

SUMMARY
Polyphosphate (polyP), a several billion year old biopolymer, is produced in every cell, tissue, and organism studied. Structurally extremely simple, polyP consists of long chains of covalently linked inorganic phosphate groups. We report here the surprising discovery that polyP shows a remarkable efficacy in accelerating amyloid fibril formation. We found that polyP serves as an effective nucleation source for various different amyloid proteins, ranging from bacterial CsgA to human α-Synuclein, Aβ1-40/42 and Tau. PolyP-associated α-Synuclein fibrils show distinct differences in seeding behavior, morphology and fibril stability compared to fibrils formed in the absence of polyP. In vivo, the amyloid-stimulating and fibril-stabilizing effects of polyP have wide-reaching consequences, increasing the rate of biofilm formation in pathogenic bacteria and mitigating amyloid toxicity in differentiated neuroblastoma cells and C. elegans strains that serve as models for human folding diseases. These results suggest that we have discovered a conserved cytoprotective modifier of amyloidogenic processes.
Graphical Abstract
INTRODUCTION
Formation of insoluble protein fibrils plays a crucial role in many different processes, ranging from bacterial biofilm formation to human folding diseases, including Alzheimer’s and Parkinson’s disease (Galvin et al., 2001; Hufnagel et al., 2013; Selkoe, 2001). The amyloidogenic proteins that are involved in these processes have little in common apart from their ability to convert from soluble proteins into insoluble cross β-sheet-rich fibrils (Eichner and Radford, 2011). The rate-limiting step in amyloid fibril formation appears to be the initial nucleation step, when monomeric peptides or proteins adopt a β-sheet-rich, nucleation-prone conformation. Other monomers are then converted into this conformation, eventually culminating in the formation of fibrils, which are deposited in either the intra- or extracellular space. The observed cytotoxic and neurological effects that accompany disease amyloid fibril formation appear to be less due to the final products but due to the accumulation of toxic oligomers that are transiently present prior to the formation of mature amyloid fibrils or generated by the shedding off of mature fibrils (Chen et al., 2015). The toxicity of these oligomers may arise from their ability to increase membrane permeability, affect mitochondrial function, and/or alter the cytoskeleton (Roberts and Brown, 2015). Therapeutic measures targeting amyloid-related protein folding diseases currently under investigation focus primarily on stabilizing non-toxic early intermediates (Hefti et al., 2013). However, acceleration and stabilization of the fibril-forming process should also act to mitigate toxicity by reducing the accumulation of toxic oligomers. This idea is consistent with reports of amyloid fibril depositions in patients with no discernable cerebral degeneration (Ingelsson et al., 2004). Therefore, it appears that amyloids can polymerize in situ without eliciting cellular toxicity.
Polyphosphate (polyP), which consists of up to 1000 phosphoanhydride bond-linked phosphate monomers, has been found in all prokaryotic and eukaryotic organisms studied so far (Kumble and Kornberg, 1995; Rao et al., 2009). Present in the cytosol and most major organelles, polyP has also been shown to be secreted into the extracellular space by platelets, astrocytes and bacteria (Holmstrom et al., 2013; Muller et al., 2009; Sakatani et al., 2016). PolyP is known for its general stress protective functions and has been recognized for specific roles in virulence, biofilm formation, and blood clotting (Morrissey, 2012; Rao et al., 2009). Yet, despite the universal presence and highly conserved nature of polyP, the mechanism(s) by which this simply structured polymer affects these diverse processes has remained largely enigmatic. Recent work from our lab revealed that polyP stabilizes unfolding proteins, thereby protecting bacteria against stress conditions that cause widespread protein unfolding and aggregation (Gray et al., 2014). In vitro studies revealed the surprising result that low micromolar concentrations of polyP-chains effectively convert various thermolabile, mainly α-helical proteins into thermostable β-sheet-rich intermediates (Gray et al., 2014) (Figures S1A, S1B). These results prompted us to test the intriguing idea that polyP might positively affect amyloidogenic processes by accelerating the transition of amyloid proteins to their fibril-forming β-sheet conformation. Here, we show that polyP does indeed serve as an effective amyloid-binding scaffold, which stabilizes amyloidogenic polypeptides in a fibril-competent β-sheet conformation. We show that physiological concentrations of polyP accelerate the transition of both functional and disease-associated amyloid monomers into cross β-sheet insoluble fibrils and effectively prevent shedding off of pre-formed fibrils. PolyP’s influence on fibril formation and stability has wide-reaching consequences, ranging from increasing biofilm formation in pathogenic bacteria to reducing amyloid cytotoxicity in differentiated neuroblastoma cells. These results suggest that we have discovered a widely conserved and cytoprotective modifier of amyloidogenic processes.
RESULTS
PolyP Accelerates Curli-Dependent Biofilm Formation in Uropathogenic E. coli (UPEC)
PolyP has long been known for its stimulatory effects on the formation of bacterial biofilms (Rashid et al., 2000). However, the mechanistic underpinnings of this effect are currently unknown. Bacterial biofilms formed in pathogenic Escherichia coli strains, such as UPEC, depend on the production of the Curli-Specific Gene A (CsgA). CsgA is synthesized in an unfolded conformation and rapidly degraded unless it is able to interact with its specific nucleator protein on the outer bacterial surface. Here CsgA forms protease-resistant, cross β-sheet fibrils termed curli, which display all the characteristics of classical amyloid fibrils (Hufnagel et al., 2013). CsgA deletion strains and other bacteria that are unable to produce curli show a severe delay in biofilm formation, a higher sensitivity toward clearing from the bladder during infection, and decreased in situ survival rates (Hufnagel et al., 2013).
To test the hypothesis that polyP increases biofilm formation by stimulating curli formation, we purified E. coli CsgA and monitored its fibrillation in the absence and presence of polyP by following thioflavin T (ThT) fluorescence. Addition of increasing amounts of short heterogeneous polyP chains (polyPSH, average chain length = 50 Pi) notably accelerated CsgA fibril formation (Figure 1A). Increasing the concentration of polyPSH during fibrillation increasingly shortened the initial nucleation phase and accelerated rates of fibril elongation (Figure 1B). Maximal stimulation of CsgA fibril formation was achieved in the presence of about 0.5 mM polyPSH chains (25 mM Pi), which is well within the physiologically relevant polyP range (up to 50 mM Pi) in E. coli (Kornberg et al., 1999). Any further increase in polyPSH levels did not accelerate kinetics or yield of CsgA fibril formation, indicative of the presence of a polyP-independent rate-determining step. Analysis of the secondary structure of CsgA during the polymerization process confirmed that polyPSH accelerates the conversion of intrinsically disordered CsgA into β-sheet-rich structures (Figures 1C, S1C).
Figure 1. PolyP Promotes Bacterial Amyloid and Biofilm Formation.

(A) ThT fluorescence measurements of CsgA (15 μM) in the absence (black) or presence of 20 μM (red), 0.1 mM (light green), 0.2 mM (cyan), 1 mM (blue), or 2 mM (purple) polyPSH-chains (average length 50 Pi) at 26°C. (B) Effect of polyPSH-chains on half-time (T1/2) (black line) and rate (red line) of CsgA fibril formation. (C) Changes in secondary structure of CsgA during fibril formation in the absence (left) or presence (right) of 1 mM polyPSH-chains. (D) Wrinkled colony phenotype of E. coli UTI89 wild-type, Δppk, and ΔcsgA mutant strains in the absence (left) or presence (right) of 100 μM polyPSH-chains after 28 hr at 26°C. Cells were harvested and endogenous CsgA was visualized by western blot analysis. σ70 was used as a loading control. See also Figure S1.
To investigate whether polyP’s influence on CsgA fibril formation is directly responsible for its effects on bacterial biofilm formation, we used a mutant variant of the UPEC strain UTI89 that lacks the polyP-generating enzyme polyphosphate kinase (Δppk) and thus produces no detectable amounts of polyP. This strain contained substantially fewer CsgA curli fibrils and showed a significant delay in macrocolony biofilm formation as compared to wild-type UTI89 (Figures 1D, S1D). Addition of 0.1 mM polyPSH (5 mM Pi) to the agar plates, however, restored CsgA fibril concentrations to wild-type levels and rescued the delay in macrocolony biofilm formation. We found no such acceleration of biofilm formation in response to polyP supplementation when we tested the UTI89 mutant strain lacking CsgA (ΔcsgA) (Figures 1D, S1D). In conclusion, these results demonstrate that physiological concentrations of polyP reduce the T1/2 of CsgA fibril formation in vitro and stimulate CsgA fibril formation in vivo, suggesting that polyP’s influence on pathogenic biofilms is at least in part due to its ability to enhance bacterial amyloid fibril formation.
PolyP Accelerates Disease-Related Amyloid Formation In Vitro
PolyP is found in many different mammalian tissues, including the brain, with concentrations between 25 and 120 μM in Pi units and an average polyP chain length of about 100 Pi units (Kumble and Kornberg, 1995). To determine whether polyP exerts similar effects on amyloid proteins associated with neurodegenerative diseases, we tested its influence on the fibrillation of α-Synuclein, which is involved in Parkinson’s disease, as well as the two amyloid proteins most commonly associated with Alzheimer’s disease, i.e. Aβ1-40/42 and Tau. For all three disease-associated amyloids, we found that presence of physiological concentrations of polyP chains (1–10 μM chains) substantially decreased the lag phase and increased the rate of nucleation (Figures 2A–I, S2A–G), suggesting that polyP is more generally involved in modifying amyloidogenic processes.
Figure 2. PolyP Accelerates Disease-Associated Amyloid Fibril Formation.

(A–I) ThT fluorescence measurements of (A) 100 μM α-Synuclein incubated in the absence (black) or presence of 1 μM (red), 5 μM (cyan) or 10 μM (light green) polyPSH-chains; (B) 5 μM α-Synuclein in the absence (black) or presence of 4 μM polyPSH-chains (red); (C) 100 μM α-Synuclein incubated in the absence (left) or presence of 20 μM polyPSH (right), supplemented with two glass beads (cyan), continuously mixed (red), or supplemented with two glass beads and continuously mixed (black); (D) 5 μM α-Synuclein incubated in the absence (black) or presence of various polyanions at 5 μM total negative charge: polyP (0.1 μM polyPSH-chains) dark red; Heparin (0.09 μM; MW 17,000) dark blue; or at 100 μM total negative charge: polyP (2 μM polyPSH-chains) red; Heparin (1.8 μM; MW 17,000) blue; DNA (33 ng/μl) green; RNA (34 ng/μl) cyan; (E) Half-time (T1/2) of fibril formation of 100 μM α-Synuclein in the presence of the indicated concentrations of various defined-length of polyPs (see also Table S1). n = 8 independent experiments. ThT fluorescence measurements of (F) 75 μM α-Synuclein without (dark blue) or with either 5 μM heparin (black) or 20 μM polyPSH (dark red) in buffer, or without (blue) or with either 5 μM heparin (grey) or 20 μM polyPSH (red) in buffer supplemented with cleared brain homogenate (1 mg/ml in terms of protein concentration); (G) 8 μM Aβ1-42 without (black) or with 1 μM (red), 5 μM (cyan), or 10 μM (green) polyP60mer. (H) Half-time of fibril formation of 8 μM Aβ1-42 in the presence of various defined-length of polyPs (see also Table S1). n = 3 independent experiments; (I) ThT fluorescence measurements of 125 μM hTau40 without (black) or with 25 μM polyPSH-chains (red). See also Figure S2.
In vitro fibril formation of α-Synuclein is an intrinsically slow process, which typically requires a combination of non-physiologically high amyloid concentrations (i.e., 70–100 μM), artificial nucleation sources such as glass beads, and constant agitation of the samples (Giehm et al., 2011). Yet, even under such optimized conditions, fibril formation of α-Synuclein proceeds only very slowly with a T1/2 of more than 20 h (Figure 2A). In the presence of micromolar levels of polyPSH chains, however, the T1/2 of α-Synuclein fibril formation was reduced to less than 5 hours (Figure 2A), fibrils formed even when 20-fold lower, physiologically relevant concentrations of α-Synuclein (i.e. 5 μM) (Iwai et al., 1995) were used (Figure 2B), and fibrillation occurred even without nucleation sources (Figure 2C). In addition, we observed that with increasing polyPSH concentrations present in the reaction, the final ThT signals of α-Synuclein significantly increased (Figures 2A–C), suggesting that presence of polyP might also increase fibril yields. Indeed, spin down experiments after 24h of fibril formation in the absence or presence of increasing concentrations of polyPSH revealed a clear correlation between the measured ThT fluorescence and the amount of α-Synuclein fibrils present (Figure S2A). From these results we concluded that polyP not only stimulates the rate of fibril formation but also increases the number of amyloid monomers transitioning into cross β-sheet fibrils, suggesting that presence of polyP chains reduces the amyloid concentration critical for nucleation.
Several other polyanions, including sulfated glucosaminoglycans such as heparin or polynucleotides such as DNA and RNA have been previously shown to influence α-Synuclein fibril formation (Cohlberg et al., 2002; Munishkina et al., 2009). To compare their efficacies, we tested the influence of all four compounds on the fibril formation of 5 μM α-Synuclein. In the absence of any other common denominator, we used our polyanions at concentrations that yield the same absolute amount of negative charges (i.e., 5 or 100 μM), calculating one charge per Pi or nucleotide unit for polyP or DNA/RNA, respectively, or two charges per glucosaminoglycan unit (MW 600) for heparin. Compared to polyP, which effectively stimulated α-Synuclein fibrillation at a charge concentration of 5 μM (equiv. to 0.1 μM polyPSH-chain), heparin only minimally stimulated fibril formation even when present at 100 μM charge concentration (equiv. to 1.8 μM heparin) (Figures 2D, S2C). Neither DNA nor RNA affected α-Synuclein fibrillation at all. When 100 μM α-Synuclein was used instead, heparin showed the previously observed stimulation of fibril formation while DNA or RNA still failed to exert any effects (Figures S2D, E). This result was consistent with some other reports, which also failed to show any fibril stimulation by DNA or RNA (Hegde and Rao, 2007). Analysis of the influence of polyP chain length on α-Synuclein fibril formation further supported our conclusion that simple presence of polyanions is not sufficient to explain the observed effects of polyP. In contrast to the presence of 1 μM long polyP130 chains (i.e., 130 μM Pi), which reduced the T1/2 of α-Synuclein (100 μM) from over 20 h to less than 2.5 h, 10 μM short polyP16-chains (i.e., 160 μM Pi) only reduced the T1/2 to 5.5 h, even though the absolute Pi-concentration was very similar (Figure 2E, Table S1). These results suggest that on a Pi basis, longer chains are more effective than shorter ones and work even when present at substoichiometric amounts. Finally, we tested whether polyP was able to maintain its stimulatory effect even within the complex environment of cell lysates, where potentially many competing compounds differentially affect amyloid fibrillation. We incubated α-Synuclein with cleared brain homogenate from a healthy individual, and monitored fibril formation by ThT fluorescence in the absence of any additives, or in the presence of either polyPSH or heparin. Whereas addition of brain homogenate completely abrogated the stimulatory effect of heparin on α-Synuclein fibril formation, polyPSH still accelerated fibril formation (Figure 2F). We conclude from these results that the polyP-mediated stimulation of α-Synuclein fibril formation is not simply a non-specific charged-based effect but represents a specific function of polyPSH chains, maintained even within the complex environment of a cell lysate.
A recent report demonstrated that astrocytes secrete polyP into the extracellular space (Holmstrom et al., 2013), prompting us to investigate the effects of polyP on the fibril formation of Aβ1-42 and Aβ1-40, whose extracellular deposition as insoluble amyloid fibrils serves as hallmark of Alzheimer’s disease. Indeed, as seen with CsgA and α-Synuclein, presence of polyP accelerated the initial nucleation and fibril elongation phase of both peptides (Figures 2G, S2F), and longer polyP-chains were slightly more effective than shorter ones (Figure 2H, S2G, Table S1). Lastly, we tested human Tau (hTau40), a protein that does not form fibrils for many months even at very high protein concentrations (Goedert et al., 1996). Yet, presence of polyPSH also promoted its fibril formation, reducing the T1/2 to less than 2 days (Figure 2I). Taken together, these results demonstrate that polyP effectively promotes the transition of a variety of different soluble amyloid monomers into insoluble β-sheet-rich fibrils.
PolyP Binding Affects Seeding Capacity and Fibril Morphology of α-Synuclein
To characterize the interaction between polyP chains and amyloids in more detail, we first tested whether amyloid fibrils remain associated with polyP. We therefore allowed 100 μM α-Synuclein fibrils to form in the presence of various amounts of polyPSH, and subsequently measured ThT fluorescence and polyP levels in the soluble supernatant and fibril-containing pellet fractions. We found that most of the otherwise soluble polyP partitioned into the fibril-containing pellet, suggesting that polyP forms apparently stable complexes with amyloid fibrils (Figure 3A).
Figure 3. Influence of PolyP on Seeding Efficiency of Fibrils and Fibril Morphology.

(A) ThT fluorescence (left panel) and polyP content (right panel) in pellet and supernatant after 24h fibril formation of 100 μM α-Synuclein in the presence of polyPSH. In the absence of fibrils, polyP remains in the supernatant (n=4). (B) ThT fluorescence of .50 μM α-Synuclein incubated without (left) or with 20 μM polyPSH (right) in the absence of any seeds (black) or in the presence of 10% w/v α-Synuclein fibrils (red) or α-SynucleinpolyP fibrils (cyan). ThT fluorescence of 10% w/v α-Synuclein fibrils (yellow) or α-SynucleinpolyP fibrils (blue). (C) TEM images of α-Synuclein fibrils formed in the absence or presence of polyPSH. (D) Bar graph of the widths of α-Synuclein fibrils (black) and α-SynucleinpolyP fibrils (grey) grouped into width of 10–15 and greater than 15 nm based on measurements of the 2D class averages (see Figure S3 for details).
To determine whether the fibrils that were formed in the presence of polyP exhibit any differences in reactivity toward non-polyP containing amyloidogenic proteins and vice versa, we conducted cross-seeding experiments using pre-formed α-Synuclein and α-SynucleinpolyP fibrils. We found that pre-formed seeds of α-Synuclein stimulate α-Synuclein fibrillation both in the absence and presence of polyP (Figure 3B). Surprisingly, however, α-SynucleinpolyP seeds were less effective in stimulating α-Synuclein fibrillation in the absence of polyP, and showed lower capacity to seed α-Synuclein fibrillation even in the presence of polyP (Figure 3B). Spin down experiments confirmed these results (Figure S3A). These results suggest that α-Synuclein and α-SynucleinpolyP fibrils might represent distinct amyloid species with α-SynucleinpolyP fibrils being a less seeding-effective species.
To directly compare fibril morphology, we analyzed the structure of α-Synuclein and α-SynucleinpolyP fibrils by transmission electron microscopy (TEM). In contrast to α-Synuclein fibrils, α-SynucleinpolyP fibrils are mostly present as single-stranded filaments, less broken up and less often twisted (Figures 3C, S3B). These results suggested that presence of polyP might stabilize the single fibril strand. 2D classification of 838 and 1432 α-Synuclein and α-SynucleinpolyP particles, respectively, confirmed these qualitative conclusions. By comparison of the fibril widths in the class averages, we determined that over 80% of analyzed α-Synuclein fibrils had a width larger than 15 nm whereas the majority (~80%) of α-SynucleinpolyP fibrils had a width of only 10 to 15 nm (Figures 3D, S3B). While we cannot conclude from these results that polyP binding alters the core cross-beta sheet structure of the α-Synuclein fibrils, our data indicate that polyP binding stabilizes a compact single-stranded filament, which appears morphologically different from the majority of α-Synuclein fibrils formed in absence of polyP. Proteinase K digests agreed with these suggested structural differences by revealing that α-SynucleinpolyP fibrils are more susceptible to proteolytic digest than fibrils formed in the absence of polyP (Figure S3C). We conclude from these results that polyP binding to α-Synuclein alters fibril morphology, which likely contributes to strain-specific seeding behavior and altered proteolytic stability.
PolyP-Associated α-Synuclein Fibrils are Stabilized Towards Molecular Shedding
Recent studies suggested that amyloid fibrils, while themselves non-toxic, might serve as reservoirs for toxic oligomers that shed off of the mature fibrils over time (Cremades et al., 2012; Tipping et al., 2015). To directly test whether polyP has any effect on the stability of mature fibrils, we prepared α-Synuclein or SynucleinpolyP fibrils for 24h, separated the fibrils from any non-polymerized material by centrifugation and extensive washing, and incubated the fibrils in the absence or presence of polyP (Figure 4A). At defined time points, we took samples and analyzed the solubility of the proteins on SDS-PAGE. We found that within 6 h of incubation, almost 50% of previously insoluble α-Synuclein fibrils had disassembled into soluble oligomeric species, and by 48h of incubation, almost 90% of α-Synuclein soluble (Figure 4A). In contrast, α-SynucleinpolyP fibrils were substantially more stable and disassembled to less than 60% over the same 48h time frame. Presence of additional polyP during the disassembly further stabilized α-SynucleinpolyP and even slowed disassembly of α-Synuclein fibrils. These results suggest that polyP either directly associates with preformed α-Synuclein fibrils, or supports re-association of α-Synuclein oligomers into more stable fibrils. TEM of the samples revealed the appearance of smaller globular structures during the disassembly process that were particularly abundant in the images taken from disassembled α-Synuclein fibrils in the absence of polyPSH (Figures 4B, S4). These results show that polyP binding not only shortens the nucleation time and accelerates fibril elongation, but also alters α-Synuclein fibril morphology and increases apparent fibril stability.
Figure 4. PolyP Delays Shedding of Mature α-Synuclein Fibrils.

Preformed α-Synuclein or α-SynucleinpolyP fibrils were incubated in the absence or presence of 20 μM polyPSH at 4°C. At the indicated time points, samples were taken and either (A) separated into soluble and insoluble fraction by centrifugation and visualized by SDS-PAGE or (B) analyzed by TEM. Representative images are shown. See also Figure S4.
PolyP Treatment Diminishes α-Synuclein Cytotoxicity
To directly test whether polyP affects amyloid toxicity, we incubated differentiated neuroblastoma SH-SY5Y cells with preformed α-Synuclein and α-SynucleinpolyP fibrils and conducted cytotoxicity measurements after 40h of incubation. Whereas incubation with α-Synuclein fibrils exerted the expected cytotoxic effect, incubation of cells with α-SynucleinpolyP fibrils caused no significant change in live/dead cell counts compared to untreated cells (Figure 5A). Moreover, α-Synuclein fibrils that were defibrillated for 24 h showed very similar cytotoxic effects on differentiated neuroblastoma cells while no amount of cell killing above background was noted when we treated the cells with α-SynucleinpolyP-fibrils that were treated with polyP for 24h (Figure 5A). We obtained similar results when we tested the effects of increasing amounts of preformed α-Synuclein and α-SynucleinpolyP fibrils on rat adrenal gland cells (PC-12). Although PC12 cells are not very susceptible to amyloid-mediated cell killing, we still found that α-SynucleinpolyP fibrils were reproducibly less toxic than α-Synuclein fibrils formed in the absence of polyP (Figures S5A).
Figure 5. Effects of PolyP on Amyloid Toxicity.

(A) 10 μM α-Synuclein and α-SynucleinpolyP fibrils either freshly prepared or incubated without or with 20 μM polyPSH for 24 h at 4°C were added to differentiated human neuroblastoma cells (SH-SY5Y) and toxicity was determined after 40 h using Trypan blue exclusion and cell count (n = 4–5, ANOVA; *p<0.05, **p<0.005,*** p<0.0005). (B) Spent media of either un-transfected (SM) HeLa cell or HeLa cells transfected with a plasmid carrying α-Synuclein that were cultivated for 48h with or without 20 μM polyPSH, was added to differentiated SH-SY5Y cells. After 40 h of incubation, toxicity was determined as before (n = 4–6; ANOVA; **p<0.005, *** p<0.0005). (C) DIC images of differentiated SH-SY5Y cells upon incubation with spent media of untransfected or cells transfected with either α-Synuclein after 48 h of incubation. (D) Same as under (B) except that spent media of CHO-cells stably transfected with the APP gene was used. (E) Same as under (C) except that spent media of CHO-cells stably transfected with the APP gene was used. (F) Uptake of fluorescently-labeled polyP (polyP-AF647) by C. elegans was visualized by microscopy. C. elegans autofluorescence was used to demonstrate polyP-AF647 specificity. (G) C. elegans GMC101 expressing Aβ1-42 were treated with polyPSH at 25°C for 5.5 hr. Then, worms were transferred onto NGM plates and incubated at 25°C for 24 h before movement was assessed (n = 4 ; two-tailed paired Student’s t-test; *p<0.05). (H) Endogenous polyP levels in the brains of transgenic hAPP(J20) mice modeling Alzheimer’s disease and non-transgenic control mice (n = 13 per group, unpaired two-tailed Student’s t-test; **p<0.005). See also Figure S5.
To further investigate the cytoprotective effects of polyP, we cultivated transiently transfected HeLa cells expressing and secreting α-Synuclein in the absence or presence of 20 μM polyPSH-chains for 48h. Presence of polyP did not affect the amount of secreted amyloids (Figure S5B). We then supplemented the cultivation media of differentiated SH-SY5Y cells with the spent media, and analyzed cell killing after 40 h of incubation. Like in published work (Emmanouilidou et al., 2010), we found that secreted α-Synuclein killed ~20% of cells. Presence of polyP, however, diminished this toxicity and incubation with α-Synuclein caused no more cell death than incubation of the differentiated SH-SY5Y cells with spent media of untransfected cells (Figure 5B). Finally, we studied the morphology of the differentiated neuroblastoma cells after treatment with freshly secreted α-Synuclein. As observed before, neurons undergo morphological changes in response to amyloids, including axon retraction (Emmanouilidou et al., 2010) (Figure 5C). In contrast, when we tested the effects of α-Synuclein that was secreted into media containing polyP, we discovered that even after 40 h of incubation, the cells looked healthy and unaltered. These results fully agreed with our cytotoxicity assays and suggest that polyP protects cells against amyloid-mediated toxicity.
PolyP Reduces Aβ1-40/42 Cytotoxicity and Delays Paralysis in Aβ1-42 - Expressing Caenorhabditis elegans
Our in vitro fibrillation studies revealed a major stimulatory effect of polyP on α-Synuclein nucleation rates and fibril yields. In contrast, the effects of polyP on Aβ1-40/42 fibrillation were more moderate, and presence of polyP did not substantially alter fibril yield. To determine whether polyP exerts any protective effect against Aβ1-40/42 toxicity in cell culture, we cultured stably transfected CHO cells that express and secrete Aβ1-40/42 in the absence or presence of 20 μM polyPSH-chains for 48h, and tested the spent media on the survival of differentiated SH-SY5Y cells. Consistent with our α-Synuclein results, we found that when polyP was present, secreted Aβ1-40/42 was substantially less toxic (Figure 5D) and no longer caused any major morphological changes on the differentiated SH-SY5Y cells (Figure 5E). These results suggest that polyP protects cells also against the cytotoxic effects of the Aβ1-40/42-peptide.
To determine whether the protective effect of polyP plays any role in amyloid diseases, we aimed to find a disease model that would allow us to the directly assess the in vivo effects of polyP. However, no polyP-synthesizing enzyme(s) has been identified in any multicellular eukaryotic organism so far, making it challenging to genetically alter polyP levels. However, we considered the possibility that organisms like C. elegans, for which established amyloid disease models exist, can take up polyanions (i.e. small inhibitory RNAs) directly through feeding. Indeed, when we added fluorescently labeled polyP to the food source (i.e., dead bacteria) of C. elegans, we saw significant amounts of polyP accumulating in the intestine and surrounding tissue within hours of feeding (Figure 5F). Importantly, transient (12 h) treatment of wild-type C. elegans with 1 mM polyPSH-chains (50 mM Pi) had no adverse effect on lifespan. Moreover, transient treatment with 5 mM polyPSH-chains did not induce the unfolded protein response or the heat shock response (Figures S5C, D). This discovery prompted us to test whether polyP exposure had any effect on the C. elegans Alzheimer’s disease model strain GMC101, which accumulates Aβ1-42 fibrils in the muscle cells, causing toxicity as determined by paralysis in early adulthood (McColl et al., 2012). We therefore exposed a synchronized population of GMC101 L4 larvae to various concentrations of polyPSH-chains for 5.5 h, washed the worms, placed them onto NGM plates containing OP50 and assessed their movement 24h later. As shown in Figure 5G, about 45% of GMC101 worms showed significant movement defects when left untreated, while less than 30% of worms pretreated with polyP were paralyzed. These results suggest that polyP protects cells against the toxic effects of amyloids also within the context of eukaryotic cells.
PolyP Levels Are Decreased in Mouse Models of Alzheimer’s Disease
Previous studies reported that polyP levels in the brain of rats decrease with age (Lorenz et al., 1997). Based on our findings, we reasoned that such a decline might contribute to the known predisposition of the elderly to neurodegenerative amyloid diseases. To determine whether deposition of amyloid plaques affects brain polyP levels even further, we directly compared polyP levels in the brain of mice modeling Alzheimer’s disease (i.e., hAPP(J20) mice) with those in age-matched non-transgenic littermates. J20 mice express a human amyloid precursor protein (hAPP) transgene hemizygously carrying the familial Alzheimer’s disease KM670/671NL (Swedish) and V717F (Indiana) mutations (Hsia et al., 1999; Mucke et al., 2000). These mice accumulate Aβ fibrils, display Aβ pathology and develop robust synaptic and cognitive deficits prior to the initiation of plaque deposition. We sacrificed plaque-depositing 15 month-old J20 mice and their non-transgenic littermates, homogenized their brains and tested for polyP levels. We found significantly lower polyP levels in the brains of plaque-depositing mice than in the brains of non-transgenic littermates (Figure 5H). These results suggest that Aβ fibril formation may either lead to a global reduction in brain polyP levels, or might sequester polyP so tightly into amyloid plaques that they are no longer accessible to the enzymes that we use to degrade and measure polyP. In either case, these results point towards the possibility that the observed decrease in readily available levels of polyP in plaque depositing mouse brains might potentially enhance the cytotoxicity of newly formed soluble Aβ oligomers, and contribute to the progressive nature of this disease.
DISCUSSION
PolyP: A β-Sheet Stabilizing Scaffold?
PolyP’s structure is extremely simple. This immediately raises the question as to how polyP can influence the diverse set of seemingly unrelated processes that it does, which range from increasing stress resistance and pathogenesis in bacteria, to supporting bone mineralization, affecting blood clotting and regulating mTOR signaling in mammals (Morrissey, 2012; Rao et al., 2009; Wang et al., 2003). We previously found that polyP stabilizes unfolding proteins in a β-sheet-rich conformation (Gray et al., 2014). These results, together with those presented here showing that polyP accelerates amyloid formation by promoting the conversion of amyloidogenic proteins and peptides into β-sheet rich fibrils, suggest that we have identified a unifying feature of polyP’s mechanism - serving as a β-sheet-stabilizing scaffold.
PolyP carries a net charge of −1 per Pi unit at physiological pH, making it one of the most densely negatively charged molecules known. It is therefore most likely that polyP interacts with proteins via electrostatic interactions. For instance, it is conceivable that polyP binding to positively charged regions of amyloid monomers stabilizes appropriate amyloidogenic conformations that are involved in fibril formation (Cohlberg et al., 2002). It is also possible that polyP binding might interfere with electrostatic interactions that otherwise stabilize amyloid monomers, making them more likely to undergo conformational changes. Alternatively, however, polyP might bind “in register” with backbone amides, thereby contributing to the stabilization of amyloidogenic proteins in fibril-competent β-sheet structures. This mechanism would also explain how polyP binding triggers conversion of amyloids like CsgA, which harbor very few positively charged residues in their amyloidogenic regions or stabilizes completely unrelated, purely α-helical enzymes, such as luciferase and citrate synthase, in a β-sheet conformation upon thermal unfolding (Gray et al., 2014). It is also tempting to speculate that polyP’s known role in other processes such as fibrin fibril formation during blood clotting is similarly based on its ability to stabilize proteins such as fibrin in a β-sheet-like conformation (Litvinov et al., 2012). Unfortunately, investigating the precise nature of polyP-polypeptide interactions will remain challenging, given that polyP lacks chemical features that can be employed by common spectroscopic techniques, and many of the polyP binding partners that we have now identified are either metastable folding intermediates or amyloid-forming fibrils.
PolyP Forms a Nucleator for Amyloidogenic Processes
Our studies revealed that substoichiometric amounts of medium-length polyP chains stimulate amyloid nucleation and fibril elongation. Together with the observation that polyP remains associated with mature fibrils, these results strongly argue that individual polyP chains associate with several amyloid monomers. In fact, simply calculating the phosphate to amyloid ratio under conditions of maximal fibril stimulation indicates that one amyloid monomer can bind to every 10–20 Pi units. Binding of several amyloid monomers to any one chain would effectively increase the local amyloid concentration, making polyP an effective nucleator of amyloidogenic processes. These results are consistent with the observation that very short polyP chains (i.e., polyP16,) are less effective in promoting fibril formation in vitro, and could explain why longer polyP chains replace nucleation sources and promote fibril formation at physiologically low amyloid concentrations. We did notice, however, that the degree to which polyP stimulated kinetics of fibril formation and final fibril yield varied with the client proteins, and seemed to correlate with the ability of client proteins to fibrillate on their own. The most pronounced stimulating effects of polyP were observed with α-Synuclein and hTau40, both of which form fibrils on their own at a very slow time scale (days to months). In contrast, CsgA and Aβ1-40/42 fibrils form quite rapidly in vitro, and their rate of fibril formation was only moderately stimulated by the additional presence of polyP. Since the rate-limiting step in fibril formation is the conversion of a largely unfolded/α-helical amyloid into a nucleation-competent β-sheet conformation, these results further support our conclusion that the major mechanism by which polyP accelerates fibril formation is by increasing the rate at which nucleation seeds form.
PolyP Mitigates Amyloid Cytotoxicity
The toxicity associated with amyloid fibrils is thought to arise primarily from toxic oligomeric and pre-fibrillar intermediates, which form either on- or off-pathway to fibril formation (Chen et al., 2015; Cremades et al., 2012) (Figure 6). Evidence suggests that mature fibrils are toxic as well, either by providing surfaces that catalyze secondary nucleation processes or by serving as sources of toxic oligomers (Tipping et al., 2015). In a process termed ‘shedding’, mature fibrils were found to disassemble, thereby increasing the number of free oligomers and fibril ends, and potentially nucleating further fibril formation (Desplats et al., 2009; Tipping et al., 2015). Our studies on α-Synuclein revealed that the presence of polyP affects all of these processes (Figure 6). Firstly, polyP accelerates nucleation and fibril formation. This acceleration is predicted to reduce the amount of toxic oligomers that accumulate during fibril formation. Secondly, α-Synuclein fibrils formed in the presence of polyP appear to be morphologically distinct, and less able to seed non-polyP amyloids, suggesting that they might also be less likely to catalyze secondary nucleation processes. Thirdly, polyP causes an apparent stabilization of the mature α-Synuclein fibrils, protecting them against disassembly over at least 48 h, even when fibrils were originally formed in the absence of polyP. This effect might be particularly relevant for cytotoxicity assays, in which either pre-formed or freshly secreted amyloid fibrils are added to cell cultures, and survival is tested 24 – 48 h later. Finally, we found polyP-associated α-Synuclein fibrils to be more proteinase K sensitive than fibrils formed in the absence of polyP. These effects individually or in concert likely contribute to the observed cytoprotective effect of polyP treatment, which becomes particularly obvious in differentiated neuroblastoma cells. At this point, we cannot exclude the possibility that some of the observed effects are indirectly caused by polyP-mediated changes in gene expression, proteostasis, or stress resistance although our C. elegans experiments argued against a polyP-mediated induction of the UPR or heat shock response. Further studies are needed to address these potential additional aspects of polyP action. In any case, our studies clearly indicate that increased levels of polyP have beneficial effects and protect neuronal cells. This is intriguing given that polyP levels in the brain decrease with age (Lorenz et al., 1997). This decrease in polyP levels might predispose older individuals to become more vulnerable to the cytotoxic effects of amyloids. Further studies of polyP and its mechanism of action may lead to the development of novel therapeutic intervention schemes to treat these devastating amyloid-linked diseases, such as Alzheimer’s and Parkinson’s disease.
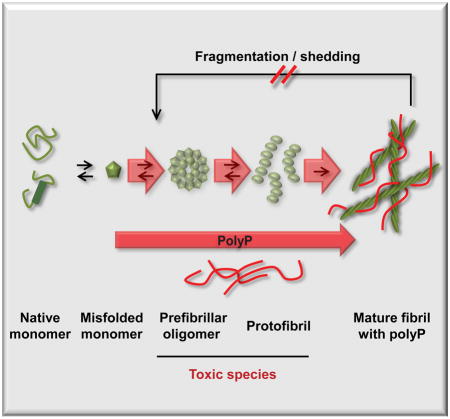
Figure 6. PolyP accelerates fibril formation and increases fibril stability.

Alpha-helical or natively unfolded amyloid monomers undergo structural rearrangements, leading to the formation of β-sheet rich oligomers and protofibrils, which are considered to be the primary cytotoxic species. Further assembly leads to the formation of cross-β-sheet fibrils, which are thought to serve as reservoirs for toxic intermediates as they disassemble back into oligomeric species in a process termed shedding. PolyP dramatically accelerates initial fibril formation, and increases fibril stability, thereby reducing the formation of toxic oligomeric species. This mechanism likely explains how presence of polyP mitigates amyloid cytotoxicity.
EXPERIMENTAL PROCEDURES
Proteins, PolyP Preparations and PolyP Labeling
Aβ1–42 was purchased from AnaSpec Inc. All other proteins were purified as described in the Supplemental Experimental Procedures. Defined length polyP chains were kindly provided by Dr. Toshikazu Shiba (Regenetiss, Japan). Short heterogeneous polyP (polyPSH; average length ~50 Pi) were purchased from Acros. For labeling proced.re, please see Supplements.
Thioflavin T Fluorescence and Circular Dichroism
10 μM thioflavin T (ThT; Sigma) was mixed with the appropriate concentrations of amyloidogenic proteins. The secondary structure of proteins was monitored using far-UV circular dichroism spectroscopy. For protein concentration, buffer conditions and instrument settings, please see Supplemental Material and Method.
Macrocolony Phenotype
UTI89 ΔppK was constructed using the lambda Red recombinase method as described (Datsenko and Wanner, 2000). To monitor macrocolony biofilms, overnight cultures of E. coli UTI89 or its mutant variants were diluted to an OD600 of ~1 in YESCA broth. 4 μl of the cultures were spotted onto YESCA agar plates (1 g/l of yeast extract, 10 g/l of casamino acids, 20 g/l of agar) containing either no or 0.1 mM polyPSH. The plates were incubated at 26°C and biofilm development was followed over time. Pictures were taken with an Olympus DP72 camera mounted on an Olympus SZX16 research stereomicroscope using bright-field microscopy. Detection of in vivo curli formation is described in the Supplemental Experimental Procedures.
Seeding Experiments, Fibril Stability Measurements, and Negative Stain of Fibrils
Fibrils of α-Synuclein (200 μM) alone or in complex with 40 μM polyPSH were prepared as described in Supplemental Material and Methods. Then, 5 μM α-Synuclein or α-SynucleinpolyP fibrils were added to 50 μM monomeric α-Synuclein in the absence or presence of 20 μM polyPSH. ThT fluorescence was monitored over time. For the TEM analysis, α-Synuclein (300 μM) alone or with 50 μM polyPSH were prepared as described. After polymerization, fibrils were applied to thin carbon-layered 400 mesh copper grids (Pelco) and stained with 0.75% (w/v) uranyl formate (pH 5.5–6.0) (details for TEM analysis can be found in Supplemental Procedures). To determine fibril stability, 50 μM α-Synuclein or α-SynucleinpolyP fibrils were incubated at 4°C in 40 mM KPi, pH 7.5, 50 mM KCl and supplemented with or without 20 μM polyPSH. At defined time points sample were taken and either directly analyzed by TEM, or separated into fibrils and soluble proteins by centrifugation (20,000 × g, 30min, 25°C). Fractions were analyzed on SDS-Page and proteins were visualized by Coomassie blue staining. Band intensity was determined with a LI-COR biosciences system.
Effects of polyP on C. elegans
PolyP uptake and the effects of polyP on GMC101 transgenic worms expressing Aβ1-42 were conducted as described in Supplemental Experimental Procedures.
Amyloid Cytotoxicity Assays
Amyloid toxicity assays were conducted with differentiated human neuroblastoma cells SH-SY5Y cells (ATCC CRL-2266). For culturing conditions and details on the cytoxicity assays, please see Supplemental Experimental Procedures.
PolyP Determination in Brain Tissues
Please see Supplemental Experimental Procedures for detailed explanations on the hemizygous transgenic PDAPP [hAPP(J20)] used in this study. Animals 14.5–16 months of age were euthanized by isoflurane overdose followed by cervical dislocation. Hemibrains were dissected and flash frozen and homogenized in liquid N2. All animal experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Texas Health Science Center at San Antonio (Animal Welfare Assurance Number: A3345-01). The level of polyP in brain homogenates was determined using a modified method originally developed by Kumble and Kornberg (Kumble and Kornberg, 1995). For details see Supplemental Experimental Procedures.
Statistical Analyses
Statistical analyses were performed using GraphPad Prism 5 software. Unless otherwise noted, data are presented as mean ± SEM, with significance calculations determined by a paired or unpaired two-tailed Student’s t test or one-sided ANOVA. A value of p > 0.05 was deemed not statistically significant (n.s.); *p < 0.05, **p < 0.005, ***p < 0.0005.
Supplementary Material
Acknowledgments
We thank M. Ivanova for expression plasmids, and purification protocols and J. Bardwell and A. Syed for critically reading the manuscript. We thank N. Wagner, M. Gray, and W. DePas for their experimental help. Defined length polyP chains were kindly provided by T. Shiba (Regenetiss, Japan). CHO cells expressing APP were kindly provided by Y. Wang (University of Michigan), and the α-Synuclein expressing plasmid pCDNA3.1 was a kind gift of T. Outerio, University of Göttingen. We thank ATCC for PC-12 and SH-SY5Y cells. C. elegans strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). This work was funded by the NIH grants AG046799 and GM065318 to U.J., AI073847 to M.R.C., and I01 BX002211-01A2 Veterans Administration Research and Development Merit Award, NIA 2 P30 AG013319-21, the Robert L. Bailey and Daughter Lisa K. Bailey Alzheimer’s Fund, and the William & Ella Owens Medical Research Foundation to V.G. J.-U.D. is funded by a postdoctoral fellowship from the German Research foundation.
Footnotes
AUTHOR CONTRIBUTIONS - C.M.C. conceived and conducted experiments, analyzed data and wrote the manuscript. D.K., N.M., L.X., J.-U.D., J.L., and S.G. conducted experiments and analyzed data. M.R.C. and D.S. conceived experiments and analyzed data. V.G. provided essential material. U.J. conceived experiments, analyzed data and wrote the manuscript.
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Chen SW, Drakulic S, Deas E, Ouberai M, Aprile FA, Arranz R, Ness S, Roodveldt C, Guilliams T, De-Genst EJ, et al. Structural characterization of toxic oligomers that are kinetically trapped during α-synuclein fibril formation. Proc Natl Acad Sci U S A. 2015;112:E1994–E2003. doi: 10.1073/pnas.1421204112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohlberg JA, Li J, Uversky VN, Fink AL. Heparin and other glycosaminoglycans stimulate the formation of amyloid fibrils from alpha-synuclein in vitro. Biochemistry. 2002;41:1502–1511. doi: 10.1021/bi011711s. [DOI] [PubMed] [Google Scholar]
- Cremades N, Cohen SI, Deas E, Abramov AY, Chen AY, Orte A, Sandal M, Clarke RW, Dunne P, Aprile FA, et al. Direct observation of the interconversion of normal and toxic forms of alpha-synuclein. Cell. 2012;149:1048–1059. doi: 10.1016/j.cell.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichner T, Radford SE. A Diversity of Assembly Mechanisms of a Generic Amyloid Fold. Molecular Cell. 2011;43:8–18. doi: 10.1016/j.molcel.2011.05.012. [DOI] [PubMed] [Google Scholar]
- Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci. 2010;30:6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvin JE, Lee VM, Trojanowski JQ. Synucleinopathies: clinical and pathological implications. Arch Neurol. 2001;58:186–190. doi: 10.1001/archneur.58.2.186. [DOI] [PubMed] [Google Scholar]
- Giehm L, Lorenzen N, Otzen DE. Assays for alpha-synuclein aggregation. Methods. 2011;53:295–305. doi: 10.1016/j.ymeth.2010.12.008. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature. 1996;383:550–553. doi: 10.1038/383550a0. [DOI] [PubMed] [Google Scholar]
- Gray MJ, Wholey WY, Wagner NO, Cremers CM, Mueller-Schickert A, Hock NT, Krieger AG, Smith EM, Bender RA, Bardwell JC, et al. Polyphosphate is a primordial chaperone. Mol Cell. 2014;53:689–699. doi: 10.1016/j.molcel.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefti F, Goure WF, Jerecic J, Iverson KS, Walicke PA, Krafft GA. The case for soluble A beta oligomers as a drug target in Alzheimer’s disease. Trends Pharmacol Sci. 2013;34:261–266. doi: 10.1016/j.tips.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Hegde ML, Rao KSJ. DNA induces folding in alpha-synuclein: Understanding the mechanism using chaperone property of osmolytes. Arch Biochem Biophys. 2007;464:57–69. doi: 10.1016/j.abb.2007.03.042. [DOI] [PubMed] [Google Scholar]
- Holmstrom KM, Marina N, Baev AY, Wood NW, Gourine AV, Abramov AY. Signalling properties of inorganic polyphosphate in the mammalian brain. Nat Commun. 2013;4:1362. doi: 10.1038/ncomms2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci U S A. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hufnagel DA, Tukel C, Chapman MR. Disease to dirt: the biology of microbial amyloids. PLoS Pathog. 2013;9:e1003740. doi: 10.1371/journal.ppat.1003740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT, Irizarry MC. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–931. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–475. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- Kornberg A, Rao NN, Ault-Riche D. Inorganic polyphosphate: a molecule of many functions. Annual Review of Biochemistry. 1999;68:89–125. doi: 10.1146/annurev.biochem.68.1.89. [DOI] [PubMed] [Google Scholar]
- Kumble KD, Kornberg A. Inorganic polyphosphate in mammalian cells and tissues. J Biol Chem. 1995;270:5818–5822. doi: 10.1074/jbc.270.11.5818. [DOI] [PubMed] [Google Scholar]
- Litvinov RI, Faizullin DA, Zuev YF, Weisel JW. The alpha-helix to beta-sheet transition in stretched and compressed hydrated fibrin clots. Biophys J. 2012;103:1020–1027. doi: 10.1016/j.bpj.2012.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz B, Munkner J, Oliveira MP, Kuusksalu A, Leitao JM, Muller WE, Schroder HC. Changes in metabolism of inorganic polyphosphate in rat tissues and human cells during development and apoptosis. Biochim Biophys Acta. 1997;1335:51–60. doi: 10.1016/s0304-4165(96)00121-3. [DOI] [PubMed] [Google Scholar]
- McColl G, Roberts BR, Pukala TL, Kenche VB, Roberts CM, Link CD, Ryan TM, Masters CL, Barnham KJ, Bush AI, et al. Utility of an improved model of amyloid-beta (Aβ1-42) toxicity in Caenorhabditis elegans for drug screening for Alzheimer’s disease. Molecular Neurodegeneration. 2012;7:57. doi: 10.1186/1750-1326-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissey JH. Polyphosphate: a link between platelets, coagulation and inflammation. Int J Hematol. 2012;95:346–352. doi: 10.1007/s12185-012-1054-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller F, Mutch NJ, Schenk WA, Smith SA, Esterl L, Spronk HM, Schmidbauer S, Gahl WA, Morrissey JH, Renne T. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. 2009;139:1143–1156. doi: 10.1016/j.cell.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munishkina LA, Fink AL, Uversky VN. Accelerated fibrillation of alpha-synuclein induced by the combined action of macromolecular crowding and factors inducing partial folding. Curr Alzheimer Res. 2009;6:252–260. doi: 10.2174/156720509788486491. [DOI] [PubMed] [Google Scholar]
- Rao NN, Gomez-Garcia MR, Kornberg A. Inorganic polyphosphate: essential for growth and survival. Annual Review of Biochemistry. 2009;78:605–647. doi: 10.1146/annurev.biochem.77.083007.093039. [DOI] [PubMed] [Google Scholar]
- Rashid MH, Rumbaugh K, Passador L, Davies DG, Hamood AN, Iglewski BH, Kornberg A. Polyphosphate kinase is essential for biofilm development, quorum sensing, and virulence of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2000;97:9636–9641. doi: 10.1073/pnas.170283397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts H, Brown D. Seeking a Mechanism for the Toxicity of Oligomeric α-Synuclein. Biomolecules. 2015;5:282–305. doi: 10.3390/biom5020282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakatani A, Fujiya M, Ueno N, Kashima S, Sasajima J, Moriichi K, Ikuta K, Tanabe H, Kohgo Y. Polyphosphate Derived from Lactobacillus brevis Inhibits Colon Cancer Progression Through Induction of Cell Apoptosis. Anticancer Res. 2016;36:591–598. [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Tipping KW, Karamanos TK, Jakhria T, Iadanza MG, Goodchild SC, Tuma R, Ranson NA, Hewitt EW, Radford SE. pH-induced molecular shedding drives the formation of amyloid fibril-derived oligomers. Proc Natl Acad Sci U S A. 2015;112:5691–5696. doi: 10.1073/pnas.1423174112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Fraley CD, Faridi J, Kornberg A, Roth RA. Inorganic polyphosphate stimulates mammalian TOR, a kinase involved in the proliferation of mammary cancer cells. Proc Natl Acad Sci U S A. 2003;100:11249–11254. doi: 10.1073/pnas.1534805100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.