated inwardly rectifying potassium current in dissociated hippocampal CA3 neurons. J Neurosci. 1996;16:6374–6385. doi: 10.1523/JNEUROSCI.16-20-06374.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodickson DL, Bean BP. Neurotransmitter activation of inwardly rectifying potassium current in dissociated hippocampal CA3 neurons: interactions among multiple receptors. J Neurosci. 1998;18:8153–8162. doi: 10.1523/JNEUROSCI.18-20-08153.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub C, Tritsch NX, Hagan NA, Gu C, Sabatini BL. Multiphasic modulation of cholinergic interneurons by nigrostriatal afferents. J Neurosci. 2014;34:8557–8569. doi: 10.1523/JNEUROSCI.0589-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MA, Chen H, Morikawa H. Recurrent inhibitory network among striatal cholinergic interneurons. J Neurosci. 2008;28:8682–8690. doi: 10.1523/JNEUROSCI.2411-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swapna I, Bondy B, Morikawa H. Differential Dopamine Regulation of Ca(2+) Signaling and Its Timing Dependence in the Nucleus Accumbens. Cell reports. 2016;15:563–573. doi: 10.1016/j.celrep.2016.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna S, Ilijic E, Surmeier DJ. Recurrent collateral connections of striatal medium spiny neurons are disrupted in models of Parkinson’s disease. J Neurosci. 2008;28:5504–5512. doi: 10.1523/JNEUROSCI.5493-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecuapetla F, Carrillo-Reid L, Bargas J, Galarraga E. Dopaminergic modulation of short-term synaptic plasticity at striatal inhibitory synapses. Proc Natl Acad Sci U S A. 2007;104:10258–10263. doi: 10.1073/pnas.0703813104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threlfell S, Lalic T, Platt NJ, Jennings KA, Deisseroth K, Cragg SJ. Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron. 2012;75:58–64. doi: 10.1016/j.neuron.2012.04.038. [DOI] [PubMed] [Google Scholar]
- Tritsch NX, Ding JB, Sabatini BL. Dopaminergic neurons inhibit striatal output through non-canonical release of GABA. Nature. 2012;490:262–266. doi: 10.1038/nature11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall NR, De La Parra M, Callaway EM, Kreitzer AC. Differential innervation of direct- and indirect-pathway striatal projection neurons. Neuron. 2013;79:347–360. doi: 10.1016/j.neuron.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CJ. The mechanism of intrinsic amplification of hyperpolarizations and spontaneous bursting in striatal cholinergic interneurons. Neuron. 2005;45:575–585. doi: 10.1016/j.neuron.2004.12.053. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, Chang HT, Kitai ST. Firing patterns and synaptic potentials of identified giant aspiny interneurons in the rat neostriatum. J Neurosci. 1990;10:508–519. doi: 10.1523/JNEUROSCI.10-02-00508.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagishita S, Hayashi-Takagi A, Ellis-Davies GC, Urakubo H, Ishii S, Kasai H. A critical time window for dopamine actions on the structural plasticity of dendritic spines. Science. 2014;345:1616–1620. doi: 10.1126/science.1255514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Ebihara K, Koshikawa N, Kobayashi M. Reciprocal regulation of inhibitory synaptic transmission by nicotinic and muscarinic receptors in rat nucleus accumbens shell. J Physiol. 2013;591:5745–5763. doi: 10.1113/jphysiol.2013.258558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Flores-Hernandez J, Surmeier DJ. Coordinated expression of muscarinic receptor messenger RNAs in striatal medium spiny neurons. Neuroscience. 2001;103:1017–1024. doi: 10.1016/s0306-4522(01)00039-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.