

Abstract
Background
Cardiac fibrosis after primary infarction is a type of pathological phenomena as shown by increased collagen in myocardial cells. Transforming growth factor (TGF)-β1 is a critical factor participating in myocardial fibrosis. A previous study has shown the inhibitory role on TGF-β1 by microRNA-24 (miR-24) via targeting Furin. This study thus investigated the role of miR-24 and Furin/TGF-β1 in rat myocardial fibrosis.
Material/Methods
A total of 40 adult SD rats (both males and females) were prepared for myocardial infarction model by ligating the descending branch of left coronary artery after anesthesia. HE staining was performed to observe myocardial fibrosis after 1, 2, and 4 weeks. Tissue RNA was extracted to detect mRNA levels of Furin, TGF-β1, and miR-24 by real-time PCR. Western blotting was used to quantify protein expression of Furin and TGF-β1 in myocardial tissues.
Results
Increased connective tissues were observed in myocardial tissues at 4 weeks after infarction by HE staining, which also revealed widening of the intra-myocardial cleft, along with more inflammatory cells and fibroblast hypertrophy. miR-24 expression was significantly depressed at 2 and 4 weeks after cardiac infarction (p<0.05). mRNA levels of Furin and TGF-β1 were elevated after infarction (p<0.05). With prolonged time periods of myocardial infarction, protein levels of Furin and TGF-β1 were further increased. The level of miR-24 was positively correlated with left ventricular end-diastolic diameter, left ventricular systolic diameter, and left ventricular ejection fraction. However, the level of Furin or TGF-b1 was negatively correlated with the above parameters.
Conclusions
This study demonstrated the important role of abnormal expression of miR-24 in myocardial fibrosis after infarction, and may provide drug targets for treating myocardial fibrosis.
MeSH Keywords: Anterior Wall Myocardial Infarction, Cystic Fibrosis, Furin, MicroRNAs
Background
Cardiovascular disease severely affects public health. Myocardial fibrosis is a pathology abnormality caused by increased collagen in cardiomyocytes. The fibrosis of myocardial tissue may lead to heart dilation, and decreased heart compliance, eventually leading to heart failure [1]. In cardiac infarction tissue, it is common to find the synthesis and deposition of multiple extracellular matrix. After several months or even years after cardiac infarction, such extracellular matrix is still in active status. Therefore, the successful management of the progression of cardiac fibrosis can significantly improve the prognosis of patients.
Various signal molecules are involved in the progression of cardiac fibrosis. For example, interleukin-1 (IL-1) and angiotensin II (Ang-II) can stimulate the differentiation of cardiac fibroblasts into muscle-like fibroblasts, which has potent ability for proliferation, migration, and collagen secretion [2]. Transforming growth factor β1 (TGF-β1) is an important member of the TGF-β family, is important for mediating cell growth and differentiation, and plays an important role in the occurrence and progression of multiple diseases. Present in multiple tissues, TGF-β1 can be secreted by various somatic cells, such as cellular secretions during inflammation. TGF-β1 induces synthesis and deposition of extracellular matrix, and participates in the occurrence of fibrosis in various tissues. A previous study reported the involvement of TGF-β1 in the fibrosis of multiple organs, such as heart, kidney, and liver [3]. TGF-β can activate transcriptional factor SMAD4, which further elevates Fn14 expression for the participation of extracellular matrix and activation of fibroblast [4]. TGF-β1 can facilitate the activation and collagen secretion of myocardial fibroblasts involved in the progression of cardiac fibrosis along with other risk factors [5]. For example, connective tissue growth factor (CTGF) is abnormally enhanced in the progression of cardiac fibrosis, largely due to induction by TGF-β1. Moreover, TGF-β1 plays a crucial role in the differentiation of cardiac fibrosis via regulating FoxO1 gene [6].
MicroRNA (miR) is a type of non-coding RNA molecule with 19~25 nucleic acid length. It can bind with 3′untranslational region (3′-UTR) to cause mRNA degradation or translational inhibition, both of which modify target gene expression and thus participate in occurrence and progression of various diseases. The highly conserved sequence during evolution and specific expression depending on the disease course endow the potency of miR as a predictive factor for classification and treatment target of various genes. Various studies showed the involvement of miR in cardiac fibrosis. For example, miR-29 can regulate collagen synthesis by inhibiting the synthesis of collagen to retard the progression of myocardial infarction [7]. miR-30 and miR-133 can decrease collagen synthesis via regulating CTGF expression [8], and miR-34a can regulate the fibrosis after myocardial infarction via targeting Smad4 [9]. MiR-24 is a hypoxia-sensitive miR in vascular endothelial cells, and is involved in cardiac vascular changes after infarction [10]. The enhancement of miR-24 expression can decrease the function of smooth muscle cells [11].
A previous study showed that Furin regulated the activation of TGF-β via angiotensin II (ANGII) [12], and a recent study revealed that Furin exerted its functions via facilitating TGF-β activation [13,14]. In other studies using cardiac fibroblasts of neonatal mice, miR-24 exerted its function via targeting Furin and hence inhibiting TGF-β1 activation. The present study thus investigated the role of miR-24 and Furin/TGF-β1 in rat myocardial fibrosis.
Material and Methods
Reagent and equipment
We used the following reagents and equipment: Real-time fluorescent quantitative RT-PCR kit (TaKaRa, Japan); TRIzol reagent (Invitrogen), ABI 7500 fluorescent quantitative PCR cycler (Applied Biosystem, USA); High-speed cold centrifuge (Eppendorf, USA); SYBR Green qPCR mix (ToYoBo, Japan); Hematoxylin-eosin dye (Qiyi Bio, China); Protein electrophoresis and transfer apparatus (Invitrogen, USA); and BCA protein quantification kit (Sigma-Aldrich, USA).
Animals and cardiac infarction model
We used 40 healthy SPF grade SD adult rats (20 males and 20 females, provided by Shangdong University) with body weight between 200 and 280 g. After anesthesia (intraperitoneal injection) by 10% chloral hydrate, tracheal intubation was performed before exposing the heart. The descending branch of the left coronary artery was blocked for 6 s to whiten the myocardial tissue below, accompanied with elevated S-T segment on ECG. Chest walls were sutured by layers. Rats were then randomly assigned into 4 groups (N=10 each). The sham group consisted of rats that underwent the surgery but without ligation, while the other 30 rats were killed at 1 week, 2 weeks, and 4 weeks after surgery (N=10, 9, and 9, respectively). Tissues samples were obtained at 3 times points, while control rats were killed and harvested at 4 weeks.
Rats were used for all experiments, and all procedures were approved by the Animal Ethics Committee of Shandong Provincial Qianfoshan Hospital.
Hematoxylin-eosin staining
Heart tissue were dehydrated, fixed, and embedded in paraffin, then 4-μm slices were prepared and de-waxed. Hematoxylin dye was added after re-hydration, followed by eosin staining. Tissue sections were mounted with coverslips and observed under a light field microscope.
Cardiac tissue RNA extraction
Harvested myocardial tissues were homogenized in liquid nitrogen and lysed in TRIzol reagent. Phenol-chloroform was used to extract total RNA, which was re-suspended in DEPC-treated water. UV spectrometry was employed to determine the purity and concentration of RNA, whose integrity was determined in 1% agarose gel electrophoresis. RNA was stored at −80°C for further use.
Real-time PCR for mRNA levels of miR-24, Furin, and TGF-β1
The TaKaRa reverse transcription kit was used to synthesize DNA using miRNA-24, Furin, and TGF-β1 mRNA following the manual’s instructions. The PCR system (total 20 μl) consisted of 9 μl 2XSYBR Green Mixture; 2 μl Primer 1 (5 μM); 2 μl Primer 2 (5 μM); 2 μl cDNA, and 5 μl ddH2O. The reaction was performed under the following conditions: 95°C denaturing for 30 s, followed by 40 cycles each of 95°C 30 s, 55°C 30 s, and 72°C 60 s on an ABI 7500 fluorescent quantitative PCR cycler. Each experiment was performed in triplicate. Primer sequences were: Furin-forward: 5′-ACTAA CACTG TGCCC TGGTG GAG-3′; Furin-reverse: 5′-ACCCT GGACA GGTAG GTTGG GTA-3′; TGF-β1-forward: 5′-TGCGC CTGCA GAGAT TCAAG-3′; TGF-β1-reverse: 5′-AGGTA ACGCC AGGAA TTGTT GCTA-3′; GAPDH-forward: 5′-GGCAC AGTCA AGGCT GAGAA TG-3′; GAPDH-reverse: 5′-ATGGT GGTGA AGACG CCAGT A-3′.
Western blotting for protein levels of Furin and TGF-β1
Myocardial tissues were homogenized and mixed with protein lysis buffer. At 4°C, the mixture was vibrated every 5 min (1 min each) for 5 times, followed by 12 000 rpm centrifugation for 20 min. The supernatant was saved protein quantification using the BCA kit. We loaded 50 ng proteins and separated them in SDS-PAGE under a 40-V electrical field. After 5 h, proteins were transferred to the membrane under 60V for 2h, followed by Ponceau red staining for 5 min. The membrane was then blocked, and incubated in primary and secondary antibody for developing. The film was scanned for analysis.
Statistical methods
The SPSS13.0 software package was used in statistical analysis and the t test was employed in 2-sample independent tests. Enumeration data were compared by chi-square test. Pearson correlation was performed to analyze the relationship between expression of miR-24, Furin, or TGF-β1 with cardiac function parameters. A significant level was defined as 0.05.
Results
Pathology of rat myocardial tissues
By HE staining, we observed the dynamic change of rat myocardial tissues within 4 weeks of cardiac infarction (Figure 1). In the control group, cells were arranged regularly, with clear and sharp muscle stripes, without significant numbers of inflammatory cells. At the first and second week of myocardial infarction, the boundary between myocardial cells disappeared, with abundant inflammatory cells and monocytes infiltration. After 4 weeks, it was clear that increasing connective tissues existed and widened the intra-myocardial cleft, accompanied with more inflammatory cells and fibroblasts. Masson staining demonstrated the longer time after myocardial infarction, and the increased collagen fibrils in the outer spaces (Figure 2).
Figure 1.

Pathology alternation of myocardial tissues at different time periods after infarction (HE staining, ×400).
Figure 2.

Pathology alternation of myocardial tissues at different time periods after infarction (Masson staining, ×400).
mRNA expression level of miR-24, Furin, and TGF-β1
Real-time PCR results showed significantly decreased miR-24 at 2 and 4 weeks after myocardial infarction. With prolonged time of infarction, miR-24 level was progressively down-regulated. mRNA levels of Furin and TGF-β1 were, however, elevated at 2 and 4 weeks after infarction, when compared to those in the control group (p<0.05, Figure 3), suggesting that miR-24, Furin, and TGF-β1 are involved in the formation of myocardial fibrosis.
Figure 3.
Gene expression level of miR-24, Furin, and TGF-β1 at different time points of rat myocardial infarction, p<0.05 compared to those in control group.
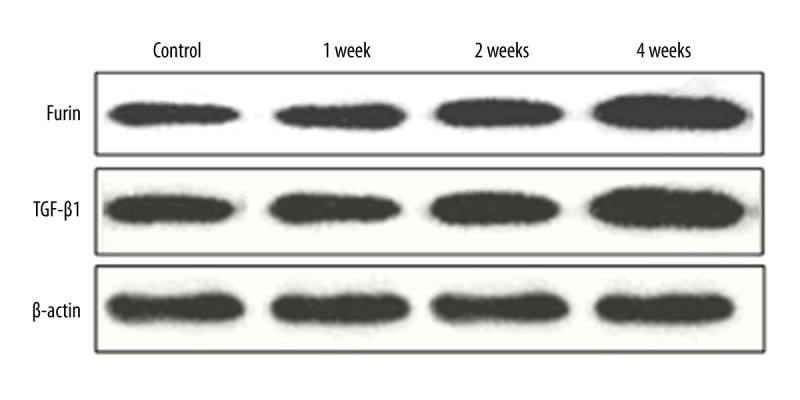
Furin and TGF-β1 protein levels in myocardial tissues
Western blotting results showed that longer time after myocardial infarction was associated with higher Furin and TGF-β1 protein levels. A significant increase was observed at 4 weeks after myocardial infarction, as Furin and TGF-β1 shared consistent patterns.
Correlation analysis of miR-24, Furin, and TGF-β1 expression with cardiac function
Through correlation analysis, expression of miR-24 was found to be positively correlated with left ventricular end-diastolic diameter (r=0.43, P<0.05), left ventricular systolic diameter (r=0.51, P<0.05), and left ventricular ejection fraction (r=0.39, P<0.05). However, expression of Furin and TGF-β1 was negatively correlated with left ventricular end-diastolic diameter (r=−0.45 or r=−0.37, P<0.05), left ventricular systolic diameter (r=−0.65 or r=−0.45, P<0.05), and left ventricular ejection fraction (r=−0.32 or r=−0.39, P<0.05). No correlation of the level of miR-24, Furin, or TGF-β1 with left ventricular mass index was observed (P>0.05).
Discussion
The most abundantly distributed cell type inside cardiac tissues is myocardial fibroblast, which, under normal physiological conditions, is at rest status. With certain pathological alternations, it can secrete high levels of collagen to damage cardiac functions [15]. The TGF-β1 signal pathway is important for myocardial fibrosis. A previous study showed the high expression of miR-24 in myocardial fibroblasts, which can affect the synthesis of extracellular matrix collagen, possibly via interfering with the TGF-β1 signal pathway via mediating Furin [16]. Another study also found the activation of TGF-β1 by Furin enhanced MMP-2 expression and further tumor cell invasion [17].
The present study revealed significant myocardial fibrosis with prolonged time of infarction, as shown by connective tissues in the inter-myocardial fiber cleft, whose width was increased, with more inflammatory cells and fibroblasts, suggesting the successful preparation of this rat myocardial infarction mode, on which further experiments were performed.
In myocardial tissues of infarction rats, miR-24 level was gradually decreased with prolonged time of infarction. Furin, which is a possible target gene for miR-24, was also up-regulated, further supporting a previous study by Wang et al. [16]. A study showed the possible involvement of Furin in the regulation of multiple important TGF-β1 protein, such as lefty-1 and lefty-2 [18]. As a pluripotent miRNA, miR-124 is correlated with blood cell differentiation, tumor formation, cell proliferation, and differentiation [19]. In human liver cancer cells and skeletal muscle cells, miR-24 regulates the TGF-β signal pathway [20,21]. The assay for mRNA expression level of TGF-β1 in myocardial tissues at different time points after infarction found the significant elevation of TGF-β1 mRNA at 2 weeks after myocardial infarction. Western blotting showed similar results, with prolonged time of infarction associated with higher level of TGF-β1 protein. A previous study also revealed the thickening of myocardial muscle and mesenchymal fibrosis in TGF-β1-over-expressed mice [22], while TGF-β1-deficient mice had significant improvement of cardiac compliance [23]. As TGF-β could facilitate myocardial fibrosis via stimulating the proliferation of fibroblast [24], the inhibition of TGF-β1 expression in myocardial tissues could improve cardiac fibrosis. Actually, the anti-fibrotic treatment after myocardial infarction including anti-TGF-β1 agent has been shown to decrease the infarction area [25].
The Smads signal pathway is the major pathway participating in myocardial fibrosis [26]. The understanding of how Furin/TGF-β1 is involved in this process, however, is still incomplete. The present study has certain inherent limitations, including the fact that a single miRNA molecule can modulate multiple target gene expressions, and a single gene can be mediated by various miRNA molecules. Further research in warranted on the role of miR-24 in the progression of myocardial fibrosis and whether miR-24 targets multiple genes, in order to provide more evidence for use in developing anti-fibrotic medicine. Results of this study provide new insights for illustrating the role of Furin/TGF-β1 signal pathway in the process of myocardial fibrosis and related mechanisms.
Conclusions
This study demonstrated the important role of abnormal expression of miR-24 in myocardial fibrosis after infarction, and may provide drug targets for treating myocardial fibrosis.
Footnotes
Disclosure of conflict of interest
None.
Source of support: This work was supported by the National Natural Science Foundation (No. 81541021 and No. 81400843), Shandong Provincial Natural Science Foundation (No. ZR2014HP033), and Shandong Provincial Science and Technology Research Project (2012YD18046)
References
- 1.Heimberg AM, Sempere LF, Moy VN, et al. MicroRNAs and the advent of vertebrate morphological complexity. Proc Natl Acad Sci USA. 2008;105(8):2946–50. doi: 10.1073/pnas.0712259105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tomasek JJ, Gabbiani G, Hinz B, et al. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3(5):349–63. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 3.Evans RA, Tian YC, Steadman R, Phillips AO, et al. TGF-beta1-mediated fibroblast-myofibroblast terminal differentiation-the role of Smad proteins. Exp Cell Res. 2003;282(2):90–100. doi: 10.1016/s0014-4827(02)00015-0. [DOI] [PubMed] [Google Scholar]
- 4.Chen S, Liu J, Yang M, et al. Fn14, a downstream target of the TGF-beta signaling pathway, regulates fibroblast activation. PLoS One. 2015;10(12):e0143802. doi: 10.1371/journal.pone.0143802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meng XM, Tang PM, Li J, Lan HY. TGF-beta/Smad signaling in renal fibrosis. Front Physiol. 2015;6:82. doi: 10.3389/fphys.2015.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vivar R, Humeres C, Muñoz C, et al. FoxO1 mediates TGF-beta1-dependent cardiac myofibroblast differentiation. Biochim Biophys Acta. 2016;1863(1):128–38. doi: 10.1016/j.bbamcr.2015.10.019. [DOI] [PubMed] [Google Scholar]
- 7.van Rooij E, Sutherland LB, Thatcher JE, et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA. 2008;105(35):13027–32. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duisters RF, Tijsen AJ, Schroen B, et al. miR-133 and miR-30 regulate connective tissue growth factor: Implications for a role of microRNAs in myocardial matrix remodeling. Circ Res. 2009;104(2):170–78. doi: 10.1161/CIRCRESAHA.108.182535. 6p following 178. [DOI] [PubMed] [Google Scholar]
- 9.Huang Y, Qi Y, Du JQ, Zhang DF. MicroRNA-34a regulates cardiac fibrosis after myocardial infarction by targeting Smad4. Expert Opin Ther Targets. 2014;18(12):1355–65. doi: 10.1517/14728222.2014.961424. [DOI] [PubMed] [Google Scholar]
- 10.Fiedler J, Jazbutyte V, Kirchmaier BC, et al. MicroRNA-24 regulates vascularity after myocardial infarction. Circulation. 2011;124(6):720–30. doi: 10.1161/CIRCULATIONAHA.111.039008. [DOI] [PubMed] [Google Scholar]
- 11.Fiedler J, Stöhr A, Gupta SK, et al. Functional microRNA library screening identifies the hypoxamir miR-24 as a potent regulator of smooth muscle cell proliferation and vascularization. Antioxid Redox Signal. 2014;21(8):1167–76. doi: 10.1089/ars.2013.5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stawowy P, Margeta C, Kallisch H, et al. Regulation of matrix metalloproteinase MT1-MMP/MMP-2 in cardiac fibroblasts by TGF-beta1 involves furin-convertase. Cardiovasc Res. 2004;63(1):87–97. doi: 10.1016/j.cardiores.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 13.Blakytny R, Ludlow A, Martin GE, et al. Latent TGF-beta1 activation by platelets. J Cell Physiol. 2004;199(1):67–76. doi: 10.1002/jcp.10454. [DOI] [PubMed] [Google Scholar]
- 14.Dogar AM, Towbin H, Hall J. Suppression of latent transforming growth factor (TGF)-beta1 restores growth inhibitory TGF-beta signaling through microRNAs. J Biol Chem. 2011;286(18):16447–58. doi: 10.1074/jbc.M110.208652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358(13):1370–80. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 16.Wang J, et al. MicroRNA-24 regulates cardiac fibrosis after myocardial infarction. J Cell Mol Med. 2012;16(9):2150–60. doi: 10.1111/j.1582-4934.2012.01523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McMahon S, Laprise MH, Dubois CM. Alternative pathway for the role of furin in tumor cell invasion process. Enhanced MMP-2 levels through bioactive TGFbeta. Exp Cell Res. 2003;291(2):326–39. doi: 10.1016/s0014-4827(03)00407-5. [DOI] [PubMed] [Google Scholar]
- 18.Constam DB, Robertson EJ. Tissue-specific requirements for the proprotein convertase furin/SPC1 during embryonic turning and heart looping. Development. 2000;127(2):245–54. doi: 10.1242/dev.127.2.245. [DOI] [PubMed] [Google Scholar]
- 19.Chhabra R, Dubey R, Saini N. Cooperative and individualistic functions of the microRNAs in the miR-23a~27a~24-2 cluster and its implication in human diseases. Mol Cancer. 2010;9:232. doi: 10.1186/1476-4598-9-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshimoto N, Inaba Y, Yamada S, et al. 2-Methylene 19-nor-25-dehydro-1alpha-hydroxyvitamin D3 26,23-lactones: Synthesis, biological activities and molecular basis of passive antagonism. Bioorg Med Chem. 2008l;16(1):457–73. doi: 10.1016/j.bmc.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 21.Sun Q, Zhang Y, Yang G, et al. Transforming growth factor-beta-regulated miR-24 promotes skeletal muscle differentiation. Nucleic Acids Res. 2008;36(8):2690–99. doi: 10.1093/nar/gkn032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosenkranz S, Flesch M, Amann K, et al. Alterations of beta-adrenergic signaling and cardiac hypertrophy in transgenic mice overexpressing TGF-beta(1) Am J Physiol Heart Circ Physiol. 2002;283(3):H1253–62. doi: 10.1152/ajpheart.00578.2001. [DOI] [PubMed] [Google Scholar]
- 23.Brooks WW, Conrad CH. Myocardial fibrosis in transforming growth factor beta(1)heterozygous mice. J Mol Cell Cardiol. 2000;32(2):187–95. doi: 10.1006/jmcc.1999.1065. [DOI] [PubMed] [Google Scholar]
- 24.Dobaczewski M, Bujak M, Li N, et al. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ Res. 2010;107(3):418–28. doi: 10.1161/CIRCRESAHA.109.216101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okada H, Takemura G, Kosai K, et al. Postinfarction gene therapy against transforming growth factor-beta signal modulates infarct tissue dynamics and attenuates left ventricular remodeling and heart failure. Circulation. 2005;111(19):2430–37. doi: 10.1161/01.CIR.0000165066.71481.8E. [DOI] [PubMed] [Google Scholar]
- 26.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]