

Abstract
The Nicholas reaction has been applied to the installation of alkyne ligation handles. Acid-promoted propargylation of hydroxyl, sulfhydryl, amino, and carboxyl groups using dicobalt hexacarbonyl-stabilized propargylium ions is reported within. This method is especially useful for the introduction of propargyl groups into base-sensitive molecules, thereby expanding the toolbox of methods for the incorporation of alkynes for bioorthogonal reactions. High-value molecules are used as the limiting reagent and various propargylium ion precursors are compared.
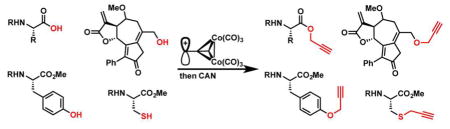
Graphical Abstract
Widespread use of the Huisgen 1,3-dipolar cycloaddition between azides and alkynes to form 1,2,3-triazoles, a click reaction,1 has led to increased interest in transformations used to synthesize and/or install alkynyl groups.2 Typically, when readying substrates for a click reaction, late-stage propargylation or 5-hexynoylation reactions of hydroxyl or amino groups are used to attach the desired alkynes.2 Propargylation of a hydroxyl group is usually achieved by a Williamson ether synthesis under basic conditions where the corresponding alkoxide is reacted with propargyl bromide (Figure 1, eq 1). Src-directed probe 1, was prepared using this approach, but required protection of the 3′- and 5′-hydroxyl groups and 6-amino group to avoid over-propargylation.3 Propargylation has also been accomplished by converting a hydroxyl group into a leaving group (i.e. a mesylate) and replacing it with propargyl amine.4
Figure 1.

Synthetic methods for alkyne incorporation.
Another commonly used protocol for installing an alkynyl group is a carbodiimide-mediated coupling reaction between 5-hexynoic acid and a hydroxyl or amino group (Figure 1, eq 2).2b,5 The Duocarmycin probe 2 exemplifies a product obtained from an EDC6-mediated coupling reaction between a cyclic, secondary amino group and 5-hexynoic acid.7 While carbodiimide couplings offer non-basic, neutral conditions, they require expensive reagents and/or the tedious removal of urea-related byproducts.
Many other methods are available for the functionalization of a compound with an alkynyl group;2,8 however, despite these options, challenges still arise when alkynylating functionally dense natural products and chemical probes for applications such as activity-based protein profiling9 for target identification.10 For example, during investigations to label two different sesquiterpene analogs with alkynyl groups (vide infra), these analogs were unstable to the basic conditions required for propargylation. Although the hexynoylation reaction could serve as an alternative for appendage of an alkyne ligation handle via an allylic ester linkage, concerns about the metabolic stability of ester-containing probes in cell culture lowered enthusiasm for this approach.11 Consequently, a method for propargylation of these sesquiterpene analogs and other biomechanistic probes under non-basic conditions was needed.
Herein, we report our studies to establish the Nicholas reaction as an alternative protocol for the propargylation of high-value small molecules. The Nicholas reaction involves the addition of a nucleophile to the cobalt-stabilized propargylic carbocation 3, generated by treating the corresponding dicobalt hexacarbonyl complexed (Co2(CO)6-) propargyl alcohol with acid. Alkyne 4 is formed after oxidative decomplexation (Figure 1, eq 3).12 While it is well known that the Nicholas reaction can be used to effect propargylation reactions of hetero-nucleophiles, classical conditions require excess nucleophile relative to the cobalt-carbonyl complex, even as the solvent in some cases, limiting its utility in the preparation of alkyne ligation handles.13 Conditions where the nucleophile is the limiting reagent would expand the utility of this approach.
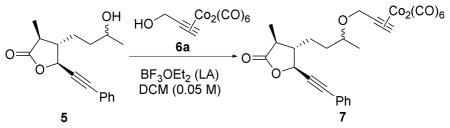
With the goal of increasing the efficiency of the Nicholas reaction, we began our reaction condition investigations using molecularly complex alcohol 5 as the limiting reagent. Initially, a reaction was carried out with a 1:1.1:1.4 ratio of the nucleophile 5, 6a, and BF3OEt2 respectively. However, these conditions led to a moderate yield of 40% so we focused on using higher equivalents of 6a and BF3OEt2. We varied the molar equivalencies of complex 6a and the Lewis-acid (BF3OEt2) while keeping the order of addition constant. To reduce the likelihood of the alcohol and ester groups of 5 tying up the BF3OEt2, Co2(CO)6-propargyl alcohol 6a was added to BF3OEt2 to form the propargyl cation, followed by the addition of alcohol 5. Using this addition order and a 1:2:2.5 molar ratio of alcohol 5, complex 6a, and BF3OEt2, Co2(CO)6-propargyl ether 7 was obtained in 47% yield (entry 1) after stirring for 4.5 h at 0 °C. Increasing the equivalents of BF3OEt2 and complex 6a afforded 7 in 36% yield (entry 2). Adding alcohol 5 more slowly lowered the yield of 7 to 28%, (entry 3).
In view of these unfruitful results, the order of addition was examined. Adding alcohol 5 to BF3OEt2 prior to addition of complex 6a did not affect the yield of 7, obtained in 44% yield (entry 4). Next, alcohol 5 (1 equiv) and BF3OEt2 (2.5 equiv) were added sequentially to Co2(CO)6-propargyl alcohol 6a (2 equiv), which increased the yield of 7 to 55% (entry 5). With this same order of addition, increasing the amount of complex 6a and BF3OEt2 lowered the yield of 7 to 22% (entry 6). For all of these examples, 6a was prepared, isolated, and purified, by column chromatography, before the reaction. While it is recognized that this complex is stable to air and moisture, it was reasoned that forming 6a in situ may be advantageous.13c To this end, complex 6a was formed in situ from propargyl alcohol and dicobalt octacarbonyl, followed by the sequential addition of alcohol 5 and BF3OEt2 to afford the highest yield of 7 (60%, entry 7). A final attempt to improve the reaction conditions by lowering the reaction temperature only resulted in decreased yields of 7.14 Decomplexation of cobalt complex 7 was achieved using ceric ammonium nitrate (CAN) in acetone to readily afford alkyne 8 in 97% yield without the need for purification (Scheme 1). Use of N-methylmorpholine-N-oxide (NMO) as an oxidant in this transformation resulted in decomposition of 7.15
Scheme 1.

Decomplexation of Co2(CO)6-alkyne 7.
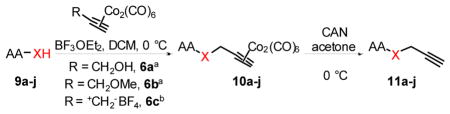
Next, the generality of these optimized reaction conditions was tested on hydroxyl, sulfhydryl, amino, and carboxyl containing amino acids; a class of compounds selected for their richness of functionality and the utility of propargylated peptides for biochemical applications.1d,2a,16 Unfortunately, when subjecting N-Boc-L-serine methyl ester (9a) to the optimized reaction conditions, 10a was obtained in 20% yield while 76% of the starting material 9a was recovered (Table 2, entry 1). Similarly, when N-Fmoc-L-serine methyl ester (9b) was subjected to the same conditions, 10b was isolated in 29% yield with 63% recovered 9b (entry 3). In both of these examples, Co2(CO)6-propargyl alcohol 6a was fully consumed and the dimerized Co2(CO)6-propargyl alcohol was obtained, resulting from the propargylium cation reacting more readily with the hydroxyl group of 6a. To overcome this competing homodimerization reaction, Co2(CO)6-methyl propargyl ether 6b was examined.12c Reaction of N-Boc-L-serine methyl ester (9a) with 6b afforded 10a in 97% yield (entry 2). The yield of 10b also increased significantly to 54% when using complex 6b (entry 4). Use of propargyl acetate for the synthesis of 10a and 10b gave yields comparable to complex 6a (See Supporting Information (SI)).
Table 2.
Synthesis of alkyne modified amino acids (AA).
| |||||
|---|---|---|---|---|---|
| entry | AA-XH, 9 | 6 | time (h) | 10, yield (%) | 11, yield (%) |
| 1 |
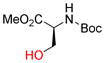 9a |
6a | 1 | 10a, 20 |
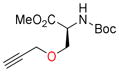 11a, 90 |
| 2 | 9a | 6b | 1 | 10a, 97 | |
|
| |||||
| 3 |
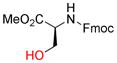 9b |
6a | 1 | 10b, 29 |
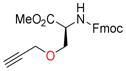 11b, 90 |
| 4 | 9b | 6b | 1 | 10b, 54 | |
|
| |||||
| 5 |
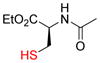 9c |
6a | 2 | 10c, 86 |
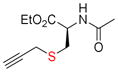 11c, 83 |
| 6 |
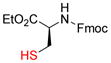 9d |
6a | 0.75 | 10d, 71 |
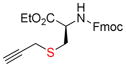 11d, 92 |
| 7 | 9d | 6bc | 2 | 10d, 67 | |
|
| |||||
| 8 |
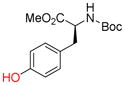 9e |
6a | 1 | 10e, 45 |
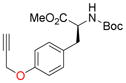 11e, 75 |
| 9 | 9e | 6b | 3 | 10e, 23 | |
|
| |||||
| 10 |
 9f |
6a | 1 | 10f, 6 |
 11f, 81 |
| 11 | 9f | 6b | 1 | 10f, 73 | |
|
| |||||
| 12 |
 9g |
6a | 0.25 | 10g, 0 |
 11gd |
| 13 | 9g | 6c | 1.5 | 10g, 46 | |
|
| |||||
| 14 |
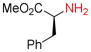 9h |
6c | 1.5 | 10h, 59 |
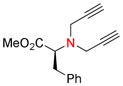 11h, 56 |
|
| |||||
| 15 |
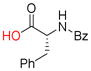 9i |
6a | 2 | 10i, 60 |
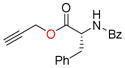 11i, 90 |
| 16 | 9i | 6c | 10i, 18 | ||
Nucleophilic group (-XH) of amino acid (AA) highlighted in red.
Complexes 6a and 6b were formed in situ.
BF3OEt2 is not used when using 6c.
Use of isolated 6b gave highest yield.
11g is unstable. For additional examples, see SI.
Next, we tested this method for the propargylation of cysteine thiols, a transformation typically accomplished using basic alkylation conditions.2a,17 Thiols react efficiently in the Nicholas reaction; however, application has been limited to the synthesis of sulfur containing macrocycles.12c,18 N-acetyl- and N-Fmoc-L-cysteine ethyl ester (9c and 9d) were reacted with complex 6a giving the corresponding Co2(CO)6-alkynes 10c and 10d in high yields of 86% and 71% (Entries 5, 6). N-Fmoc cysteine 9d was also reacted with methyl propargyl ether complex 6b, which gave a comparable yield of 67% for 10d (entry 7).
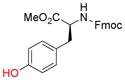
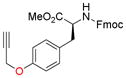
To evaluate the phenolic side chain of tyrosine in the Nicholas reaction, N-Boc-L-tyrosine methyl ester (9e) was reacted with complex 6a.13a Two major products were observed; the desired product, 10e, was isolated in 45% yield (57% based on recovered 9e) (entry 8), while an unstable byproduct was obtained in trace amounts. 1H NMR analysis of this byproduct revealed aromatic signals integrating for three protons, resulting from electrophilic aromatic substitution (see SI, S5).12c Because 9e was recovered along with complete consumption of 6a, complex 6b was tested. This reaction required a longer reaction time and did not improve the yield of 10e (23% yield, entry 9) due to Boc instability.19 When the N-Fmoc tyrosine ester 9f was reacted with complex 6a, 10f was formed in 6% yield (56% based on recovered starting material) (entry 10). Employing complex 6b resulted in a significantly improved yield to 73% (entry 11). A byproduct, presumably formed by electrophilic aromatic substitution, was also observed by TLC for these reactions.
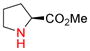
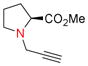
Amino groups were tested by subjecting L-proline methyl ester (9g) to the Nicholas reaction with 6a. Consumption of 9g was observed by TLC within 15 min with no evidence of 10g (entry 12). We presume the BF3OEt2 coordinates with the nitrogen of proline. To circumvent this issue, the cationic propargylium ion was prepared as tetrafluoroborate salt 6c by reacting complex 6a with tetrafluoroboric acid in diethyl ether at 0 °C.12b,20 Reaction of 6c with proline ester 9g in DCM at 0°C afforded Co2(CO)6-alkyne 10g in 46% yield (entry 13). The primary amine of L-phenylalanine methyl ester (9h) also proved to be an effective nucleophile; when reacted with 6c, dialkylation afforded amine 10h in 59% yield (entry 14).
Carboxyl groups were also subjected to the Nicholas reaction conditions. Only a few examples of carboxyl groups serving as a nucleophile in the Nicholas reaction have been reported. 21 Reaction of N-Bz-D-phenylalanine (9i) with complex 6a and BF3OEt2 afforded Co2(CO)6-propargyl ester 10i in 60% yield (entry 15). Reaction of 9i with complex 6c afforded a lower yield for 10i (18%, entry 16); thus, the utility of preformed propargylium salt is not necessarily general.
Co2(CO)6-alkyne modified amino acids 10a-j underwent oxidative decomplexation with CAN. The propargyl derivatives of serine, cysteine, tyrosine, and phenylalanine 11a-f, i were afforded in high yields (75–94%). A moderate yield of 56% was observed for the formation of dipropargylamine 11h (entry 14). Proline alkyne derivative 11g appeared to be unstable, permitting isolation and NMR characterization only once prior to decomposition (entry 12).
To effect mono-alkynylation of primary amines, an alternative tetrafluoroborate salt 12 was prepared from Co2(CO)6-2-methyl-3-butyn-2-ol (Scheme 2). Reaction of 12 with phenylalanine ester 9h afforded the mono-alkynylated propargyl amine 13 after oxidative decomplexation.
Scheme 2.

Reaction of primary amine 9h with BF4− salt 12.
Finally, to show the synthetic utility of these conditions for base-sensitive, functionally dense molecules we applied the Nicholas reaction conditions to two sesquiterpene analogs. Base-sensitive guaianolide analog 14, previously synthesized in our group, was reacted with Co2(CO)6-propargyl alcohol 6a, formed in situ, and BF3OEt2, to give the Co2(CO)6-alkyne derivative in 46% yield.22 Reaction with CAN generated alkyne probe 15 in quantitative yield.
Melampomagnolide B (MelB) (16) has been used as a parthenolide mimic for conjugation to biotin via an ester-linkage.23, 24 However, these biotinylated compounds may have metabolic stability issues for in vivo biochemical experiments. Formation of the alternative ether linkage using the allylic alcohol handle has proven to be difficult; MelB is base sensitive and the allylic hydroxyl group was unreactive in our hands towards oxidation or bromination.25 Reaction of MelB (16) with Co2(CO)6-propargyl alcohol complex 6a and BF3OEt2 afforded the corresponding Co2(CO)6-alkyne product after 1 h in 19% yield. A shortened reaction time of 10 min gave a 41% yield (45% yield based on recovered 16), suggesting the Co2(CO)6-alkyne product was unstable to the reaction conditions. Reacting MelB (16) with Co2(CO)6-methyl propargyl ether 6b gave a 39% yield of the coupled product. Cobalt decomplexation afforded the MelB alkyne probe 17 in 94% yield (Scheme 3).
Scheme 3.

Synthesis of alkyne probes 15 and 17.
In conclusion, the Nicholas reaction conditions described provide an acid-mediated alternative for propargylation of molecularly complex compounds. Reaction conditions were optimized for use of high-value nucleophiles as limiting reagents, a practice atypical for the Nicholas reaction. A number of functional groups acted as the nucleophilic species, including hydroxyl, sulfhydryl, carboxyl, and amino groups. For substrates that react slower than the competing dimerization of Co2(CO)6-propargyl alcohol 6a, use of methyl propargyl ether complex 6b improved yields. Propargylation of amino groups required the preparation of propargylium tetrafluoroborate salts. Mono- and di-alkynylation of a primary amino group was achieved selectively depending on the steric nature of the propargylium ion. Bz, Cbz, Ac, and Fmoc amine protecting groups were all tolerated. Finally, these conditions provided an alternative propargylation strategy for base-sensitive sesquiterpene analogs.
Supplementary Material
Table 1.
Optimization of Nicholas Reaction with alcohol 5.
| |||||
|---|---|---|---|---|---|
| entry | equiv (5:6a:LA) | order of addition | temp (°C) | time (h) | yield (%) |
| 1a | 1:2:2.5 | LA, 6a, 5 | 0 | 4.5 | 47 |
| 2 | 1:3:5 | LA, 6a, 5 | 0 | 4 | 36 |
| 3 | 1:2:2.5 | LA, 6a, 5c | 0 | 4 | 28 |
| 4a | 1:2:2.5 | LA, 5, 6a | 0 | 4.5 | 44 |
| 5b | 1:2:2.5 | 6a, 5, LA | 0 | 4 | 55 |
| 6 | 1:3:5 | 6a, 5, LA | 0 | 5 | 22 |
| 7b | 1:2:2.5 | 6a, 5, LAd | 0 | 3.5 | 60 |
Dimerized product of 6a was isolated in 33–34% yield.
Dimerized product of 6a was isolated in 25–28% yield.
Alcohol 5 was added dropwise over 5 min.
Complex 6a was generated in situ from propargyl alcohol and Co2(CO)8
Acknowledgments
We gratefully acknowledge the NIH (R21-CA194661 to DAH and R01-GM054161 to KMB) and the Department of Defense (PC141033 to DAH) for funding. Mass spectrometry performed at the University of Minnesota was conducted at the Masonic Cancer Center Analytical Biochemistry Core Facility, which is supported by the National Institute of Health (P30-CA77598).
Footnotes
The authors declare no competing financial interest.
The SI is available free of charge on the ACS Publications website at DOI: Full experimental details, characterization data, and 1H and 13C NMR spectra for all new compounds (PDF)
References
- 1.(a) Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; (b) Meldal M, Tornoe CW. Chem Rev. 2008;108:2952–3015. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]; (c) Sletten EM, Bertozzi CR. Angew Chem Int Ed. 2009;48:6974–6998. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Tang W, Becker ML. Chem Soc Rev. 2014;43:7013–7039. doi: 10.1039/c4cs00139g. [DOI] [PubMed] [Google Scholar]; (e) Martell J, Weerapana E. Molecules. 2014;19:1378–1393. doi: 10.3390/molecules19021378. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Tiwari VK, Mishra BB, Mishra KB, Mishra N, Singh AS, Chen X. Chem Rev. 2016;116:3086–3240. doi: 10.1021/acs.chemrev.5b00408. [DOI] [PubMed] [Google Scholar]
- 2.(a) Johansson H, Pedersen DS. Eur J Org Chem. 2012:4267–4281. [Google Scholar]; (b) Lehmann J, Wright MH, Sieber SA. Chem Eur J. 2016;22:4666–4678. doi: 10.1002/chem.201504419. [DOI] [PubMed] [Google Scholar]
- 3.Gushwa NN, Kang S, Chen J, Taunton J. J Am Chem Soc. 2012;134:20214–20217. doi: 10.1021/ja310659j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen MS, Hadjivassiliou H, Taunton J. Nat Chem Biol. 2007;3:156–160. doi: 10.1038/nchembio859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For selected examples, see: Bottcher T, Sieber SA. J Am Chem Soc. 2010;132:6964–6972. doi: 10.1021/ja909150y.Kalesh KA, Sim DSB, Wang J, Liu K, Lin Q, Yoa SQ. Chem Commun. 2010;46:1118–1120. doi: 10.1039/b919888a.Krysiak JM, Kreuzer J, Macheroux P, Hermetter A, Sieber SA, Breinbauer R. Angew Chem Int Ed. 2012;51:7035–7040. doi: 10.1002/anie.201201955.Kreuzer J, Bach NC, Forler D, Sieber SA. Chem Sci. 2015;6:237–245. doi: 10.1039/c4sc02339k.
- 6.1-ethyl-3-(3-dimethylaminopropyl)carbodiimide.
- 7.Wirth T, Pestel GF, Ganal V, Kirmeier T, Schuberth I, Rein T, Tietze LF, Sieber SA. Angew Chem Int Ed. 2013;52:6921–6925. doi: 10.1002/anie.201208941. [DOI] [PubMed] [Google Scholar]
- 8.For a C-H functionalization strategy, see: Li J, Cisar JS, Zhou C-Y, Vera B, Williams H, Rodriguez AD, Cravatt BF, Romo D. Nat Chem. 2013;5:510–517. doi: 10.1038/nchem.1653.
- 9.For selected reviews on ABPP, see: Cravatt BF, Wright AT, Kozarich JW. Annu Rev Biochem. 2008;77:383–414. doi: 10.1146/annurev.biochem.75.101304.124125.Nodwell MB, Sieber SA. ABPP Methodology: Introduction and Overview. In: Sieber SA, editor. Activity-Based Protein Profiling. Vol. 324. Springer; Berlin Heidelberg: 2012. pp. 1pp. 1–41.Willems LI, Overkleeft HS, van Kasteren SI. Bioconjug Chem. 2014;25:1181–1191. doi: 10.1021/bc500208y.Yang P, Liu K. ChemBioChem. 2015;16:712–724. doi: 10.1002/cbic.201402582.
- 10.(a) Bottcher T, Pitscheider M, Sieber SA. Angew Chem Int Ed. 2010;49:2680–2698. doi: 10.1002/anie.200905352. [DOI] [PubMed] [Google Scholar]; (b) Su Y, Ge J, Zhu B, Zheng YG, Zhu Q, Yao SQ. Cur Opin Chem Biol. 2013;17:768–775. doi: 10.1016/j.cbpa.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 11.(a) Ettmayer P, Amidon GL, Clement B, Testa B. J Med Chem. 2004;47:2393–2404. doi: 10.1021/jm0303812. [DOI] [PubMed] [Google Scholar]; (b) Li B, Sedlacek M, Manoharan I, Boopathy R, Duysen EG, Masson P, Lockridge O. Biochem Pharmacol. 2005;70:1673–1684. doi: 10.1016/j.bcp.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 12.Lockwood RF, Nicholas KM. Tetrahedron Lett. 1977;18:4163–4165.for reviews on the Nicholas Reaction, see: Nicholas KM. Acc Chem Res. 1987;20:207–214.Teobald BJ. Tetrahedron. 2002;58:4133–4170.Diaz DD, Betancort JM, Martin VS. Synlett. 2007:343–359.
- 13.(a) Diaz DD, Martin VS. Tetrahedron Lett. 2000;41:9993–9996. [Google Scholar]; (b) Hope-Weeks LJ, Mays MJ, Solan GA. Eur J Inorg Chem. 2007:3101–3114. [Google Scholar]; (c) Ortega N, Martin VS, Martin T. J Org Chem. 2010;75:6660–6672. doi: 10.1021/jo101566x. [DOI] [PubMed] [Google Scholar]
- 14.Reactions at -40 °C and -10 °C afforded 7 in 23% and 38% yield, respectively.
- 15.Hayashi Y, Yamaguchi H, Toyoshima M, Okado K, Toyo T, Shoji M. Chem Eur J. 2010;16:10150–10159. doi: 10.1002/chem.201000795. [DOI] [PubMed] [Google Scholar]
- 16.Ahmad Fuaad A, Azmi F, Skwarczynski M, Toth I. Molecules. 2013;18:13148–13174. doi: 10.3390/molecules181113148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Struthers H, Spingler B, Mindt TL, Schibli R. Chem Eur J. 2008;14:6173–6183. doi: 10.1002/chem.200702024. [DOI] [PubMed] [Google Scholar]
- 18.For selected examples, see: Hope-Weeks LJ, Mays MJ, Woods AD. J Chem Soc, Dalton Trans. 2002:1812–1819.Hagendorn T, Brase S. RSC Adv. 2014;4:15493–15495.
- 19.Evans EF, Lewis NJ, Kapfer I, Macdonald G, Taylor RJK. Synth Commun. 1997;27:1819–1825. [Google Scholar]
- 20.Amouri H, Begue J-P, Chennoufi A, Bonnet-Delpon D, Gruselle M, Malezieux B. Org Lett. 2000;2:807–809. doi: 10.1021/ol005554l. [DOI] [PubMed] [Google Scholar]
- 21.Shea KM, Closser KD, Quintal MM. J Org Chem. 2005;70:9088–9091. doi: 10.1021/jo051691q. [DOI] [PubMed] [Google Scholar]
- 22.Wen B, Hexum JK, Widen JC, Harki DA, Brummond KM. Org Lett. 2013;15:2644–2647. doi: 10.1021/ol400904y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.(a) Macias FA, Galindo JCG, Massanet GM. Phytochemistry. 1992;31:1969–1977. [Google Scholar]; (b) Nasim S, Pei S, Hagen FK, Jordan CT, Crooks PA. Bioorg Med Chem. 2011;19:1515–1519. doi: 10.1016/j.bmc.2010.12.045. [DOI] [PubMed] [Google Scholar]
- 24.(a) Kwok BHB, Koh B, Ndubuisi MI, Elofsson M, Crews CM. Chem Biol. 2001;8:759–766. doi: 10.1016/s1074-5521(01)00049-7. [DOI] [PubMed] [Google Scholar]; (b) Janganati V, Penthala NR, Madadi NR, Chen Z, Crooks PA. Bioorg Med Chem Lett. 2014;24:3499–3502. doi: 10.1016/j.bmcl.2014.05.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Attempts to manipulate the allylic alcohol of 17 included use of: PCC, PDC, Dess-Martin periodinane, and PBr3.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.