

Abstract
In the 20th century, the complications of head injuries were controlled but not eliminated. The wars of the 21st century turned attention to blast, the instant of impact and the primary injury of concussion. Computer calculations have established that in the first 5 milliseconds after the impact, four independent injuries on the brain are inflicted: 1) impact and its shockwave, 2) deceleration, 3) rotation and 4) skull deformity with vibration (or resonance). The recovery, pathology and symptoms after acute brain trauma have always been something of a puzzle. The variability of these four modes of injury, along with a variable reserve of neurones, explains some of this problem.
Keywords: Traumatic brain injury, High energy shockwaves, Recovery of function, Brain reserve, Concussion, Postconcussion syndrome
In the 20th century, the complications of head injuries were understood and controlled although not eliminated. In the 21st century, interest turned to explosive blast and the instant of impact.
The four injuries of the first 5 milliseconds
Animals are poor models for traumatic brain injury; their heads and brains are so different to ours. Rats’ heads do not swing freely on the neck, they have little frontal lobe and their midbrain does not meet the cerebrum at right angles. Their skull has nothing like the shock conducting properties and elasticity of the human skull.
The first 5 milliseconds of injury is best modelled by finite element calculations on computers1–5 validated by cadaver experiments,2,5 real and surrogate.1,4 Computer models suggest four injuries after a blow hits the head:
Impact sends a shockwave through the brain.
Deceleration swings the cerebrum on the brain stem, stretching it.
Rotation stretches the connections between the brain’s two hemispheres.
Vibration (resonance) of the skull after impact bruises the underlying cortex.
This combination of movements has been recognised.6
What does each of the four injuries do to the brain?
1. Impact is common to nearly all traumatic brain injuries, even the mildest concussion. (Injury without impact does happen occasionally such as airbag injuries or shaken baby syndrome.)
Impact sends a shockwave (lasting a millisecond or so)1 throughout the brain, transmitted by the ventricles, then repeatedly reflected and refracted by the skull and the boundaries between structures such as the cortex–white matter boundary or the basal ganglia. These many reflections and refractions jumble the waves so their crossings augment and cancel each other (Fig 1). All over the brain (cerebellum and brain stem included), patches of neurons are damaged where negative pressure waves happen to coincide. Consequently, even the most minor concussions are marked by widespread death of neurones.7
Figure 1.
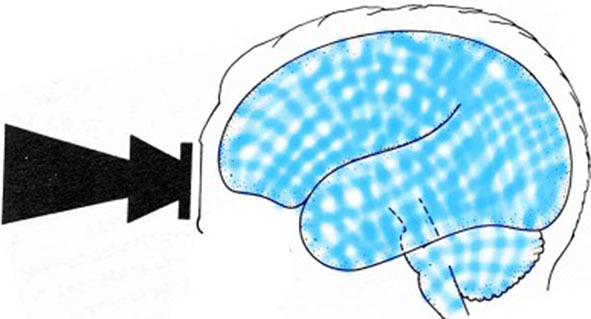
Multiple reflected waves criss-cross and overlap everywhere so negative phases may overlap and reinforce each other, enough to kill neurones. Consequently, any particular neurone’s fate is random.
The negative phase of the wave is the most damaging, pulling apart or stretching structures such as cell walls, proteins and deoxyribonucleic acid (DNA) chains. The strongest negative waves (-100kPa, -750mmHg pressure) form microsecond bubbles, cavities that tear blood vessels and strip tissues apart, thereby creating potential cavities for extradural clots.3
2. Deceleration is usual (but not essential) to traumatic brain injury. The cerebrum swings on the brainstem, stretching and tearing the reticular formation and the corticospinal tracts. Consciousness is therefore lost and spasticity starts. Brain stem traction alone, without impact, can kill shaken babies.8
3. Rotation. Impact is usually not central. As a result, it adds rotation to the impact and stretches axons crossing between the hemispheres.
4. Vibration (resonance) of the skull after impact bruises the underlying cortex. The skull deforms and vibrates, like a bell9 for 3–5 milliseconds,1 bruising the cortex.
Deceleration has already swung the frontal and temporal cortex hard up against the skull, eliminating a layer of cerebrospinal fluid. When the skull vibrates, the force spreads directly to the cortex, with no layer of cerebrospinal fluid to reflect the wave or cushion its force.
The inevitable consequences after the first 5 milliseconds
Metabolic injury
Mildly injured cells suffer metabolic lesions from stretched protein or DNA chains but keep intact cell membranes, nuclei and mitochondria. Most will recover (though temporarily stop working) while a few will slowly die.10
The mitochondria all cluster around the nucleus so the cell’s energy supply, acetyl coenzyme A, must pass down the protein chains of intracellular neurofibrils to the distant synapse. Shockwaves disrupt these and, without energy, synapses fail, as does the sodium pump. The membrane potential falls and the cell cannot convey impulses.
Membrane injury
Where cell membrane or synapse is torn, cytoplasm spills, thereby starting the ‘cascade of consequences’.11
The cascade of consequences
The cascade of consequences for glutamate, the most common neurotransmitter, is shown in Figure 2 but each of about 20 neurotransmitters has its own cascade. Glutamate is an excitatory neurotransmitter, and so probably accounts for the occasional acute epilepsy (not permanent) soon after injury and the strange combativeness of some concussed individuals. During the next week, cells die off and the brain swells from cell breakdown, letting sodium flow into the cell; without energy, the sodium pump cannot work. The cascade makes the full effects of a concussion develop over 48 hours.
Figure 2.

The cascade of consequences
Late neurone death
Even with intact cell membranes, over the two or three months after impact, some scattered neurones all over the brain die a slow metabolic death because the random overlay of jumbled waves has disrupted nuclear or mitochondrial nucleic acid chains irreparably. Microglia phagocytose cerebral debris and repair the myelin sheaths of the axons. They too suffer shockwave damage and immediate or late death. As a result, they may be late in starting repair. Brain scarring may therefore continue beyond six months.12
Evidence of damage in minor concussion
Diffusion tensor imaging is a magnetic resonance imaging (MRI) technique that tracks the movement of water along the axon from the nucleus distally; damage to the axon lets water leak out, in other directions. This technique shows white matter lesions in minor concussions,13 where they would be predicted, from rotation in the fibres crossing the midline, particularly the anterior corpus callosum, and from deceleration pulling on the long reticulospinal tracts. Ordinary MRI abnormalities were not seen in these minor concussions.
MRI spectroscopy shows the relative concentration of some metabolites in a region. It demonstrates a block to energy production for 3–4 weeks in minor sporting concussions,14 when no other MRI abnormalities are seen.
Cognitive function and white matter lesions
Executive function is impaired in proportion to the white matter damage, especially in the frontal lobe and corpus callosum.13 Memory loss is due to disruption of the fornix.13,15 The fornix (Latin for ‘arch’) links the hippocampus to the thalamus but it arches right around and over the third ventricle so ventricular shockwaves stretch it (Fig 3). Memory and the fornix are triply at risk from ventricular shockwaves, not only from arching over the ventricle but also at either end. Both the hippocampal synapses at the beginning (pyramidal cells) and the end of the arch (mammillary bodies) lie exposed in the floor of the ventricle.
Figure 3.
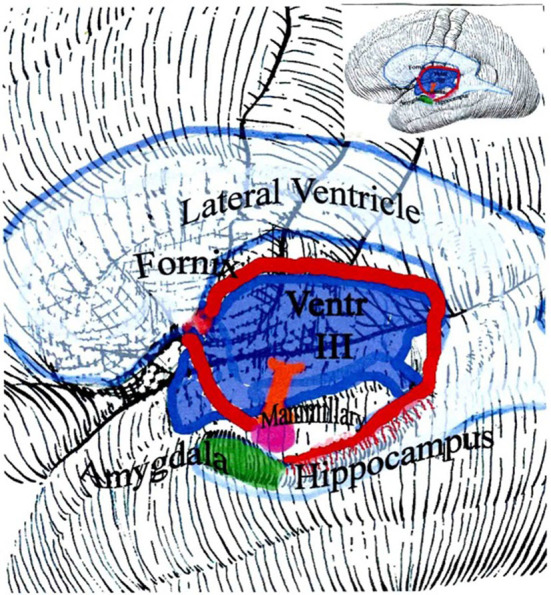
The fornix connects the hippocampus to the thalamus. Through the thalamus, the fornix also connects to the rest of the brain. Memory output goes from the hippocampus, beginning at the amygdala, to the rest of the brain. Owing to the way the fornix arches around the ventricle, it is readily disrupted by a ventricular shockwave.
In a pig model of concussion, some pyramidal cells in the hippocampus, originating in the fornix, had become ‘dark cells’ at 48 hours following injury.16 No detail of these neurones was seen: no nucleus, no mitochondria, no cytoplasm; dark debris only.
Other psychological features of concussion
Slow thinking does not relate to white matter damage13 but perhaps to the number of alternative pathways interrupted by neuronal drop-out from spreading shockwaves. Slowed thinking may persist surprisingly long after concussion. Perhaps the later gradual deaths of neurones explains why.
Fatigue is usual after all sorts of cerebral injury, from fewer neural pathways17 and limited resources of nerve, muscle or mental will, or all three. Like hunger, it is a decision based on bodily sensations but censored, suppressed or exaggerated by emotions and circumstances. The brain usually rotates the cells and pathways it uses, if possible using a fresh path. When injury causes widespread neuronal disconnection and death, alternate paths are few and so fatigue is normal. As alternate circuits recover and new ones are trained, fatigue lessens.
Post-traumatic stress disorder is another psychological feature of concussion. Memory is not a record but a reconstruction, using the frontal lobe to detect gaps and fill in details. After wounding, post-traumatic stress disorder affects about 1 in 4 soldiers but with concussion added, the probability of post-traumatic stress increases by another half.18 Reconstruction of pathways can change memories and beliefs about the accident, according to emotions; with damaged frontal connections, new beliefs are easily implanted.19 Paradoxically, with severe brain injury, post-traumatic stress disorder is rare.
The brain’s defences
The skull, scalp and face spread the impact. The face crumples slowly, absorbing impact, and the neck limits deceleration and rotation. A helmet modifies all four events.
Recovery, cell repair and new neurones
Many cells can restore their damaged metabolism and new neurones can grow in the subependymal zone, just below the lining of the ventricles,20 from where they migrate out to other areas of the brain, or possibly, stem cells proliferate locally.21
The cerebral reserve
The idea of a cerebral reserve has been suggested for other neurone losing disorders such as multiple sclerosis and Alzheimer’s disease.22 The reserve is that stock of spare neurones and underused pathways, replacing neurones lost from existing pathways and circuits. In teenage years, the brain matures by discarding underused pathways so then the reserve of spare pathways is large.23 Luckily, these are also the years in which brain injuries are most common (ages 10–25 years).
Reserves vary, from person to person and with age
Aging reduces (without eliminating) the cerebral reserves of pathways and neurons. Consequently, the older recover less readily. A prospective survey in Denmark showed that minor concussion (<12 days in hospital) before the age of 12 years has little risk of permanent damage.24 Whether the skull was fractured did not influence the outcome. Individual case follow-up confirms this.23 Serious damage before the age of three years, however, will prevent normal development of the brain.25
Repeated concussions (or disease) in the several months before the reserve is re-established may exhaust the reserve. Occasionally, a second concussion is disastrous.
Genes influence the maturation and size of the reserve.23 The apolipoprotein E4 gene seems to account for much of the poor recovery from severe brain injury but not all of it.26 About one in five Westerners carries a single copy of this gene. For minor brain injuries, being heterozygous does not make much difference;27 minor injuries can obtain enough neurons from even a reduced reserve. For major injuries, the genetic size of the reserve does matter.
Past depletion of the reserve, recovering from past injury or disease, impairs recovery. Brain shrinkage (from illness or malnutrition, alcoholism or drugs) also reduces the reserve.
Those with traumatic brain injuries are not an ‘average’ sample of the population; the risk takers, the incautious, the impaired, the addicted and the intoxicated suffer brain injury disproportionately often. Brain reserves are an educational and social quantity too. National servicemen in the Swedish army who did poorly in the initial intelligence test were more likely later to suffer subdural haematomas.28 This has been confirmed in the Danish army.29
Conclusions
Recovery from brain injury has been notoriously unpredictable; it reflects the sum of four independent injuries from the initial impact, with a varying cerebral reserve. These four injuries (the shockwave of impact, rotation, deceleration and skull vibration) can each vary in their severity and pathology. The shockwave causes cell necrosis, rotation stresses the connections between the hemispheres, deceleration of the head pulls on the brain stem and vibrations of the skull damage the cortex. The brain’s reserve for recovery reflects age, genetics and previous use or abuse of the brain.
Declaration
The opinions expressed in this paper are the personal views of the author and not those of his employer, the Accident Compensation Corporation.
References
- 1.Ganpule S, Alai A, Plougonven E, Chandra N. . Mechanics of blast loading on the head models in the study of traumatic brain injury using experimental and computational approaches. Biomech Model Mechanobiol 2013; : 511–531. [DOI] [PubMed] [Google Scholar]
- 2.Katagiri M, Katagata K, Pramudita JA, Ujihashi S. . Development and application of stress-based skull fracture criteria using a head finite element model. J Biomech Sci Eng 2012; : 449–462. [Google Scholar]
- 3.Panzer MB, Myers BS, Capeheart BP, Bass CR. . Development of a finite element model for blast brain injury and the effects of CSF cavitation. Ann Biomed Eng 2012; : 1,1530–1,544. [DOI] [PubMed] [Google Scholar]
- 4.Chafi MS, Karami G, Zielewski M. . Biomechanical assessment of brain dynamic responses due to blast pressure waves. Ann Biomed Eng 2010; : 490–504. [DOI] [PubMed] [Google Scholar]
- 5.Hardy WN, Mason MJ, Foster CD et al. . A study of the response of the human cadaver head to impact. Stapp Car Crash J 2007; : 17–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeKosky ST, Ikonomovic MD, Gandy S. . Traumatic brain injury – football, warfare, and long-term effects. N Engl J Med 2010; : 1,293–1,296. [DOI] [PubMed] [Google Scholar]
- 7.Oppenheimer DR. . Microscopic lesions in the brain following head injury. J Neurol Neurosurg Psychiatry 1968; : 299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geddes JF, Vowles GH, Hackshaw AK et al. . Neuropathology of inflicted head injury in children. II. Microscopic brain injury in infants. Brain 2001; : 1,299–1,306. [DOI] [PubMed] [Google Scholar]
- 9.Moss WC, King MJ, Blackman EG. . Skull flexure from blast waves: a mechanism for brain injury with implications for helmet design. Phys Rev Lett 2009; : 108702. [DOI] [PubMed] [Google Scholar]
- 10.Povlishock JT, Katz DI. . Update of neuropathology and neurological recovery after traumatic brain injury. J Head Trauma Rehabil 2005; : 76–94. [DOI] [PubMed] [Google Scholar]
- 11.Blennow K, Hardy J, Zetterberg H. . The neuropathology and neurobiology of traumatic brain injury. Neuron 2012; : 886–899. [DOI] [PubMed] [Google Scholar]
- 12.Bigler ED. . Quantitative magnetic resonance imaging in traumatic brain injury. J Head Trauma Rehabil 2001; : 117–134. [DOI] [PubMed] [Google Scholar]
- 13.Kinnunen KM, Greenwood R, Powell JH et al. . White matter damage and cognitive impairment after traumatic brain injury. Brain 2011; : 449–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vagnozzi R, Signoretti S, Cristofori L et al. . Assessment of metabolic brain damage and recovery following mild traumatic brain injury: a multicentre, proton magnetic resonance spectroscopic study in concussed patients. Brain 2010; : 3,232–3,242. [DOI] [PubMed] [Google Scholar]
- 15.Jorge RE, Acion L, White T et al. . White matter abnormalities in veterans with mild traumatic brain injury. Am J Psychiatry 2012; : 1,284–1,291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suneson A, Hansson HA, Seeman T. . Pressure wave injuries to the nervous system caused by high-energy missile extremity impact: Part II. Distant effects on the central nervous system – a light and electron microscopic study on pigs. J Trauma 1990; : 295–306. [DOI] [PubMed] [Google Scholar]
- 17.Filippi M, Rocca MA. Cortical reorganisation in patients with MS. . J Neurol Neurosurg Psychiatry 2004; : 1,087–1,089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenfeldt JV, McFarlane AC, Bragge P et al. . Blast-related traumatic brain injury. Lancet Neurol 2013; : 882–893. [DOI] [PubMed] [Google Scholar]
- 19.Dockree PM, O'Keeffe FM, Moloney P et al. . Capture by misleading information and its false acceptance in patients with traumatic brain injury. Brain 2006; : 128–140. [DOI] [PubMed] [Google Scholar]
- 20.Eriksson PS, Perfilieva E, . Björk-Eriksson T et al. Neurogenesis in the adult human hippocampus. Nat Med 1998; : 1,313–1,317. [DOI] [PubMed] [Google Scholar]
- 21.Curtis MA, Penney EB, Pearson J et al. . The distribution of progenitor cells in the subependymal layer of the lateral ventricle in the normal and Huntington's disease human brain. Neuroscience 2005; : 777–788. [DOI] [PubMed] [Google Scholar]
- 22.Mosconi L, Herholz K, Prohovnik I et al. . Metabolic interaction between ApoE genotype and onset age in Alzheimer's disease: implications for brain reserve. J Neurol Neurosurg Psychiatry 2005; : 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giedd JN, Stockman M, Weddle C et al. . Anatomic magnetic resonance imaging of the developing child and adolescent brain and the effects of genetic variation. Neuropsychol Rev 2010; : 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Teasdale TW, Engberg AW. . Cognitive dysfunction in young men following head injury in childhood and adolescence: a population study. J Neurol Neurosurg Psychiatry 2003; : 933–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson V, Godfrey C, Rosenfeld JV, Caltroppa C. . Predictors of cognitive function and recovery 10 years after traumatic brain injury in young children. Pediatrics 2012; : e254–e256. [DOI] [PubMed] [Google Scholar]
- 26.Teasdale GM, Nicoll JA, Murray G, Fiddes M. . Association of apolipoprotein E polymorphism with outcome after head injury. Lancet 1997; : 1,069–1,071. [DOI] [PubMed] [Google Scholar]
- 27.Liberman JN, Stewart WF, Wesnes K, Troncoso J. . Apolipoprotein E e4 and short-term recovery from predominantly mild brain injury. Neurology 2002; : 1,038–1,044. [DOI] [PubMed] [Google Scholar]
- 28.Nordström A, Edin BB, Lindström S, NordstrÖm P. . Cognitive function and other risk factors for mild traumatic brain injury in young men: nationwide cohort study. BMJ 2013; : f723. [DOI] [PubMed] [Google Scholar]
- 29.Teasdale TW, Frøsig AJ. Cognitive ability and educational level in relation to concussion: a population study of young men. BMJ Open 2013; : e002321. [DOI] [PMC free article] [PubMed] [Google Scholar]