

Abstract
Rationale
(−)-Stepholidine is a tetrahydroberberine alkaloid that is known to interact with dopamine receptors and has also been proposed as a novel antipsychotic agent. Its suggested novelty lies in the fact that it has been proposed to have D1-like receptor agonist and D2-like receptor antagonist properties. Thus, it might be effective in treating both positive and negative (cognition) symptoms of schizophrenia. However, its activity on specific dopamine receptor subtypes has not been clarified, especially with respect to its ability to activate D1-like receptors.
Objectives
We wished to examine the affinity and functional activity of (−)-stepholidine at each of the human dopamine receptor subtypes expressed in a defined cellular environment.
Methods
D1–D5 dopamine receptors were stably expressed in cell lines and their interactions with (−)-stepholidine were examined using radioligand binding and various functional signaling assays. Radioligand binding assays were also performed using bovine striatal membranes.
Results
(−)-Stepholidine exhibited high (nM) affinity for D1 and D5 receptors, somewhat lower (two- to four-fold) affinity for D2 and D3 receptors, and low micromolar affinity for D4 receptors. Functionally, (−)-stepholidine was ineffective in activating G protein-mediated signaling of D1-like and D2 receptors and was also ineffective in stimulating β-arrestin recruitment to any dopamine receptor subtype. It did, however, antagonize all of these responses. It also antagonized D1–D2 heteromer-mediated Ca2+ mobilization. Radioligand binding assays of D1-like receptors in brain membranes also indicated that (−)-stepholidine binds to the D1 receptor with antagonist-like properties.
Conclusions
(−)-Stepholidine is a pan-dopamine receptor antagonist and its in vivo effects are largely mediated through dopamine receptor blockade with potential cross-talk to other receptors or signaling proteins.
Keywords: Stepholidine, Dopamine, Antipsychotic, β-arrestin, Receptor
Introduction
The neurotransmitter dopamine is a known regulator of many important neuronal processes that include motor control, cognition, reward, and memory. Changes in dopaminergic signaling are also associated with various neuropsychiatric disorders such as Parkinson’s disease, schizophrenia, drug addiction, and others. The actions of dopamine are mediated by five dopamine receptor subtypes (D1R–D5R), which are grouped by structure and function into the D1-like (D1R and D5R) and D2-like (D2R, D3R, and D4R) subfamilies. The D1-like receptors couple to Gs/olf proteins that activate adenylyl cyclase, leading to increased cAMP accumulation, whereas the D2-like receptors couple to Gi/o proteins that inhibit adenylyl cyclase activity (Beaulieu and Gainetdinov 2011; Missale et al. 1998; Sibley and Monsma 1992). It is not surprising that compounds targeting dopamine receptors are of great interest from the viewpoint of drug development and clinical use. In fact, one of the most highly validated drug targets in neurology and psychiatry is the D2R. However, most drugs targeting dopamine receptors are problematic, typically exhibiting side effects due to the activation or blockade of other receptor-mediated signaling pathways. More selective and efficacious agents that target dopamine receptors are thus desired.
(−)-Stepholidine is a tetrahydroberberine alkaloid, originally isolated from the Chinese herb Stephania intermedia, which has been used in traditional Chinese medicine to treat various maladies (Mo et al. 2007; Yang et al. 2007). The effects of (−)-stepholidine on regulating various dopaminergic systems involved in the treatment of disease have also been examined. (−)-Stepholidine has been purported to display a variety of therapeutic actions including enhancing the effects of typical antipsychotics, and improvement of antipsychotic-induced dyskinesias (Mo et al. 2007). Furthermore, it has been reported that (−)-stepholidine relieves motor symptoms of Parkinson’s disease when co-administered with L-DOPA (Yang et al. 2007) or bromocriptine (Jin et al. 2002), and may slow the progression of neuronal degeneration in Parkinson’s patients (Yang et al. 2007). (−)-Stepholidine has also been reported to have antipsychotic-like effects in reducing amphetamine- and phencyclidine-induced locomotion as well as in conditioned avoidance response (Natesan et al. 2008) and prepulse inhibition paradigms (Ellenbroek et al. 2006).
With respect to the actions of (−)-stepholidine on specific dopamine receptor subtypes, all evidence to date suggests that it is an antagonist of D2-like receptors, however, since most studies were performed using behavioral outputs or ex vivo tissue assays, the effects on individual receptor subtypes could not be determined (Jin et al. 2002; Mo et al. 2007; Yang et al. 2007). The actions of (−)-stepholidine on D1-like receptors have been significantly more controversial and difficult to discern. It has variously been reported as an antagonist (Sun and Jin 1992; Xu et al. 1989; Zou et al. 1997), a partial agonist (Sun et al. 1996; Yang et al. 2007), or a full agonist (Jin et al. 2002; Mo et al. 2007; Natesan et al. 2008) at D1-like receptor responses. Behaviorally, in unilateral 6-hydroxy-dopamine lesioned rats, (−)-stepholidine was shown to induce contralateral rotation, which was attributed to its stimulation of super-sensitive D1Rs as the response was blocked by a D1-like-selective antagonist (Jin et al. 1992; Jin et al. 2002). Biochemically, the primary functional assay used to characterize (−)-stepholidine has been adenylyl cyclase stimulation, mostly using membranes prepared from brain tissue. Here, the results have also been mixed, although some studies have shown that (−)-stepholidine stimulation of cAMP production could be blocked by a D1-like-selective antagonist (Dong et al. 1997b; Jin et al. 2002). On the basis of these collective studies, it has been widely assumed that (−)-stepholidine has dual actions on dopamine receptors functioning as an agonist at D1Rs and an antagonist at D2Rs (Jin et al. 2002; Yang et al. 2007). Such a receptor profile might be particularly useful as an antipsychotic as it would block mesocortical and mesolimbic D2Rs to diminish the positive symptoms of schizophrenia (Kapur and Mamo 2003) and stimulate prefrontal cortical D1Rs to reverse disease-related decreases in cognition (Abi-Dargham et al. 2002).
Importantly, as noted above, most of the effects of (−)-stepholidine on dopamine receptors have been studied in animals or animal tissue, where the actions on specific receptor subtypes cannot be discerned. This raises the possibility that (−)-stepholidine might interact with other receptors, or signaling proteins, which are either responsible for eliciting the measured response or involved in modifying the response through indirect means. In this regard, it should be noted that (−)-stepholidine has been shown to be a potent agonist of 5-HT1A serotonin receptors (Gao et al. 2011; Mo et al. 2010). A potential further complication is that (−)-stepholidine likely interacts with both D1-like and D2-like receptors in vivo, which may produce counteracting effects. Finally, the main biochemical pathway that has been used to characterize the signaling activities of (−)-stepholidine is the adenylyl cyclase-cAMP system. However, it is now known that dopamine receptors can signal through alternate pathways, such as β-arrestin-mediated outputs (Beaulieu et al. 2009; Shenoy and Lefkowitz 2011; Urs et al. 2011). Moreover, it is now well accepted that agonist ligands can be biased to selectively activate one signaling pathway over another, even when mediated by the same receptor (Rominger et al. 2014; Urban et al. 2007; Violin et al. 2014). This phenomenon has been referred to as biased agonism or functional selectivity and has been observed for dopamine receptors (Allen et al. 2011; Chen et al. 2012; Free et al. 2014; Lane et al. 2008). This raises the possibility that some of the disparate effects observed for (−)-stepholidine could be due to biased signaling, especially within the D1-like receptor subtypes.
In this study, we have taken the approach of investigating the actions of (−)-stepholidine using homogenous dopamine receptor subtypes (D1R–D5R) expressed heterologously in cell lines. Using these defined environments, we have evaluated multiple signaling outputs, especially those mediated by the D1R. We also investigated if (−)-stepholidine would bind in an agonist-like fashion to D1-like receptors in membranes prepared from bovine brain. Using these receptor-specific signaling paradigms, as well as the brain membrane studies, we were unable to identify any agonist-like activity in response to (−)-stepholidine stimulation. These data serve to clarify the actions of (−)-stepholidine by ruling out signaling bias within dopamine receptors, and suggest that previously observed responses with (−)-stepholidine are due to pan-dopamine receptor antagonism or interactions with other receptors or signaling molecules.
Materials and methods
Materials
(−)-Stepholidine was obtained from two independent commercial suppliers, Stanford Chemical (Irvine, CA), and Biorbyt (Cambridgeshire, UK). Compounds were tested in-house via NMR to confirm purity (>99 %) and structural accuracy. Identical results were obtained with batches from both suppliers. All other chemicals were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated. All tissue culture media and components were obtained from Mediatech, Inc. (Manassas, VA).
Cell culture
HEK293T cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with a final concentration of 10 % fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin, 1 mM sodium pyruvate, and 10 μg/mL gentamicin. CHO-K1 cells were maintained in Ham’s F12 media supplemented with a final concentration of 10 % fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin, 800 μg/mL geneticin, and 300 μg/mL hygromycin. Cells were incubated at 37 °C with 5 % CO2 and 90 % humidity. They were dissociated using either calcium-free Earle’s balanced salt solution (EBSS) for HEK293T cells or Cellstripper (Corning, Manassas, VA) for CHO-K1 cells, collected by centrifugation at 900×g for 10 min, and plated mechanically.
Radioligand binding assays
Radioligand competition-binding assays were conducted with slight modifications as previously described by our laboratory (Chun et al. 2013) using the same cell lines as those used for the functional assays. HEK293 cells stably transfected with human D1R, D2R, D3R (Codex Biosolutions, Inc., Gaithersburg, MD), or CHO cells stably transfected with human D4R or D5R (DiscoveRx Inc., Fremont, CA), were dissociated from plates using calcium-free EBSS (D1R-D3R) or Cellstripper (Corning, Manassas, VA) (D4R and D5R) and collected by centrifugation at 900×g for 10 min. Cells were resuspended in 5 mM Tris–HCl and 5 mM MgCl2, pH 7.4 at 4 °C, and lysed using Dounce homogenization. Cell lysate was centrifuged at 30,000×g for 30 min and the membranes were resuspended in EBSS with calcium at pH 7.4. Resuspended membranes (4–18 μg protein depending on receptor subtype) were incubated for 90 min at room temperature with competing ligands and either 0.5 nM [3H]-SCH23390 (D1R and D5R), or 0.5 nM [3H]-methylspiperone (D2R, D3R, and D4R) in a final reaction volume of 250 μL. Non-specific binding was determined in the presence of 4 μM (+)-butaclamol. Bound ligand was separated from free by filtration through a PerkinElmer Unifilter-96 GF/C 96 well micro-plate using the PerkinElmer Unifilter-96 Harvester, followed by three 1-mL washes per well of ice-cold EBSS. After drying, 50 μL of liquid scintillation cocktail (MicroScint PS, Perkin Elmer, Waltham, MA) was added to each well, plates were sealed and analyzed on a PerkinElmer Topcount NXT™. Ki values were determined using the Cheng-Prusoff equation (Cheng and Prusoff 1973) using IC50 values from individual experiments.
β-Arrestin-2 recruitment assay
The ability of compounds to recruit β-arrestin-2 was determined using DiscoveRx PathHunter technology (DiscoveRx Inc., Fremont, CA), which exploits the enzyme complementation of fusion-tagged receptor and β-arrestin proteins. Assays were conducted with minor modifications as previously published by our laboratory (Banala et al. 2011; Free et al. 2014). Briefly, CHO-K1 cells stably expressing D1R, D2R, D3R, D4R, or D5R (DiscoveRx, Inc.) were seeded in CP2 media (DiscoveRx) at a density of 2,625 cells/well in 384-well black, clear-bottom plates. Following 24 h of incubation, the cells were treated with various concentrations of compounds in PBS and incubated at 37 °C for 90 min. DiscoveRx reagent was then added to the cells according to the manufacturer’s recommendations followed by a 30–60 min incubation at room temperature. Luminescence was measured on a Hamamatsu FDSS μCELL reader (Hamamatsu, Bridgewater, NJ). Data were collected as relative luminescence units (RLU) and subsequently normalized to a percentage of the control luminescence seen with a maximum concentration of dopamine, with zero percent being RLU seen in the absence of any compound.
DiscoveRx cAMP accumulation assay
cAMP assays were conducted using the DiscoveRx HitHunter assay kit (DiscoveRx Inc.), according to the manufacturer’s recommendations. Cells stably expressing the human D1R, D5R (HEK293, Codex Biosolutions Inc., Gaithersburg, MD), or D2R (DiscoveRx, Inc.) were seeded at a density of 5,000 cells/well in 384-well black, clear-bottom plates. After 16–24 h of incubation at 37 °C, 5 % CO2, and 90 % humidity, the media was removed and replaced with 5 μL/well PBS. Cells were treated with 2.5 μL of various concentrations of compound diluted in PBS, either alone (D1R and D5R) or in the presence (D2R) of an EC80 concentration of forskolin (100 μM) plus 0.2 mM sodium metabisulfite and incubated for 60 min at 37 °C, in 5 % CO2 and 90 % humidity. DiscoveRx HitHunter reagents were then added, and cells incubated in the dark at room temperature, according to manufacturer recommendations. Luminescence was measured on a Hamamatsu FDSS μCELL (Hamamatsu Photonics K. K., Bridgewater, NJ) for 10 s. Data were collected as RLUs and values were normalized to a percentage of the maximum forskolin-stimulated cAMP signal (D2R), or the maximum cAMP accumulation seen with dopamine stimulation (D1R, D5R).
[35S]GTPγS binding assays
Membranes were prepared from HEK293 cells stably expressing the human D1R (Codex Biosolutions Inc., Gaithersburg, MD). Cells were mechanically dissociated from plates by scraping in ice-cold lysis buffer (1 mM EDTA, 20 mM HEPES, pH 7.4), and then lysed using Dounce homogenization. Cell lysate was centrifuged at 30,000×g and the membranes were resuspended in buffer containing 20 mM HEPES, 150 mM NaCl, 10 mM MgCl2, and 1 mM EDTA, pH 7.4. Membranes were diluted to 25 μg protein per assay reaction and incubated with vehicle or dopamine and/or (−)-stepholidine with 0.1 nM [35S]GTPγS in a total volume of 100 μL for 1 h at 30 °C. Stimulated binding that was not blocked by inclusion of 4 μM (+)-butaclamol was defined as non-dopamine receptor-mediated binding and was ~15 % of total binding. Non-specific binding was determined in the presence of 10 μM cold GTPγS, and subtracted from all values. Assay reactions were filtered through a PerkinElmer Unifilter-96 GF/C 96 well micro-plate using the PerkinElmer Unifilter-96 Harvester, followed by three 1-mL washes using ice-cold 50 mM Tris–HCl, pH 7.4 containing 0.9 % NaCl. After drying, 50 μL of liquid scintillation cocktail (MicroScint PS, Perkin Elmer, Waltham, MA) was added to each well, plates were sealed, and analyzed on a PerkinElmer Topcount NXT™.
Calcium mobilization assays
D1R–D2R-mediated calcium mobilization was assessed as previously described (Chun et al. 2013). Briefly, HEK293T cells were transiently transfected with the human D1R and D2R and 24 h later the cells were replated into 384-well, optical, clear-bottom, black-walled plates at 30,000 cells/well (ThermoScientific, Waltham, MA). At 48-h post-transfection, the cells were incubated for 60 min at room temperature in the dark with Fluo-8 NW calcium dye and a quencher to block any signal from extracellular calcium (Screen Quest™ Fluo-8 NW Calcium Assay Kit, AAT Bioquest, Inc., Sunnyvale, CA), as recommended by the manufacturer. Compounds (diluted in the presence of 0.2 mM sodium metabisulfite) were then added to the plates followed by reading on a FlexStation-3 (Molecular Devices, Sunnyvale, CA) using an excitation wavelength of 488 nm and an emission wavelength of 525 nm. Data were recorded and quantified as maximum minus minimum (max−min) RFU within the assay window using SoftMax Pro Software (Molecular Devices, Sunnyvale, CA) and normalized to the maximum response seen with the control agonist dopamine.
Bovine striatal radioligand binding assays
Flash-frozen bovine corpus striata were obtained from Rockland Immunochemicals (Gilbertsville, PA). Small (~2.5 g) pieces of frozen striata were placed in ~20 mL of ice-cold lysis buffer containing 5 mM Tris–HCl and 5 mM MgCl2, pH 7.4. The tissue was disrupted using a Polytron homogenizer with two 5-s bursts, and then further lysed using a glass Dounce homogenizer with 20 strokes. The lysate was then centrifuged at 30,000×g for 10 min. Membrane pellets were resuspended in wash buffer containing 50 mM Tris–HCl, 5 mM MgCl2, pH 7.4 in a volume of 30 mL, incubated at 37 °C for 15 min, and centrifuged at 30,000×g for 10 min. The membrane pellet was resuspended in 20 mL of the wash buffer and recentrifuged as above. Following the last wash, the pellet was again suspended in a small volume of assay buffer and diluted to a protein concentration of 60 μg per reaction as measured via a Bradford protein assay (Biorad). Assays were run in assay buffer, which was identical to the wash buffer but with the addition of 50 nM ketanserin, to block the radioligand from binding to 5-HT2-like serotonin receptors, and 0.2 mM of the antioxidant sodium metabisulfite. Membranes were incubated in triplicate for 90 min at room temperature with the indicated compounds and 0.5 nM [3H]-SCH23390 in a final reaction volume of 0.5 mL. Non-specific binding was determined in the presence of 4 μM (+)-butaclamol. Bound ligand was separated from free by filtration onto pretreated (0.6 % polyethylenimine) GF/B filter membranes using a Brandel harvester (Gaithersburg, MD), followed by washing three times with 2 mL of ice-cold wash buffer. Radioactivity trapped on the filters was determined after incubation with Complete Counting Cocktail liquid solution (RPI, Mt Prospect, IL) and counting in a Beckman scintillation counter. Competition curves were analyzed using non-linear regression analysis (GraphPad Prism) and fit to either one or two binding sites. In the case of a one-site fit, the Ki values were determined using the Cheng-Prusoff equation (Cheng and Prusoff 1973) using IC50 values from individual experiments.
Results
While it has previously been demonstrated that (−)-stepholidine is capable of interacting with both D1R and D2Rs, the relative selectivity, and/or ability to bind to other subtypes of dopamine receptors is unknown outside of a limited screen by the NIMH psychoactive drug-screening program (PDSP) (Table 1). To characterize the affinity of (−)-stepholidine for all recombinant, and thus homogeneous, dopamine receptor subtypes, we conducted full displacement curves using competition-binding assays to both D1-like (D1R and D5R) and D2-like (D2R, D3R, and D4R) receptors using the D1-like- or D2-like-selective antagonists [3H]-SCH23390 or [3H]-methylspiperone, respectively. As demonstrated in Fig. 1a, (−)-stepholidine is able to fully displace radioligand binding to both the D1R and D5R. These competition curves were best fit to single binding sites with Ki values of 5.1±2.3 nM, and 5.8±3.1 nM (n=3) for the D1R and D5R respectively. Figure 1b shows competition curves to the D2-like dopamine receptors, D2R, D3R, and D4R. (−)-Stepholidine completely, and uniformly, displaced specific [3H]-methylspiperone binding to all receptors with Ki values of 11.6±4.2, 23.4±8.7, and 1,453±301 nM (n=3) for the D2R, D3R, and D4R respectively. These affinities are largely consistent with those reported in the PDSP database (Table 1), with the exception of the D2R for which we observed higher potency for (−)-stepholidine. The reasons for this are unclear, but the rank order of potency is similar. Taken together, these data indicate that (−)-stepholidine displays the highest affinity for D1-like receptors and does not differentiate between the D1R and D5R. (−)-Stepholidine also exhibits relatively high affinity for the D2R and D3R, with a slight preference for the D2R. In contrast, (−)-stepholidine exhibits relatively low affinity for the D4R.
Table 1.
Affinities of (−)-stepholidine for a variety of receptors and other drug targets as documented in the PDSP database
| Receptor/target | Ki (nM) | Receptor/target | Ki (nM) |
|---|---|---|---|
| SERT | >10,000 | M2 | >10,000 |
| 5-HT1A | 143.4 | M5 | >10,000 |
| 5-HT1B | 6,984 | D1 | 5.9 |
| 5-HT1D | >10,000 | D2 | 974.3 |
| 5-HT1E | >10,000 | D3 | 30.1 |
| 5-HT2A | >10,000 | D4 | 3,748 |
| 5-HT2B | 226 | D5 | 4.4 |
| 5-HT2C | >10,000 | DAT | >10,000 |
| 5-HT3 | >10,000 | GABA-A | >10,000 |
| 5-HT5 | >10,000 | Glutamate-NMDA | >10,000 |
| 5-HT6 | >10,000 | H1 | >10,000 |
| 5-HT7 | >10,000 | H2 | >10,000 |
| Alpha1A | >10,000 | H3 | >10,000 |
| Alpha1B | >10,000 | H4 | >10,000 |
| Alpha2A | >10,000 | Imidazoline-1 | 8,909 |
| Alpha2B | 6,884 | NET | >10,000 |
| Alpha2C | 214.5 | DOR | >10,000 |
| Beta1 | >10,000 | KOR | >10,000 |
| Beta2 | >10,000 | MOR | >10,000 |
| Calcium channel | >10,000 | Sigma-1 | 1,600 |
| CB1 | >10,000 | Sigma-2 | 53.4 |
| CB2 | >10,000 |
Affinity (Ki) determinations were obtained from the National Institute of Mental Health’s Psychoactive Drug Screening Program (PDSP). For experimental details please refer to the PDSP web site: http://pdsp.med.unc.edu/. Ki values >10,000 nM showed no or minimal displacement of an orthosteric radioligand at the highest concentration tested. Ki values between 1 and 10 μM are indicated in boldface whereas Ki values that are less than 1 μM are indicated in italics
Fig. 1.
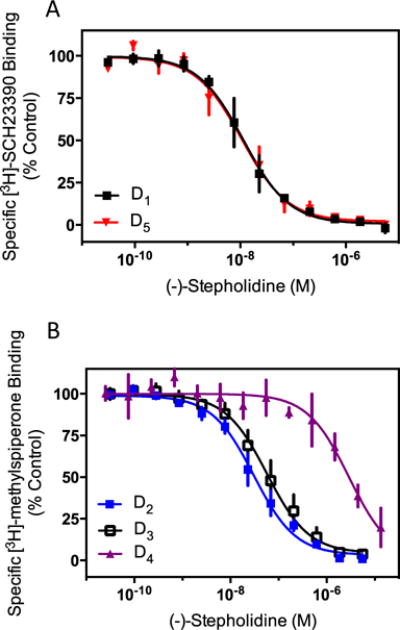
(−)-Stepholidine displays relatively high affinity for dopamine receptor subtypes. Membranes from cells stably expressing dopamine receptors were harvested as described in the “Materials and methods” section. a D1R or D5R containing membranes were incubated with the indicated concentrations of (−)-stepholidine and 0.5 nM [3H]-SCH23390. b D2R, D3R or D4R containing cell membranes were incubated with the indicated concentrations of (−)-stepholidine and 0.5 nM [3H]-methylspiperone. Radioligand concentrations were at or near the Kd of the ligand for its representative receptor as determined by independent saturation binding isotherms (data not shown). Data show means of three independent experiments performed in triplicate and expressed as a percentage of specific binding seen in the absence of any competing ligand (mean±SEM, n=3). Ki values of (−)-stepholidine were determined using the Cheng-Prusoff equation and experimentally determined IC50 values, and are given in the text
Since (−)-stepholidine has previously been suggested to have agonist activity at D1Rs in vivo (or unknowingly at D5Rs), we initially evaluated its effects in cAMP accumulation assays. Both the D1R and D5R are coupled to Gs/olf proteins that stimulate adenylyl cyclase activity and cAMP accumulation. Figure 2a shows that in cells stably transfected with the D1R, dopamine robustly stimulates cAMP accumulation. Surprisingly, (−)-stepholidine has no effect on cAMP accumulation, even when tested using a very high concentration (0.1 mM). As (−)-stepholidine has no apparent agonist activity in this assay, we examined its ability to antagonize the dopamine response. In this approach, the cells were incubated with an EC80 concentration of dopamine along with various concentrations of (−)-stepholidine, or the reference D1R antagonist, SCH23390 (Fig. 2b). As can be seen, both (−)-stepholidine and SCH23390 potently and fully antagonize dopamine stimulation of the D1R. The IC50 values were determined to be 20.5±14, and 22.3±13 nM (n=3), for SCH23390 and (−)-stepholidine, respectively. Because of the importance of these findings, we replicated these experiments using a different type of cAMP assay (HTRF) (Supplemental Fig. 1). Importantly, we observed identical results in that (−)-stepholidine did not stimulate D1R-mediated cAMP accumulation, but potently and fully blocked the dopamine-stimulated cAMP response. The IC50 value for (−)-stepholidine in this assay was determined to be 58±19 nM (n=4), a close approximation to that determined in Fig. 2b. Taken together, these results indicate that (−)-stepholidine does not stimulate D1R-mediated cAMP accumulation, but rather antagonizes this response.
Fig. 2.
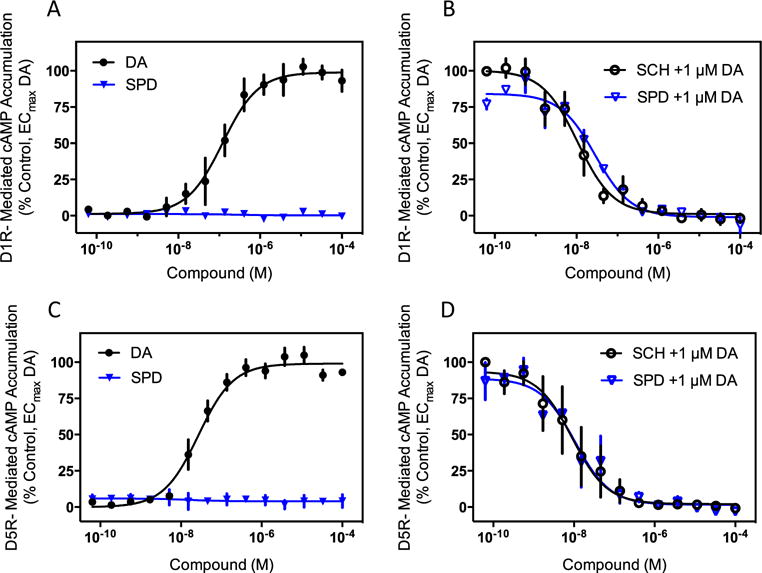
(−)-Stepholidine is an antagonist of dopamine-mediated G protein activation at D1-like receptors. a HEK293 cells stably expressing the D1R were assayed for cAMP accumulation using the DiscoveRx HitHunter cAMP assay as described in the “Materials and methods” section. Cells were incubated with the indicated concentrations of dopamine (DA) or (−)-stepholidine (SPD) to determine agonist activity. No measurable agonist activity was seen with (−)-stepholidine whereas dopamine gave a robust response (EC50=0.5±0.3 μM, n=3). b Cells were incubated with the indicated concentrations of (−)-stepholidine, or the reference D1R antagonist SCH23390 (SCH), in the presence of an EC80 concentration of dopamine (1 μM) to determine antagonist activity. IC50 values were 20.5±14 nM, and 22.3±13 nM, for SCH23390 and (−)-stepholidine, respectively. Data are normalized to the percentage of stimulation seen with the EC80 concentration of dopamine and are means±SEM of three independent experiments, each performed in triplicate. c HEK293 cells stably expressing the D5R were assayed for cAMP accumulation using the DiscoveRx cAMP assay as described above. Cells were incubated with the indicated concentrations of dopamine or (−)-stepholidine to determine agonist activity. No measurable agonist activity was seen with (−)-stepholidine while dopamine gave a robust agonist response (EC50=27.3±0.7 nM, n=3). d Cells were incubated with the indicated concentrations of (−)-stepholidine, or the reference D5R antagonist SCH23390 (SCH), in the presence of an EC80 concentration of dopamine (1 μM) to determine antagonist activity. IC50 values were 20.7±16.6 nM, and 27.1±22 nM, for SCH23390 and (−)-stepholidine, respectively. Data are expressed as described in panel b and are means±SEM of three independent experiments, each performed in triplicate
Similar results were obtained for the D5R, which is also coupled to stimulation of cAMP accumulation. Figure 2c shows that, when cells expressing the D5R were stimulated with (−)-stepholidine, it failed to produce any measurable cAMP accumulation, despite the observation of a robust dopamine response. In contrast, when evaluated as an antagonist, (−)-stepholidine blocked dopamine stimulation of the D5R in a manner similar to that of the reference antagonist, SCH23390 (Fig. 2d). The IC50 values for (−)-stepholidine and SCH23390 were 27.1±22 and 20.7±16.6 nM (n=3), respectively. Notably, (−)-stepholidine’s potency for inhibiting the D5R is essentially identical to that for inhibiting the D1R (22.3±13 nM), which agrees with its equivalent potency for inhibiting D1R and D5R radioligand binding (Fig. 1). Taken together, these results indicate that (−)-stepholidine is not an agonist at D1-like receptor-mediated cAMP accumulation, but instead functions as an antagonist of these responses.
Since (−)-stepholidine has been reported to act as an antagonist of the D2R in animal experimentation, we wished to evaluate its activity at the D2R in vitro. The D2R couples to Gi/o proteins that inhibit adenylyl cyclase and reduce intracellular of cAMP levels (Beaulieu and Gainetdinov 2011). Activating adenylyl cyclase with a stimulator such as forskolin and then examining inhibition of cAMP accumulation can easily test this. Figure 3a shows that in cells stably transfected with the D2R, dopamine potently and fully suppresses forskolin-stimulated cAMP accumulation. In contrast, (−)-stepholidine has no effect on inhibiting cAMP accumulation even when tested at very high concentrations. To examine the ability of (−)-stepholidine to antagonize this response, the cells were incubated with forskolin plus an EC80 concentration (1 μM) of dopamine to inhibit the forskolin activity, and either (−)-stepholidine or the reference D2R antagonist sulpiride. Both sulpiride and (−)-stepholidine potently and completely reverse dopamine’s ability to inhibit forskolin-stimulated cAMP accumulation (Fig. 3b). The IC50 values were 128±57 and 36.5±2.1 nM (n=3) for sulpiride and (−)-stepholidine, respectively. These data confirm the notion that (−)-stepholidine acts an antagonist of the D2R.
Fig. 3.
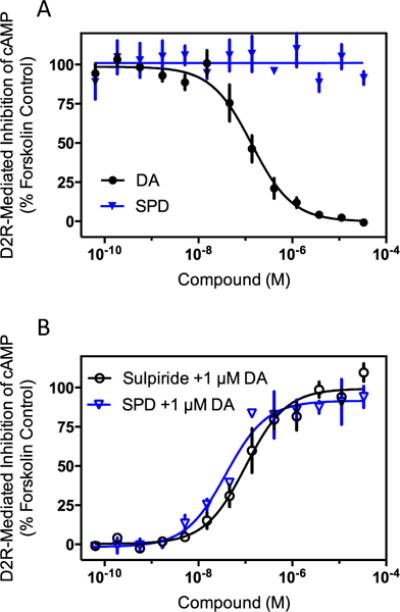
(−)-Stepholidine is an antagonist of dopamine-mediated G protein signaling at the D2R. a CHO-K1 cells stably expressing the D2R were assayed for agonist inhibition of forskolin-stimulated cAMP accumulation as described in the “Materials and methods” section. Cells were stimulated with the indicated concentrations of dopamine (DA) or (−)-stepholidine (SPD) resulting in an EC50 of 150±43 nM, n=4, for dopamine, whereas (−)-stepholidine failed to produce any measurable agonist response. b For antagonist-mode assays, cells were stimulated with 100 μM forskolin, as above, along with an EC80 concentration (1 μM) of dopamine to inhibit the forskolin activity. The cells were then incubated with the indicated concentrations of the D2R reference antagonist sulpiride, or (−)-stepholidine. IC50 values were 128±57 nM, and 36.5±2.1 nM, for sulpiride and (−)-stepholidine, respectively (mean±SEM, n=4). Data are expressed as a percentage of the stimulation seen with 100 μM forskolin alone, and are average curves of four independent experiments each performed in triplicate
Given our negative results concerning the ability of (−)-stepholidine to stimulate D1R-mediated cAMP accumulation, we wished to further evaluate the activity of (−)-stepholidine on the D1R. In order to do this, we initially performed [35S]GTPγS binding assays in membranes prepared from cells expressing the D1R. This assay directly measures receptor-mediated G protein activation that occurs when GTP binds to the Gα subunit of the heterotrimeric G protein causing subunit dissociation and interaction with downstream effectors (Strange 2010). Use of the non-hydrolyzable GTP analog, [35S]GTPγS, allows for the activated Gα subunit to accumulate and be quantified using a radioligand binding assay paradigm. Notably, this assay will detect activation of any G protein irrespective of the downstream second messenger. Figure 4a shows that dopamine stimulation of D1R-containing membranes results in robust [35S]GTPγS binding with an EC50 of 2.2±0.6 μM, n=3. However, unlike dopamine, (−)-stepholidine failed to produce any measurable [35S]GTPγS binding to the membranes. (−)-Stepholidine was also tested as an antagonist of D1R-mediated [35S]GTPγS binding by stimulating the membranes with an EC80 concentration of dopamine and then measuring the ability of either (−)-stepholidine or SCH23390 to block the binding (Fig. 4b). In concurrence with our cAMP accumulation data, (−)-stepholidine was a potent and full antagonist of dopamine-stimulated [35S]GTPγS binding exhibiting an IC50 of 16.5±8.7 nM, n=3. These findings suggest that (−)-stepholidine is unable to activate G proteins coupled to the D1R, but instead acts as an antagonist of the D1R in vitro.
Fig. 4.
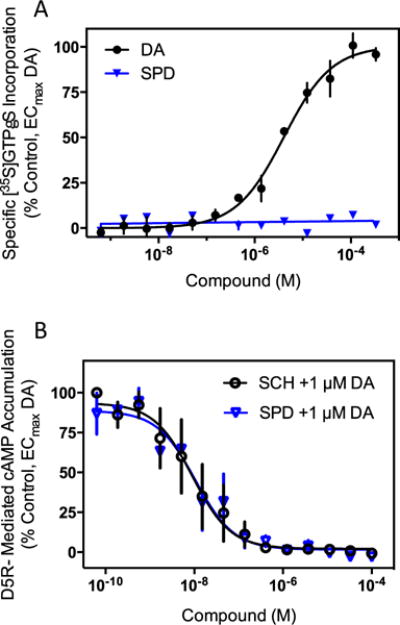
(−)-Stepholidine is an antagonist of D1R-mediated [35S]GTPγS binding to G proteins. Membranes from HEK293 cells stably expressing the D1R were harvested and assayed for receptor-mediated GTPγS binding using [35S]GTPγS incorporation as described in the “Materials and methods” section. a To measure agonist activity, membranes were incubated with the indicated concentrations of dopamine or (−)-stepholidine (SPD). Dopamine gave an agonist response with an EC50 of 2.2±0.6 μM (mean ± SEM, n=3), while (−)-stepholidine showed no measurable agonist activity. b To measure antagonist activity, membranes were incubated with the indicated concentrations of (−)-stepholidine, or the reference D1R antagonist SCH23390 (SCH), in the presence of an EC80 (10 μM) concentration of dopamine. (−)-Stepholidine acted as a full antagonist with an IC50 of 16.5±8.7 nM (n=3), whereas SCH23390 exhibited an IC50 value of 4.3±0.4 nM (n=3)
While canonical signaling pathways for dopamine receptor activation have been thought to be mediated by G proteins, a more recently characterized pathway, which is G protein-independent, occurs through agonist recruitment of β-arrestins to the receptors. β-arrestins can act as scaffolding proteins that engender signaling by various downstream effector proteins (Luttrell and Gesty-Palmer 2010; Shenoy and Lefkowitz 2011). In fact, Caron and colleagues have argued that inhibition of β-arrestin-2-mediated signaling in D2R-expressing neurons is correlated with antipsychotic properties (Masri et al. 2008; Urs et al. 2011; 2012). Moreover, agonists have been identified that are functionally selective, or biased, for activating either G protein- or β-arrestin-2-mediated signaling by the D2R (Allen et al. 2011; Chen et al. 2012; Free et al. 2014). We therefore thought that it would be important to characterize the activity of (−)-stepholidine on the recruitment of β-arrestin-2 to all dopamine receptor subtypes to determine if it exhibits any agonist activity, or functionally selective properties. We found that dopamine produced robust β-arrestin-2 recruitment to all dopamine receptors (shown in Fig. 5a for the D3R, data not shown for other subtypes). Conversely, (−)-stepholidine failed to produce any measurable recruitment of β-arrestin-2 to any dopamine receptor subtype (Fig. 5a), despite using concentrations up to 100 μM, well in excess of its Ki values determined for each receptor subtype (Fig. 1).
Fig. 5.
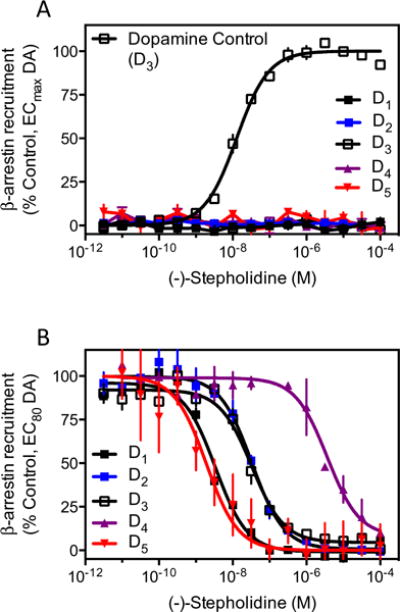
(−)-Stepholidine is an antagonist of dopamine-stimulated recruitment of β-arrestin-2 to all dopamine receptor subtypes. a Cells stably expressing each receptor subtype were assayed for agonist-stimulated β-arrestin-2 recruitment using the DiscoveRx PathHunter assay as described in the “Materials and methods” section. While all subtypes displayed robust dopamine (DA)-stimulated recruitment (data not shown), only the dopamine response for the D3R is shown for a comparative control. (−)-Stepholidine failed to stimulate any measurable recruitment of β-arrestin-2 to any of the dopamine receptor subtypes. Data are expressed as the percentage of the maximum response seen with dopamine for each individual receptor subtype and are mean values of four independent experiments performed in triplicate. b The cells were assayed for (−)-stepholidine-mediated inhibition of dopamine-stimulated β-arrestin-2 recruitment to each receptor subtype. Cells were incubated with the indicated concentrations of (−)-stepholidine followed by stimulation with an EC80 concentration of dopamine. Data are expressed as a normalized percentage of the response to an EC80 concentration of dopamine for each individual subtype of receptor and are mean values from four independent experiments. Mean IC50 values are given in the text
In contrast, when (−)-stepholidine was examined for its ability to inhibit dopamine-stimulated β-arrestin-2 recruitment, it was determined that it could fully antagonize this activity for all receptor subtypes (Fig. 5b). IC50 values for (−)-stepholidine were calculated using non-linear regression analysis of individual dose-response curves and were determined to be: D1R (4.5±1.7 nM), D2R (32.7±3.3 nM), D3R (77.7±27.8 nM), D4R (4,075±1,461 nM), and D5R (3.7±1.6 nM), expressed as mean±SEM (n=3). These values are similar to those observed for (−)-stepholidine inhibition of radioligand binding (see above) and exhibit an identical rank order of potency where D1R=D5R>D2R≥D3R>D4R. Taken together, these findings indicate that (−)-stepholidine is a relatively potent (nM) antagonist of β-arrestin-2 recruitment to all subtypes of dopamine receptors, with the exception of the D4R where it displays micromolar potency. These data suggest that (−)-stepholidine does not function as an agonist of β-arrestin-2 recruitment to any dopamine receptor subtype, but rather antagonizes dopamine stimulation of this signaling pathway.
Given the above observations, we considered other potential D1R-mediated signaling pathways where (−)-stepholidine might exhibit agonist activity. Notably, previous studies have suggested that the D1R can signal as part of a D1R–D2R heteromer complex that leads to Gq and phospholipase C (PLC) activation and intracellular Ca2+ mobilization when stimulated (Pei et al. 2010; Perreault et al. 2014; Rashid et al. 2007). Recently, however, we have shown that the mechanisms of D1R-D2R-mediated Ca2+ mobilization are more complex and multi-faceted (Chun et al. 2013). Nonetheless, we examined the effects of (−)-stepholidine on Ca2+ mobilization in D1R and D2R co-transfected cells. As seen in Fig. 6a, cells transfected with both receptors gave a robust Ca2+ response upon the addition of dopamine (EC50=32.3±13.2 nM, n=3). No response was observed in non-transfected cells or in cells transfected with only one receptor subtype ((Chun et al. 2013), and data not shown). However, when D1R and D2R co-transfected cells were stimulated with (−)-stepholidine at concentrations up to 50 μM, no Ca2+ mobilization was detected, indicating that (−)-stepholidine is devoid of agonist activity at D1R–D2R heteromer-mediated signaling. (−)-Stepholidine was also evaluated for its ability to inhibit the Ca2+ response using D1R and D2R co-transfected cells that were stimulated with an EC80 concentration of dopamine (Fig. 6b). As can be seen, (−)-stepholidine was able to fully inhibit the Ca2+ response with relatively high potency (IC50=19.3±3.7 nM, n=3). These results suggest that any effects (−)-stepholidine might have on D1R–D2R heteromer-mediated signaling in vivo would be antagonistic in nature.
Fig. 6.
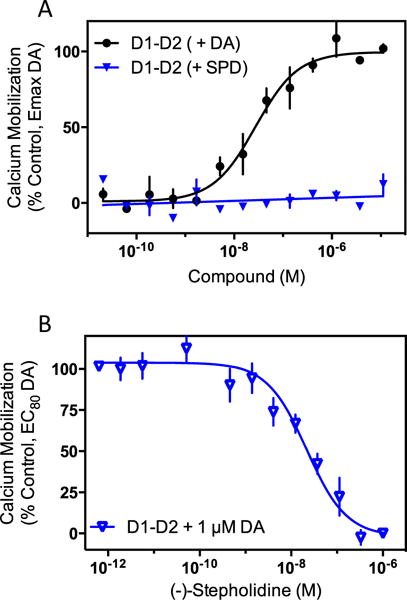
(−)-Stepholidine is an antagonist of dopamine-stimulated D1RD2R dimer-dependent intracellular calcium mobilization. a HEK293T cells were transiently transfected with D1R and D2R and then tested for calcium mobilization using a live cell-based assay as described in the “Materials and methods” section. Dopamine (DA) stimulated intracellular calcium mobilization (EC50=32.3±13.2 nM, n=3), whereas (−)-stepholidine (SPD) did not produce a calcium response. Neither compound produced a response in non-transfected, or in D1R- or D2R-only transfected cells (data not shown). Data are expressed as the % maximum dopamine response. b The same cells were assayed for inhibition of dopamine-stimulated calcium mobilization using an EC80 (1 μM) concentration of dopamine. (−)-Stepholidine inhibited dopamine-stimulated calcium mobilization in cells co-transfected with the D1R and D2R (IC50=19.3±3.7 nM, n=3). Data are means of three independent experiments and are expressed the as the percent stimulation of the EC80 concentration of dopamine
Our current data using heterologous expression systems and multiple signaling outputs have not revealed agonist activity of (−)-stepholidine at any dopamine receptor subtype. This contrasts with previously hypothesized agonist effects of (−)-stepholidine at D1-like receptors, mostly in vivo (Dong et al. 1997a; Jin et al. 2002; Natesan et al. 2008; Yang et al. 2007). Conceivably, the D1R might couple to some unknown factor in vivo that would alter the functional efficacy of (−)-stepholidine such that it would interact with the receptor as an agonist. In order to test this possibility, we examined the ability of (−)-stepholidine to directly interact with D1-like receptors using endogenous receptor-expressing brain tissue. Previous experiments using membranes prepared from corpus striatum and radiolabeled D1-selective antagonists showed that dopamine and other agonists compete for binding in a complex fashion indicative of high and low affinity agonist binding states (Hess et al. 1986; Leff et al. 1985; Schulz et al. 1985) where the high affinity state represents the D1R-Gs complex. However, if GTP (or GTP analogs) is included in the assay, the ability of agonists to compete for binding is diminished, mainly due to a reduction in the quantity of the D1R-Gs complex and/or the affinity of agonists for it (Hess et al. 1986; Leff et al. 1985; Schulz et al. 1985). In contrast, GTP does not affect the competition of antagonist ligands for binding to the D1-like receptors. Using this paradigm, we characterized the ability of (−)-stepholidine to interact with the D1-like receptors in bovine striatal membranes. Figure 7a shows that dopamine displacement of the D1-like receptor-selective radioligand [3H]-SCH23390 exhibits two sites of high and low affinity with Ki values of 5.3±1.7 nM and 1.6±0.4 μM, respectively. The high affinity site represents ~40 % percent of the total specific binding. In the presence of GTPγS, however, the dopamine curve is shifted significantly to the right (Fig. 7a), still exhibiting two binding sites, but the high affinity site is diminished to ~19 % of the total binding and the Ki values are decreased to 10.6±7 nM (high) and 5.6±0.1 μM (low). In contrast, (−)-stepholidine competition for [3H]-SCH23390 binding to D1-like receptors in the membranes is unaffected by GTPγS and exhibits only a single binding site in the absence (Ki=16.0±2.4 nM) or presence (Ki=19.2±4.4 nM) of GTPγS (Fig. 7b). These data indicate that (−)-stepholidine does not interact with D1-like receptors expressed in brain membranes in an agonist-specific fashion, but rather appears to exhibit antagonist-like properties.
Fig. 7.
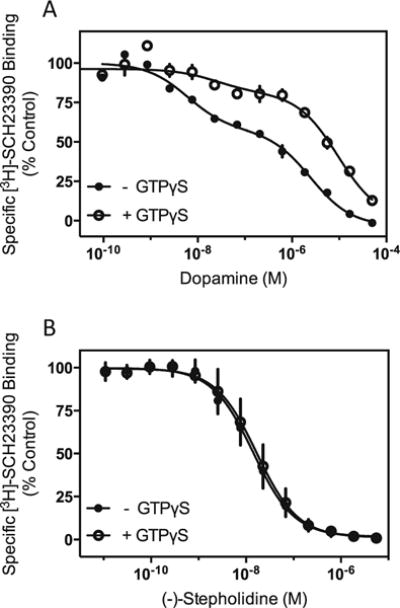
(−)-Stepholidine behaves like an antagonist in bovine striatal D1-like receptor radioligand binding assays. Competition-binding assays using the D1-like receptor-selective antagonist, [3H]-SCH23390 were conducted in neuronal membranes isolated from bovine striatum, as described in the “Materials and methods” section. a Dopamine competition curves in the absence or presence of 1 mM GTPγS. As determined using non-linear regression analysis, the control curve is best explained by two binding sites of high and low affinity with the relative proportions: 39±0.1 %RH and 61±0.1 %RL. The Ki values for the high and low affinity sites are: KH=5.3±1.7 nM and KL=1.6±0.4 μM, respectively. In the presence of GTPγS, the dopamine competition curve is also best fit to two sites, but with the following parameters: 19±0.1 %RH and 81±0.1 %RL, KH=10.6±7 nM and KL=5.6±0.1 μM. The experiment shown is representative of two, each performed in triplicate. b (−)-Stepholidine (SPD) competition curves in the absence or presence of 1 mM GTPγS. Non-linear regression analysis indicates that a one-site fit best explains the data for each curve. The Ki values are 16.0±2.4 nM for control and 19.2±4.4 nM for + GTPγS condition. The experiments shown are representative of three, each performed in triplicate
Discussion
The Chinese herbal extract (−)-stepholidine has been purported to exhibit a number of clinical effects including antipsychotic-like activity and reduction of L-DOPA induced dyskinesias, commonly seen with Parkinson’s disease treatment. Despite these intriguing in vivo effects, and clear links to dopaminergic pathways, a complete pharmacological characterization of this compound on all dopamine receptor subtypes has been lacking. This study now represents the first report of (−)-stepholidine interactions with all dopamine receptor subtypes and the first time that its activity on β-arrestin signaling has been described. Using defined, heterologously expressed receptor subtypes, we found that (−)-stepholidine has the highest affinity for the D1R and D5R receptor subtypes. Moreover, it does not distinguish between these receptors exhibiting equal affinity for both, suggesting that (−)-stepholidine will bind to both D1-like receptors when administered to animals. Similarly, (−)-stepholidine exhibited high nanomolar affinity for the D2R and D3R subtypes showing potency values that were only two- to four-fold lower than those for the D1R and D5R. In contrast, (−)-stepholidine exhibited much lower affinity, in the micromolar range, for the D4R. Taken together, our results suggest that, when administered to animals using behaviorally active doses, (−)-stepholidine likely binds to the D1R, D2R, D3R, and D5R, but not the D4R.
While current dogma mostly classifies (−)-stepholidine as a D1R agonist and a D2R antagonist, there are significant gaps in our understanding of this suggested signaling profile. Firstly, any purported effects on the D1R might also include the D5R. Similarly, any actions at the D2R could also be due to D3R blockade. Furthermore, its classification on D1R has been unclear possibly due to model systems, receptor preparations, or assays (for example, Sun and Jin 1992; Xu et al. 1989; Zou et al. 1997 vs Jin et al. 2002; Mo et al. 2007; Natesan et al. 2008). For these reasons, we re-examined the functional activity of (−)-stepholidine in defined homogeneous receptor expressing systems. We also wished to see if (−)-stepholidine might exhibit functionally selective, or biased signaling properties by evaluating its activity for signaling through a non-G protein-mediated pathway, namely β-arrestin recruitment. As expected, (−)-stepholidine exhibited full antagonist activity at the D2R in both cAMP and β-arrestin assays. Using the β-arrestin-2 recruitment assay, we also showed that (−)-stepholidine functionally antagonized both the D3R and D4R, where the potency for the D3R was similar to that of the D2R while that for the D4R was much lower, consistent with the radioligand binding experiments. These results are consistent with all prior data suggesting that (−)-stepholidine is an antagonist of D2-like receptors.
With respect to (−)-stepholidine’s activity on D1-like receptors, we examined a number of different functional outputs. Firstly, we evaluated its ability to stimulate cAMP production in either D1R or D5R expressing cell lines. Despite robust cAMP accumulation by dopamine, (−)-stepholidine was ineffective as an agonist in this assay using either receptor subtype. Instead, (−)-stepholidine functioned as a potent antagonist—consistent with its demonstrated high affinity for both D1Rs and D5Rs. Since these results appeared to conflict with some previous studies (Dong et al. 1997a; Dong et al. 1997b; Jin et al. 2002; Natesan et al. 2008), we repeated these experiments using a different type of cAMP assay. Nonetheless, (−)-stepholidine was ineffective in activating the D1R with respect to this signaling output. Given these results, we decided to investigate G protein activation directly, irrespective of any downstream second messengers, using [35S]GTPγS binding. Consistent with the cAMP data, we found that (−)-stepholidine was unable to stimulate [35S]GTPγS binding in D1R expressing cells. In contrast, (−)-stepholidine antagonized dopamine-stimulated [35S]GTPγS binding. Taken together, these results suggest that (−)-stepholidine does not directly activate G protein-mediated signaling through D1-like receptors.
Since most prior studies that functionally classified (−)-stepholidine as a D1-like receptor agonist involved in vivo experimentation, where non-cAMP-mediated signaling could be operative, we decided to investigate if (−)-stepholidine might exhibit functionally selective properties and show agonist activity in other D1R/D5R signaling assays. Importantly, since the D1R has been suggested to also signal through β-arrestin-mediated pathways (Urs et al. 2011), we examined (−)-stepholidine’s activity for recruiting β-arrestin-2 to the D1R or D5R. However, as with the G protein-mediated assays, we found that (−)-stepholidine was ineffective in stimulating D1R- or D5R-mediated β-arrestin-2 recruitment. Rather, (−)-stepholidine was a potent antagonist of this response for both of the D1-like receptors. These results, as well as those with the D2-like receptors (see above) suggest that (−)-stepholidine is not a biased agonist at β-arrestin-mediated signaling.
There have been numerous reports in the literature that D1-like receptors can couple to PLC activation, phosphotidylinositol turnover and Ca2+ mobilization—a Gq-mediated signaling pathway (Perreault et al. 2014; Rashid et al. 2007). It has been further hypothesized that this activity is due to D1R–D2R heteromerization where the dimer unit switches G protein coupling specificity from Gs (D1R) and Gi (D2R) to Gq (Perreault et al. 2014; Rashid et al. 2007). We have partially replicated these findings in that co-expression of D1R and D2R in cells engenders dopamine-stimulated Ca2+ mobilization while expression of either receptor alone does not (Chun et al. 2013). However, we also found that this response was largely blocked by specific toxins affecting the activities of Gs or Gi, thus suggesting that the mechanisms underlying D1R–D2R-mediated Ca2+ mobilization are more complex than Gq activation alone. Nonetheless, this response could represent another D1R-mediated signaling pathway where (−)-stepholidine could exhibit agonist activity. We thus tested (−)-stepholidine’s activity on the Ca2+ response in the D1R and D2R co-expressing cells. However, as with the other signaling assays, (−)-stepholidine was devoid of agonist activity, yet potently inhibited the Ca2+ response elicited by dopamine. Thus, despite the mechanisms involved, (−)-stepholidine is not an agonist of D1R-mediated PLC/Ca2+ signaling.
Despite the negative results in our survey of D1R-mediated signaling pathways, we considered the possibility that the receptor might be coupled with some unknown protein or factor that would enable (−)-stepholidine to produce an active signaling state of the D1R. While dopamine receptors are known to exist in complexes of multiple proteins (Free et al. 2007), to our knowledge, such a mechanism would be unprecedented. We therefore decided to directly examine D1-like receptor binding in membranes derived from brain tissue to see if (−)-stepholidine could produce an active binding state. This would be detected as a high affinity agonist binding site in competition curves of radiolabeled antagonist binding to the D1R (Dong et al. 1997a; Hess et al. 1986; Leff et al. 1985; Schulz et al. 1985). We observed high and low affinity agonist binding states with dopamine competition curves of [3H]-SCH23390 binding in bovine striatal membranes, in agreement with previous observations (Hess et al. 1986; Leff et al. 1985; Schulz et al. 1985). In addition, the binding of dopamine was sensitive to guanine nucleotides as expected for a G protein-coupled state of the receptor (Skinbjerg et al. 2010). In contrast, (−)-stepholidine exhibited a uniphasic, one site/state competition-binding curve that was insensitive to guanine nucleotides. These data indicated that (−)-stepholidine is incapable of producing an active G protein-coupled state of the D1R in endogenous expressing brain tissue.
Our characterization of the interactions of (−)-stepholidine with dopamine receptor subtypes suggests that it is a pan-dopamine receptor antagonist without agonist efficacy in a number of different signaling pathways. While this agrees with prior observations with respect to D2-like receptors, it conflicts with some (but not all) reports purporting agonist activity at D1-like receptors. The reasons for this are unclear, however, some possibilities exist. As discussed above, there could be an in vivo-specific signaling pathway involving the D1R or D5R that we have not examined. For instance, the D1R has been proposed to oligomerize with several other GPCRs including the D3R, histamine H3, adenosine A1, prostaglandin E1, and also the non-GPCR sigma1 and NMDA receptors (Ehrlich et al. 2013; Moreno et al. 2014; Perreault et al. 2014). However, no unique signaling pathway has been proposed for any of these heteromeric receptor complexes, nor, with the exception of the D1R-NMDA receptor complex (Wang et al. 2012), has their existence been widely confirmed.
A more likely explanation may involve (−)-stepholidine’s ability to interact with other receptors and signaling proteins. (−)-Stepholidine was found to bind to 5-HT1A and 5-HT2B serotonin, alpha2C adrenergic, and sigma-2 receptors with relatively high affinities (Ki values=143.4, 226, 214, and 53.4 nM, respectively, Table 1). Depending on the administered dose of (−)-stepholidine, it is certainly possible that it could interact with any or all of these receptor subtypes (or others that have not been identified). The functional profile of (−)-stepholidine on these other receptors has not been studied in detail, with the exception of the 5-HT1A receptor where (−)-stepholidine was shown to act as a partial agonist (Gao et al. 2011; Guo et al. 2009). Notably, it has been suggested that the difference in therapeutic-like responses between (−)-stepholidine and other atypical antipsychotic drugs may involve both D2R antagonism and partial agonist activity at 5-HT1A receptors (Gao et al. 2011; Mo et al. 2010). Interestingly, (−)-stepholidine also exhibits relatively high affinity for the 5-HT2B serotonin receptor and, while its functional actions on this receptor are not clear, chronic stimulation of the 5-HT2B receptor is known to lead to cardiac valve disease (Hutcheson et al. 2011; Setola et al. 2005). It would thus be important to characterize (−)-stepholidine’s functional effects on the 5-HT2B receptor before further consideration of its clinical use. Interestingly, (−)-stepholidine has also been shown to block Ca2+ channels (Yang et al. 2007). Conceivably, (−)-stepholidine’s interactions with other neurotransmitter receptors or channels in vivo could affect neuronal circuitry, particularly in abnormal (i.e., lesioned) brain states (Sun et al. 1996; Zou et al. 1997), that might mimic activation of D1-like receptors as observed using behavioral outputs.
Another important consideration is the potential impact of (−)-stepholidine metabolism in vivo and the possibility of active metabolites. To our knowledge, other than the report of Sun et al. (2009), the metabolism of (−)-stepholidine has not been extensively studied. In the study of Sun et al. (2009), (−)-stepholidine was found to be rapidly metabolized within minutes of administration; however, the activities of the identified metabolites on dopamine receptors were not investigated. Also noteworthy is the observation that small changes in the stepholidine scaffold can dramatically affect its activity at dopamine receptors (Mo et al. 2007). It is thus conceivable that one or more of the metabolites of (−)-stepholidine is active in stimulating some aspect of D1-like receptor signaling. Clearly, this will require further investigation.
In conclusion, using homogeneous receptor subtypes in defined in vitro expression systems, we have clarified the functional activities of (−)-stepholidine on all dopamine receptor subtypes. (−)-Stepholidine was found to be a pan-dopamine receptor antagonist with particularly high affinity at the D1R, D2R, D3R, and D5R subtypes. This agrees with and confirms previous reports of (−)-stepholidine acting as a D2-like receptor antagonist in vivo and further shows that either the D2R or D3R, or both could mediate these activities. In contrast, our results conflict with some previous assertions that (−)-stepholidine exhibits agonist activity at D1-like receptors, especially in vivo. This study provides clarity on this point, however, and suggests that the primary activity of (−)-stepholidine on all dopamine receptors is to block their signaling.
Supplementary Material
Acknowledgments
This work was supported, in part, by the Intramural Research Program of the National Institute of Neurological Disorders and Stroke (NINDS). The authors would like to thank Dr. Peter W. Dematteo (NIDA) for optical rotation analysis and NMR confirmation of (−)stepholidine batches, and Mr. Bryce Adams for technical assistance.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00213-014-3726-8) contains supplementary material, which is available to authorized users.
All authors declare no conflicts of interest.
Contributor Information
Julie A. Meade, Molecular Neuropharmacology Section, National Institute of Neurological Disorders and Stroke, National Institutes of Health, 5625 Fishers Lane, Room 4S-04, Bethesda, MD 20892-9405, USA
R. Benjamin Free, Molecular Neuropharmacology Section, National Institute of Neurological Disorders and Stroke, National Institutes of Health, 5625 Fishers Lane, Room 4S-04, Bethesda, MD 20892-9405, USA.
Nicole R. Miller, Molecular Neuropharmacology Section, National Institute of Neurological Disorders and Stroke, National Institutes of Health, 5625 Fishers Lane, Room 4S-04, Bethesda, MD 20892-9405, USA
Lani S. Chun, Molecular Neuropharmacology Section, National Institute of Neurological Disorders and Stroke, National Institutes of Health, 5625 Fishers Lane, Room 4S-04, Bethesda, MD 20892-9405, USA Cell, Molecular, Developmental Biology & Biophysics Program, Johns Hopkins University, Baltimore, MD, USA.
Trevor B. Doyle, Molecular Neuropharmacology Section, National Institute of Neurological Disorders and Stroke, National Institutes of Health, 5625 Fishers Lane, Room 4S-04, Bethesda, MD 20892-9405, USA Department of Medicinal Chemistry and Molecular Pharmacology, Purdue University, West Lafayette, IN, USA.
Amy E. Moritz, Molecular Neuropharmacology Section, National Institute of Neurological Disorders and Stroke, National Institutes of Health, 5625 Fishers Lane, Room 4S-04, Bethesda, MD 20892-9405, USA
Jennie L. Conroy, Molecular Neuropharmacology Section, National Institute of Neurological Disorders and Stroke, National Institutes of Health, 5625 Fishers Lane, Room 4S-04, Bethesda, MD 20892-9405, USA
Val J. Watts, Department of Medicinal Chemistry and Molecular Pharmacology, Purdue University, West Lafayette, IN, USA
David R. Sibley, Molecular Neuropharmacology Section, National Institute of Neurological Disorders and Stroke, National Institutes of Health, 5625 Fishers Lane, Room 4S-04, Bethesda, MD 20892-9405, USA
References
- Abi-Dargham A, Mawlawi O, Lombardo I, Gil R, Martinez D, Huang Y, Hwang DR, Keilp J, Kochan L, Van Heertum R, Gorman JM, Laruelle M. Prefrontal dopamine D1 receptors and working memory in schizophrenia. J Neurosci. 2002;22:3708–3719. doi: 10.1523/JNEUROSCI.22-09-03708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JA, Yost JM, Setola V, Chen X, Sassano MF, Chen M, Peterson S, Yadav PN, Huang XP, Feng B, Jensen NH, Che X, Bai X, Frye SV, Wetsel WC, Caron MG, Javitch JA, Roth BL, Jin J. Discovery of beta-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc Natl Acad Sci U S A. 2011;108:18488–18493. doi: 10.1073/pnas.1104807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banala AK, Levy BA, Khatri SS, Furman CA, Roof RA, Mishra Y, Griffin SA, Sibley DR, Luedtke RR, Newman AH. N-(3-fluoro-4-(4-(2-methoxy or 2,3-dichlorophenyl)piperazine-1-yl)butyl)arylcarboxamides as selective dopamine D3 receptor ligands: critical role of the carboxamide linker for D3 receptor selectivity. J Med Chem. 2011;54:3581–3594. doi: 10.1021/jm200288r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR, Caron MG. Akt/GSK3 signaling in the action of psychotropic drugs. Annu Rev Pharmacol Toxicol. 2009;49:327–347. doi: 10.1146/annurev.pharmtox.011008.145634. [DOI] [PubMed] [Google Scholar]
- Chen X, Sassano MF, Zheng L, Setola V, Chen M, Bai X, Frye SV, Wetsel WC, Roth BL, Jin J. Structure-functional selectivity relationship studies of beta-arrestin-biased dopamine D(2) receptor agonists. J Med Chem. 2012;55:7141–7153. doi: 10.1021/jm300603y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Chun LS, Free RB, Doyle TB, Huang XP, Rankin ML, Sibley DR. D1–D2 dopamine receptor synergy promotes calcium signaling via multiple mechanisms. Mol Pharmacol. 2013;84:190–200. doi: 10.1124/mol.113.085175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong ZJ, Chen LJ, Jin GZ, Creese I. GTP regulation of (−)-stepholidine binding to R(H) of D1 dopamine receptors in calf striatum. Biochem Pharmacol. 1997a;54:227–232. doi: 10.1016/s0006-2952(97)00152-4. [DOI] [PubMed] [Google Scholar]
- Dong ZJ, Guo X, Chen LJ, Han YF, Jin GZ. Dual actions of (−)-stepholidine on the dopamine receptor-mediated adenylate cyclase activity in rat corpus striatum. Life Sci. 1997b;61:465–472. doi: 10.1016/s0024-3205(97)00404-9. [DOI] [PubMed] [Google Scholar]
- Ehrlich AT, Furuyashiki T, Kitaoka S, Kakizuka A, Narumiya S. Prostaglandin E receptor EP1 forms a complex with dopamine D1 receptor and directs D1-induced cAMP production to adenylyl cyclase 7 through mobilizing G(betagamma) subunits in human embryonic kidney 293 T cells. Mol Pharmacol. 2013;84:476–486. doi: 10.1124/mol.113.087288. [DOI] [PubMed] [Google Scholar]
- Ellenbroek BA, Zhang XX, Jin GZ. Effects of (−)stepholidine in animal models for schizophrenia. Acta Pharmacol Sin. 2006;27:1111–1118. doi: 10.1111/j.1745-7254.2006.00365.x. [DOI] [PubMed] [Google Scholar]
- Free RB, Chun LS, Moritz AE, Miller BN, Doyle TB, Conroy JL, Padron A, Meade JA, Xiao J, Hu X, Dulcey AE, Han Y, Duan L, Titus S, Bryant-Genevier M, Barnaeva E, Ferrer M, Javitch JA, Beuming T, Shi L, Southall NT, Marugan JJ, Sibley DR. Discovery and characterization of a G protein-biased agonist that inhibits beta-arrestin recruitment to the D2 dopamine receptor. Mol Pharmacol. 2014;86:96–105. doi: 10.1124/mol.113.090563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Free RB, Hazelwood LA, Cabrera DM, Spalding HN, Namkung Y, Rankin ML, Sibley DR. D1 and D2 dopamine receptor expression is regulated by direct interaction with the chaperone protein calnexin. J Biol Chem. 2007;282:21285–21300. doi: 10.1074/jbc.M701555200. [DOI] [PubMed] [Google Scholar]
- Gao M, Chu HY, Jin GZ, Zhang ZJ, Wu J, Zhen XC. l-Stepholidine-induced excitation of dopamine neurons in rat ventral tegmental area is associated with its 5-HT(1A) receptor partial agonistic activity. Synapse. 2011;65:379–387. doi: 10.1002/syn.20855. [DOI] [PubMed] [Google Scholar]
- Guo Y, Zhang H, Chen X, Cai W, Cheng J, Yang Y, Jin G, Zhen X. Evaluation of the antipsychotic effect of bi-acetylated l-stepholidine (l-SPD-A), a novel dopamine and serotonin receptor dual ligand. Schizophr Res. 2009;115:41–49. doi: 10.1016/j.schres.2009.08.002. [DOI] [PubMed] [Google Scholar]
- Hess EJ, Albers LJ, Le H, Creese I. Effects of chronic SCH23390 treatment on the biochemical and behavioral properties of D1 and D2 dopamine receptors: potentiated behavioral responses to a D2 dopamine agonist after selective D1 dopamine receptor upregulation. J Pharmacol Exp Ther. 1986;238:846–854. [PubMed] [Google Scholar]
- Hutcheson JD, Setola V, Roth BL, Merryman WD. Serotonin receptors and heart valve disease—it was meant 2B. Pharmacol Ther. 2011;132:146–157. doi: 10.1016/j.pharmthera.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin GZ, Huang KX, Sun BC. Dual actions of (−)-stepholidine on dopamine receptor subtypes after substantia nigra lesion. Neurochem Int. 1992;20(Suppl):175S–178S. doi: 10.1016/0197-0186(92)90234-i. [DOI] [PubMed] [Google Scholar]
- Jin GZ, Zhu ZT, Fu Y. (−)-Stepholidine: a potential novel antipsychotic drug with dual D1 receptor agonist and D2 receptor antagonist actions. Trends Pharmacol Sci. 2002;23:4–7. doi: 10.1016/s0165-6147(00)01929-5. [DOI] [PubMed] [Google Scholar]
- Kapur S, Mamo D. Half a century of antipsychotics and still a central role for dopamine D2 receptors. Prog Neuro-Psychopharmacol Biol Psychiatry. 2003;27:1081–1090. doi: 10.1016/j.pnpbp.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Lane JR, Powney B, Wise A, Rees S, Milligan G. G protein coupling and ligand selectivity of the D2L and D3 dopamine receptors. J Pharmacol Exp Ther. 2008;325:319–330. doi: 10.1124/jpet.107.134296. [DOI] [PubMed] [Google Scholar]
- Leff SE, Hamblin MW, Creese I. Interactions of dopamine agonists with brain D1 receptors labeled by 3H-antagonists. Evidence for the presence of high and low affinity agonist-binding states. Mol Pharmacol. 1985;27:171–183. [PubMed] [Google Scholar]
- Luttrell LM, Gesty-Palmer D. Beyond desensitization: physiological relevance of arrestin-dependent signaling. Pharmacol Rev. 2010;62:305–330. doi: 10.1124/pr.109.002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masri B, Salahpour A, Didriksen M, Ghisi V, Beaulieu JM, Gainetdinov RR, Caron MG. Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc Natl Acad Sci U S A. 2008;105:13656–13661. doi: 10.1073/pnas.0803522105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiol Rev. 1998;78:189–225. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- Mo J, Guo Y, Yang YS, Shen JS, Jin GZ, Zhen X. Recent developments in studies of l-stepholidine and its analogs: chemistry, pharmacology and clinical implications. Curr Med Chem. 2007;14:2996–3002. doi: 10.2174/092986707782794050. [DOI] [PubMed] [Google Scholar]
- Mo J, Zhang H, Yu LP, Sun PH, Jin GZ, Zhen X. L-stepholidine reduced L-DOPA-induced dyskinesia in 6-OHDA-lesioned rat model of Parkinson’s disease. Neurobiol Aging. 2010;31:926–936. doi: 10.1016/j.neurobiolaging.2008.06.017. [DOI] [PubMed] [Google Scholar]
- Moreno E, Moreno-Delgado D, Navarro G, Hoffmann HM, Fuentes S, Rosell-Vilar S, Gasperini P, Rodriguez-Ruiz M, Medrano M, Mallol J, Cortes A, Casado V, Lluis C, Ferre S, Ortiz J, Canela E, McCormick PJ. Cocaine disrupts histamine H3 receptor modulation of dopamine D1 receptor signaling: sigma1-D1-H3 receptor complexes as key targets for reducing cocaine’s effects. J Neurosci. 2014;34:3545–3558. doi: 10.1523/JNEUROSCI.4147-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natesan S, Reckless GE, Barlow KB, Odontiadis J, Nobrega JN, Baker GB, George SR, Mamo D, Kapur S. The antipsychotic potential of l-stepholidine—a naturally occurring dopamine receptor D1 agonist and D2 antagonist. Psychopharmacology. 2008;199:275–289. doi: 10.1007/s00213-008-1172-1. [DOI] [PubMed] [Google Scholar]
- Pei L, Li S, Wang M, Diwan M, Anisman H, Fletcher PJ, Nobrega JN, Liu F. Uncoupling the dopamine D1–D2 receptor complex exerts antidepressant-like effects. Nat Med. 2010;16:1393–1395. doi: 10.1038/nm.2263. [DOI] [PubMed] [Google Scholar]
- Perreault ML, Hasbi A, O’Dowd BF, George SR. Heteromeric dopamine receptor signaling complexes: emerging neurobiology and disease relevance. Neuropsychopharmacology. 2014;39:156–168. doi: 10.1038/npp.2013.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid AJ, So CH, Kong MM, Furtak T, El-Ghundi M, Cheng R, O’Dowd BF, George SR. D1-D2 dopamine receptor heterooligomers with unique pharmacology are coupled to rapid activation of Gq/11 in the striatum. Proc Natl Acad Sci U S A. 2007;104:654–659. doi: 10.1073/pnas.0604049104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rominger DH, Cowan CL, Gowen-MacDonald W, Violin JD. Biased ligands: pathway validation for novel GPCR therapeutics. Curr Opin Pharmacol. 2014;16C:108–115. doi: 10.1016/j.coph.2014.04.002. [DOI] [PubMed] [Google Scholar]
- Schulz DW, Stanford EJ, Wyrick SW, Mailman RB. Binding of [3H]SCH23390 in rat brain: regional distribution and effects of assay conditions and GTP suggest interactions at a D1-like dopamine receptor. J Neurochem. 1985;45:1601–1611. doi: 10.1111/j.1471-4159.1985.tb07233.x. [DOI] [PubMed] [Google Scholar]
- Setola V, Dukat M, Glennon RA, Roth BL. Molecular determinants for the interaction of the valvulopathic anorexigen norfenfluramine with the 5-HT2B receptor. Mol Pharmacol. 2005;68:20–33. doi: 10.1124/mol.104.009266. [DOI] [PubMed] [Google Scholar]
- Shenoy SK, Lefkowitz RJ. beta-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol Sci. 2011;32:521–533. doi: 10.1016/j.tips.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley DR, Monsma FJ., Jr Molecular biology of dopamine receptors. Trends Pharmacol Sci. 1992;13:61–69. doi: 10.1016/0165-6147(92)90025-2. [DOI] [PubMed] [Google Scholar]
- Skinbjerg M, Seneca N, Liow JS, Hong J, Weinshenker D, Pike VW, Halldin C, Sibley DR, Innis RB. Dopamine beta-hydroxylase-deficient mice have normal densities of D(2) dopamine receptors in the high-affinity state based on in vivo PET imaging and in vitro radioligand binding. Synapse. 2010;64:699–703. doi: 10.1002/syn.20781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strange PG. Use of the GTPgammaS ([35S]GTPgammaS and Eu-GTPgammaS) binding assay for analysis of ligand potency and efficacy at G protein-coupled receptors. Br J Pharmacol. 2010;161:1238–1249. doi: 10.1111/j.1476-5381.2010.00963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun BC, Jin GZ. Characteristics of (−)-stepholidine on the firing activity of substantia nigral dopamine neurons after repeated reserpine treatment. Biol Signals. 1992;1:331–338. doi: 10.1159/000109338. [DOI] [PubMed] [Google Scholar]
- Sun BC, Zhang XX, Jin GZ. (−)-Stepholidine acts as a D1 partial agonist on firing activity of substantia nigra pars reticulata neurons in 6-hydroxydopamine-lesioned rats. Life Sci. 1996;59:299–306. doi: 10.1016/0024-3205(96)00298-6. [DOI] [PubMed] [Google Scholar]
- Sun Y, Dai J, Hu Z, Du F, Niu W, Wang F, Liu F, Jin G, Li C. Oral bioavailability and brain penetration of (−)-stepholidine, a tetrahydroprotoberberine agonist at dopamine D(1) and antagonist at D(2) receptors, in rats. Br J Pharmacol. 2009;158:1302–1312. doi: 10.1111/j.1476-5381.2009.00393.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, Miller KJ, Spedding M, Mailman RB. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- Urs NM, Daigle TL, Caron MG. A dopamine D1 receptor-dependent beta-arrestin signaling complex potentially regulates morphine-induced psychomotor activation but not reward in mice. Neuropsychopharmacology. 2011;36:551–558. doi: 10.1038/npp.2010.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urs NM, Snyder JC, Jacobsen JP, Peterson SM, Caron MG. Deletion of GSK3beta in D2R-expressing neurons reveals distinct roles for beta-arrestin signaling in antipsychotic and lithium action. Proc Natl Acad Sci U S A. 2012;109:20732–20737. doi: 10.1073/pnas.1215489109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violin JD, Crombie AL, Soergel DG, Lark MW. Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol Sci. 2014;35:308–316. doi: 10.1016/j.tips.2014.04.007. [DOI] [PubMed] [Google Scholar]
- Wang M, Wong AH, Liu F. Interactions between NMDA and dopamine receptors: a potential therapeutic target. Brain Res. 2012;1476:154–163. doi: 10.1016/j.brainres.2012.03.029. [DOI] [PubMed] [Google Scholar]
- Xu SX, Yu LP, Han YR, Chen Y, Jin GZ. Effects of tetrahydroprotoberberines on dopamine receptor subtypes in brain. Acta Pharmacol Sin. 1989;10:104–110. [PubMed] [Google Scholar]
- Yang K, Jin G, Wu J. The neuropharmacology of (−)-stepholidine and its potential applications. Curr Neuropharmacol. 2007;5:289–294. doi: 10.2174/157015907782793649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou LL, Liu J, Jin GZ. Involvement of receptor reserve in D1 agonistic action of (−)-stepholidine in lesioned rats. Biochem Pharmacol. 1997;54:233–240. doi: 10.1016/s0006-2952(97)00153-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.