

Abstract
The D1 dopamine receptor (D1R) has been implicated in numerous neuropsychiatric disorders, and D1R-selective ligands have potential as therapeutic agents. Previous studies have identified substituted benzazepines as D1R-selective agonists, but the in vivo effects of these compounds have not correlated well with their in vitro pharmacological activities. A series of substituted benzazepines, and structurally dissimilar D1R-selective agonists, were tested for their functional effects on D1R-mediated cAMP accumulation, D1R-promoted β-arrestin recruitment, and D1R internalization using live cell functional assays. All compounds tested elicited an increase in the level of cAMP accumulation, albeit with a range of efficacies. However, when the compounds were evaluated for β-arrestin recruitment, a subset of substituted benzazepines, SKF83959, SKF38393, SKF82957, SKF77434, and SKF75670, failed to activate this pathway, whereas the others showed similar activation efficacies as seen with cAMP accumulation. When tested as antagonists, the five biased compounds all inhibited dopamine-stimulated β-arrestin recruitment. Further, D1R internalization assays revealed a corroborating pattern of activity in that the G protein-biased compounds failed to promote D1R internalization. Interestingly, the biased signaling was unique for the D1R, as the same compounds were agonists of the related D5 dopamine receptor (D5R), but revealed no signaling bias. We have identified a group of substituted benzazepine ligands that are agonists at D1R-mediated G protein signaling, but antagonists of D1R recruitment of β-arrestin, and also devoid of agonist-induced receptor endocytosis. These data may be useful for interpreting the contrasting effects of these compounds in vitro versus in vivo, and also for the understanding of pathway-selective signaling of the D1R.
Keywords: Dopamine, D1 receptor, functional selectivity, biased agonism, G protein, β-arrestin, benzazepine
Graphical Abstract
Dopamine is a critically important transmitter in both the central and peripheral nervous systems. In the central nervous system, dopamine regulates several important functions, including movement, learning and memory, reward circuitry, cognition, and certain neuroendocrine functions.1 Dysregulation of dopaminergic signaling is also central in the etiology and/or therapy of a number of neuropsychiatric disorders.2 The effects of dopamine are mediated by five different receptor subtypes that are members of the G protein-coupled receptor (GPCR) superfamily and are divided into two subfamilies on the basis of structure, pharmacology, and signaling properties.3,4 The D1-like subfamily consists of the D1 and D5 receptors that are defined as being coupled to Gs/Golf proteins, which activate adenylate cyclase and increase cAMP levels. In contrast, the D2-like subfamily, consisting of the D2–D4 receptors, is described as being coupled to Gi/Go proteins, which inhibit adenylate cyclase and modulate certain ion channels. However, as discussed below, both classes of dopamine receptors are also known to signal through additional pathways.
GPCRs, including dopamine receptors, are extremely important drug targets and account for approximately one-third of Food and Drug Administration (FDA)-approved medications.5 Nonetheless, many of these medications can exhibit side effects that can range from bothersome to potentially toxic. Typically, these are due to off-target interactions of the drug with other receptors, channels, or transporters but sometimes are intrinsic to on-target activation by the drug itself. In these cases, the therapeutic effects of a drug targeting a GPCR may be due to stimulation of a specific signaling pathway activated by that receptor, whereas the side effect(s) is mediated by an alternative signaling pathway (e.g., ref 6). Hitherto, this has been an intractable problem as all agonist drugs for a receptor typically activate every signaling pathway associated with that receptor, as would the endogenous agonist. Recently, however, for many receptors, drugs that are capable of inducing or stabilizing unique active signaling states of the receptor such that they selectively activate one signaling pathway and not another or even inhibit a parallel signaling pathway have been identified.7–10 This observation has been termed “functional selectivity” or “biased agonism”, which obviously has enormous potential for drug discovery and development.11–14 Recently, a structural basis for the functional selectivity of several GPCRs has been suggested,15–19 indicating that the rational design of functionally selective or signaling-biased compounds may be achievable.
Within dopamine receptors, functional selectivity of signaling has been clearly observed for the D2 receptor (D2R),20–24 which signals not only through G proteins but also through the multifunctional adaptor protein, β-arrestin. Agonist activation of the D2R leads to β-arrestin recruitment and the formation of a complex of protein kinase B (Akt) and protein phosphatase 2A. This results in the dephosphorylation and inactivation of Akt. Because Akt constitutively inhibits glycogen synthase kinase-β (GSK3β), the latter is then activated, leading to downstream signaling and behavioral outcomes.25,26 Recently, ligands for the D2R that are selectively biased for stimulating either G protein27 or β-arrestin-mediated signaling28,29 have been identified. These developments may assist in determining which signaling arms of the D2R are involved in various behavioral responses to receptor agonists and the therapeutic effects of various agents used to treat neuropsychiatric disorders associated with the D2R.
With respect to D1-like receptors, the existence of functionally selective agonists has been somewhat controversial. While D1Rs are well-known to signal through activating Gs, or Golf, and elevating intracellular cAMP levels, they have also been hypothesized to signal through Gq with subsequent activation of phospholipase Cβ (PLCβ) and Ca2+ mobilization. For instance, D1R-selective agonists from the benzazepine family have been postulated to selectively link the D1R to PLCβ signaling while having negligible effects in activating adenylate cyclase, or acting as antagonists of this latter response.30,31 A prototypical agonist from this group of compounds is SKF83959, which has been shown to induce unique behavioral effects in rodents.32–35 This hypothesis of PLCβ-selective D1R agonists has subsequently been partially revised with the identification of D1–D2 receptor dimers that have been postulated to switch their coupling from Gs (D1R) and Gi/o (D2R) to Gq and thus activate PLCβ.36 Indeed, SKF83959 has been proposed as a prototypical D1–D2 receptor dimer-selective agonist.37,38 However, the selectivity of SKF83959 for either D1–D2 receptor dimers or D1 receptor-mediated PLCβ signaling has recently come under question39,40 (for a review, see ref 41). The possibility that the effects of SKF83959 on PLCβ-mediated signaling may not be mediated by D1Rs, or D1–D2 receptor dimers, but rather by interactions with other GPCRs or signaling proteins has been raised.39–44 Notably, D1Rs have also been suggested to signal through β-arrestin-mediated pathways,45 although biased signaling between β-arrestin- and G protein-mediated pathways has not been extensively investigated for the D1R, as for the D2R (see above).
We now report the identification of D1R-selective benzazepines that are functionally selective at the D1R in that they exhibit biased signaling through G proteins (increased intracellular cAMP levels) in the absence of recruiting β-arrestin. These highly biased ligands thus lack the possibility of signaling through β-arrestin-mediated pathways. Not all D1R-selective benzazepines were found to exhibit biased signaling properties, but included among the G protein-biased agonists was SKF83959, thus suggesting a possible mechanism for the unique effects of this drug in vivo. Interestingly, the benzazepines that were highly G protein-biased at the D1R exhibited less or no bias at the closely related D5R. Our results provide the first identification of G protein signaling-biased agonists for the D1R. Such compounds may prove to be useful for treating neurological disorders in which D1R stimulation is desirable, such as Parkinson’s disease46 or cognitive disorders.47,48
RESULTS AND DISCUSSION
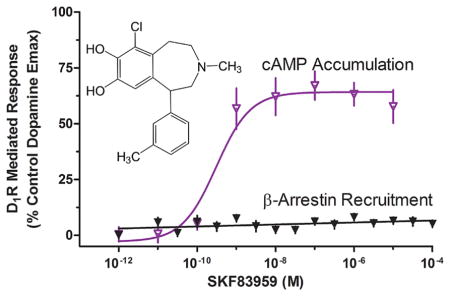
Recently, we investigated the receptor selectivity of the substituted benzazepine, SKF83959, which has been proposed as a prototypical D1–D2 heteromer receptor-selective agonist.39 We were unable to generate data in support of this hypothesis but did find that SKF83959 potently inhibited D1–D2 dimer-mediated Ca2+ mobilization in HEK293 cells.39 In other preliminary experiments, we found that when the D1R was expressed alone (in the absence of the D2R), SKF83959 potently stimulated D1R-mediated cAMP accumulation. Surprisingly, in parallel experiments, SKF83959 was incapable of promoting the recruitment of β-arrestin to the D1R, suggesting that it may exhibit bias between G protein- and β-arrestin-mediated signaling of the D1R. We thus sought to characterize biased activation of these D1R signaling pathways using a series of SKF83959-related substituted benzazepines, and other structurally dissimilar D1R-selective agonists. The structures of all of the compounds used in this study are shown in Figure 1.
Figure 1.

Chemical structures of compounds used in this study. (A) D1R-selective ligands shown include dihydrexidine, A77636, and apomorphine, as well as substituted benzazepines that were not found to be functionally selective for D1R signaling, including the D1R-selective antagonist SCH23390. (B) Substituted benzazepines that were found to exhibit D1R-mediated G protein-biased signaling.
We initially investigated the ability of the D1R-selective compounds to promote D1R-mediated cAMP formation (Figure 2 and Table 1). In Figure 2A, we tested three agonists from different structural categories that are known to exhibit selectivity for the D1R (reviewed in ref 49). Both dihydrexidine and A77636 stimulated cAMP accumulation to the same degree as dopamine, whereas apomorphine was not as efficacious, exhibiting ~75% efficacy compared to dopamine. Figure 2B shows the results with four D1R-selective substituted benzazepines that exhibit full, or nearly full, agonist activity with respect to stimulating cAMP accumulation. SKF81297 and SKF83822 are frequently used as highly efficacious D1R-selective agonists for in vitro and in vivo studies (e.g., refs 50–53), whereas fenoldopam (also known as SKF82526) is an FDA-approved antihypertensive agent.54 Figure 2C shows the results with D1R-selective substituted benzazepines that are less than full agonists for stimulating cAMP accumulation. The most efficacious of these is SKF38393, while the least efficacious is SKF75670. Notably, SKF38393 is a well-known partial agonist of D1R-mediated cAMP accumulation.55–57 The observation that this compound exhibits high, but less than full, efficacy in these experiments suggests that there is receptor reserve for D1R-mediated cAMP accumulation. Interestingly, the HEK293 cells used for these experiments express ~350 fmol/mg of D1R, which is similar to, or somewhat less than, the level of D1R expression in rodent striatum or cortex.58,59 The well-known D1R-selective antagonist, SCH23390 (Figure 1), completely blocked cAMP accumulation of all the agonists tested (data not shown). The average EC50 and Emax values for all of the agonists tested in Figure 2 are listed in Table 1.
Figure 2.

Agonist stimulation of D1R-mediated cAMP accumulation. HEK293 cells stably expressing the D1R were assayed for agonist stimulation of cAMP accumulation as described in Methods. Cells were stimulated with the indicated concentrations of dopamine or test compound. (A) Cells were stimulated with the D1R-selective agonists A77636, dihydrexidine, and apomorphine. (B) Cells were stimulated with the indicated substituted benzazepines, all of which behaved as full, or nearly full, agonists of D1R-mediated cAMP accumulation. (C) Cells were stimulated with substituted benzazepines that behaved as partial agonists of D1R-mediated cAMP accumulation. All data are means of at least three independent experiments conducted on different days in triplicate and expressed as a percentage of the maximal signal given by dopamine (N = 3–4). EC50 and Emax values were obtained for each individual experiment through nonlinear regression, and average data are reported in Table 1.
Table 1.
Stimulation of cAMP Accumulation by D1R Agonistsa
| compound | EC50 (nM) | Emax (% of DA) |
|---|---|---|
| dopamine | 3.2 ± 2 | 100 |
| A77636 | 1.6 ± 0.3 | 98.3 ± 8.5 |
| dihydrexidine | 1.5 ± 0.5 | 92.9 ± 1 |
| apomorphine | 0.45 ± 0.1 | 76.2 ± 8.9 |
| SKF81297 | 2.0 ± 0.2 | 99.9 ± 2.9 |
| chloro-APB | 0.73 ± 0.1 | 87.9 ± 5 |
| SKF83822 | 0.12 ± 0.06 | 73.9 ± 5.9 |
| fenoldopam | 2.8 ± 0.1 | 91.8 ± 3.8 |
| SKF38393 | 1.1 ± 0.4 | 86.7 ± 9.3 |
| SKF83959 | 0.2 ± 0.02 | 59.6 ± 11 |
| SKF82957 | 0.3 ± 0.09 | 60.2 ± 4.5 |
| SKF77434 | 1.0 ± 0.3 | 51.9 ± 3.8 |
| SKF75670 | 0.45 ± 0.03 | 30 ± 3.7 |
EC50 and Emax values were obtained from nonlinear regression analysis of individual dose–response curves as shown in Figure 2. Comparison to the control maximal response exhibited by dopamine (DA) derives the percent response (Emax) of a test compound.
We next investigated the ability of the selected agonist ligands to recruit β-arrestin to the D1R (Figure 3 and Table 2) using a complementation assay as previously described.27,39 Figure 3A shows that both A77636 and dihydrexidine are potent full agonists with respect to this functional response whereas apomorphine is a low-efficacy partial agonist. Apomorphine thus appears to exhibit biased efficacy with respect to G protein versus β-arrestin activation by the D1R. We were also interested in evaluating these same compounds for their ability to inhibit dopamine-stimulated β-arrestin recruitment, and the results are shown in Figure 3B. As expected, the full agonists A77636 and dihydrexidine were without effect in this assay, whereas the partial agonist apomorphine antagonized the dopamine response to approximately the level seen with stimulation by apomorphine alone. Complete inhibition of the dopamine response was observed with SCH23390. Figure 3C shows the β-arrestin recruitment stimulated by four of the substituted benzazepines that were highly efficacious in stimulating cAMP accumulation. All of these compounds potently stimulated β-arrestin recruitment but exhibited partial agonist activity, generally between 50 and 60% of the maximal dopamine response. Not surprisingly, when tested as antagonists in this assay, all of the compounds displayed partial inhibition of dopamine-stimulated β-arrestin recruitment (Figure 3D). In Figure 3E, we investigate the ability of the five substituted benzazepines that were partial agonists in stimulating cAMP accumulation to recruit β-arrestin to the D1R. Surprisingly, none of these compounds exhibited β-arrestin recruitment within the detection limits of the assay. In contrast, all of them functioned as potent antagonists of dopamine-stimulated recruitment of β-arrestin to the D1R (Figure 3F). These five compounds, SKF83959, SKF38393, SKF82957, SKF77434, and SKF75670, thus appear to be biased in their efficacy to promote D1R signaling through G proteins in the absence of β-arrestin recruitment and associated responses. The average EC50 and Emax values for all of the agonists tested in Figure 3 are listed in Table 2.
Figure 3.

Agonist stimulation of recruitment of β-arrestin to the D1R. DiscoveRx PathHunter cells were assayed for agonist-induced recruitment of β-arrestin-2 to the D1R as described in Methods. (A) Cells were stimulated with the D1R-selective agonists A77636, dihydrexidine, and apomorphine. (B) Cells were incubated with an EC80 concentration of dopamine (1 μM) along with the indicated concentrations of the test compounds shown in panel A. The known D1R antagonist SCH23390 was used as a control. (C) Cells were stimulated with the indicated substituted benzazepines, all of which behaved as partial agonists of D1R-mediated stimulation of β-arrestin recruitment. (D) Cells were incubated with an EC80 concentration of dopamine (1 μM) along with the indicated concentrations of the test compounds shown in panel C. The known D1R antagonist SCH23390 was used as a control. (E) Cells were stimulated with the indicated substituted benzazepines, none of which exhibited agonist activity of D1R-mediated stimulation of β-arrestin recruitment. (F) Cells were incubated with an EC80 concentration of dopamine (1 μM) along with the indicated concentrations of the test compounds shown in panel E. The known D1R antagonist SCH23390 was used as a control. All data are means of at least three independent experiments conducted on different days in triplicate and are expressed as a percentage of either the maximal signal given by dopamine (A, C, and E) or the signal given by an EC80 concentration (1 μM) of dopamine (B, D, and F). Average EC50/IC50 and Emax/Imax values were obtained using nonlinear regression analyses and are listed in Table 2.
Table 2.
Stimulation of β-Arrestin Recruitment by D1R Agonists
| compound |
β-arrestin recruitment stimulationa
|
β-arrestin recruitment inhibitionb
|
||
|---|---|---|---|---|
| EC50 (nM) | Emax (% of DA) | IC50 (nM) | Imax (% inhibition) | |
| dopamine | 240 ± 70 | 100 | NA | NA |
| A77636 | 3.6 ± 1.9 | 100.7 ± 8.6 | NA | <5 |
| dihydrexidine | 67.7 ± 8.2 | 106.2 ± 7.7 | NA | <5 |
| apomorphine | 22.6 ± 13.2 | 15.8 ± 3.6 | 652 ± 74 | 62.7 ± 5.3 |
| SKF81297 | 13.2 ± 6 | 61.7 ± 2.6 | 18.7 ± 0.7 | 20.4 ± 3.4 |
| chloro-APB | 2.0 ± 0.4 | 57.7 ± 11.1 | 3.9 ± 1 | 29.5 ± 4 |
| SKF83822 | 2.3 ± 1 | 61.5 ± 3.5 | 10.6 ± 2.8 | 20.4 ± 2.4 |
| fenoldopam | 31.7 ± 15 | 54.7 ± 3.8 | 42.6 ± 5.1 | 43.8 ± 1.9 |
| SKF38393 | >10000 | <5 | 404 ± 47 | >95 |
| SKF83959 | >10000 | <5 | 8.6 ± 2.8 | >95 |
| SKF82957 | >10000 | <5 | 4.6 ± 0.8 | >95 |
| SKF77434 | >10000 | <5 | 299 ± 68 | >95 |
| SKF75670 | >10000 | <5 | 36.4 ± 11 | >95 |
EC50 and Emax values were obtained from nonlinear regression analysis of individual dose–response curves as shown in panels A, C, and E of Figure 3. Comparison to the control maximal response exhibited by dopamine (DA) derives the percent response (Emax) of a test compound. Compounds that exhibited <5% stimulation of β-arrestin recruitment were considered inactive.
IC50 and maximal inhibition (Imax) values were obtained from nonlinear regression analysis of individual dose–response curves as shown in panels B, D, and F of Figure 3. Compounds that exhibited <5% inhibition of β-arrestin recruitment were considered inactive. NA indicates not applicable, as no inhibition was detected at the concentration tested.
While it might be tempting to hypothesize that spare receptors in the cAMP assay could explain the difference in efficacies observed for some of the compounds in the β-arrestin assay, it should be noted that the cellular level of D1R expression in the β-arrestin assay is ~10-fold (3200 fmol/mg of protein) higher than that of the cAMP assay (see above). However, it should also be noted that the cell background in these two assays differs (HEK293 for cAMP and CHO for β-arrestin). Thus, to rule out cell background as an explanation for the observed differences, we evaluated all of the compounds for their ability to stimulate cAMP accumulation in D1R-expressing CHO cells. Figure 1 of the Supporting Information shows that all of the G protein-biased agonists, including those that are entirely devoid of β-arrestin recruitment, are capable of potently stimulating D1R-mediated cAMP accumulation in a CHO cell background. Thus, cell background does not seem to play a role in the biased activity of the substituted benzazepines.
Because β-arrestin recruitment typically promotes receptor internalization, or endocytosis,60 we investigated the activity of the compounds for promoting D1R internalization (Figure 4 and Table 3). For this series of experiments, we used cells that are stably transfected with the D1R fused to a small fragment of β-galactosidase along with cotransfection of a complementing fragment of β-galactosidase that is fused to an endosomal marker protein.61 When the receptor is internalized into endosomes, β-galactosidase is complemented and provides a luminescent signal upon addition of substrate. Figure 4A shows that dihydrexidine is a full agonist in this assay, whereas A77636 and apomorphine are partial agonists. The substituted benzazepines that exhibited partial agonist activity for promoting β-arrestin recruitment typically showed partial agonist activity for promoting receptor internalization (Figure 4B). Similarly, with the exception of SKF38393, which showed a small, but statistically significant (Student’s t test; p = 0.004) degree of receptor internalization, all of the substituted benzazepines that were unable to recruit β-arrestin failed to promote receptor internalization (Figure 4C). The average EC50 and Emax values for all of the agonists tested in Figure 4 are listed in Table 3.
Figure 4.

Agonist-induced internalization of the D1R. Receptor internalization assays were conducted using the D1R PathHunter internalization assay system as described in Methods. Percent internalization is expressed as the maximum produced by dopamine. (A) Cells were stimulated with the reference D1R-selective agonists, A77636, dihydrexidine, and apomorphine. (B) Cells were incubated with the substituted benzazepines that showed low or no bias in the cAMP and β-arrestin recruitment assays. (C) Cells were incubated with the substituted benzazepines that were G protein-biased and showed insignificant β-arrestin recruitment. All data are means of at least three independent experiments conducted on different days in triplicate and expressed as a percentage of the maximal signal given by dopamine (N = 3–4). EC50 and Emax values were obtained for each individual experiment through nonlinear regression, and average data are reported in Table 3.
Table 3.
Stimulation of Receptor Internalization by D1R Agonistsa
| compound | EC50 (nM) | Emax (% of DA) |
|---|---|---|
| dopamine | 2086 ± 600 | 100 |
| A77636 | 0.18 ± 0.1 | 47.5 ± 2.8 |
| dihydrexidine | 428 ± 180 | 72.3 ± 1 |
| apomorphine | 14 ± 5.7 | 30.1 ± 6.7 |
| SKF81297 | 23 ± 12 | 59.5 ± 9 |
| chloro-APB | 9.6 ± 0.6 | 72.3 ± 6.8 |
| SKF83822 | 7600 ± 133 | 87.0 ± 2.5 |
| fenoldopam | 45.8 ± 26 | 52.7 ± 5.3 |
| SKF38393 | 165 ± 55 | 17.5 ± 2.7 |
| SKF83959 | NA | <5 |
| SKF82957 | NA | <5 |
| SKF77434 | NA | <5 |
| SKF75670 | NA | <5 |
EC50 and Emax values were obtained from nonlinear regression analysis of individual dose–response curves as shown in Figure 4. Comparison to the control maximal response exhibited by dopamine (DA) derives the percent response (Emax) of a test compound. Compounds that exhibited <5% stimulation of receptor internalization were considered inactive. NA indicates not applicable as no measurable internalization was detected.
Figure 5 shows a comparison of the maximal agonist-stimulated cAMP accumulation, β-arrestin recruitment, and receptor internalization produced by each agonist used in this study. In general, there is a good correlation between the ability of an agonist to promote β-arrestin recruitment and internalization of the D1R. There are, however, some exceptions. For instance, A77636 was a full agonist in promoting β-arrestin recruitment yet was a partial agonist in inducing receptor internalization (Figure 5A). Conversely, SKF38393 appeared not to recruit β-arrestin but stimulated a small degree of receptor internalization. Those agonists that were partially efficacious in promoting β-arrestin recruitment were also partial agonists for promoting D1R internalization. Most importantly, the substituted benzazepines shown in Figure 5B, SKF83959, SKF82957, SKF77434, SKF75670, and SKF38393, that showed no β-arrestin recruitment largely failed to promote receptor internalization despite the fact that they were able to stimulate cAMP accumulation. These results further suggest that these compounds are highly biased for activating G protein-mediated signaling of the D1R.
Figure 5.

Comparison of agonist-stimulated cAMP accumulation, β-arrestin recruitment, and receptor internalization. The maximal response seen using each agonist at 30 μM is compared to the maximal response produced by dopamine in each assay as determined in the assays shown in Figures 2–4. The data represent the means ± SEM values from three or four individual experiments. (A) D1R-selective agonists that exhibited low or no signaling bias. (B) Activities of highly G protein-biased substituted benzazepines.
Figure 5 also illustrates that some substituted benzazepine compounds exhibited little to no bias with respect to their efficacies for promoting cAMP accumulation versus β-arrestin recruitment and receptor internalization. For instance, the efficacies of chloro-APB for promoting β-arrestin recruitment and receptor internalization are only slightly lower than that for stimulating cAMP accumulation (Figure 5A). Similarly, for SKF83822, the efficacies for promoting cAMP accumulation and β-arrestin recruitment are similar while that for inducing receptor internalization is actually higher (Figure 5A). This suggests that the signaling bias seen for the compounds in Figure 5B cannot be explained by spare receptor or “signaling strength” phenomena.
We were also interested in evaluating the activities of the D1R G protein-biased agonists at the closely related D5 dopamine receptor (D5R). Figure 6A shows that these compounds also stimulate D5R-mediated cAMP accumulation with efficacies ranging from ~30 to 70% of the maximal dopamine response. A notable exception, however, was that SKF83959 did not exhibit agonist activity in this assay. Thus, in general, the activities for stimulating cAMP accumulation are similar to those observed with the D1R (Figure 2). However, in dramatic contrast to the D1R, all of the compounds were also partially efficacious in stimulating recruitment of β-arrestin to the D5R with efficacies ranging from 25 to 35% (Figure 6B). These results suggest that the compounds are less biased or even not biased at the D5R. In fact, SKF83959 even appears to exhibit “reverse bias” in that it is a partial agonist with respect to D5R-mediated β-arrestin recruitment while exerting no activity in stimulating cAMP accumulation. Thus, despite the structural similarities of the D1R and D5R, there are clearly agonists that can differ in their signaling biases at these two receptors.
Figure 6.

Stimulation of D5R-mediated cAMP accumulation or β-arrestin recruitment by the D1R-biased agonists. (A) D5R-expressing cells were stimulated with the five substituted benzazepines that exhibited high G protein bias at the D1R followed by cAMP measurement as described in Methods. (B) Recruitment of β-arrestin to the D5R following stimulation with the D1R-biased agonists was measured as described in Methods. All data are means of at least three independent experiments conducted on different days in triplicate and expressed as a percentage of the maximal signal given by dopamine (N = 3–4). EC50 and Emax values were obtained for each individual experiment through nonlinear regression, and average data are reported in Table 4.
The major finding of this investigation is the first identification of G protein-biased agonists of the D1R. We investigated a series of compounds based on the substituted benzazepine scaffold that had previously been shown to exhibit selectivity for the D1R. All of these compounds exhibited agonist activity for stimulating D1R-mediated cAMP accumulation, albeit with a variety of efficacies. In contrast, when we examined the ability of these compounds to stimulate the recruitment of β-arrestin to the receptor, some of the compounds (SKF83959, SKF38393, SKF82957, SKF77434, and SKF75670) were completely inactive. This biased activity cannot be explained by less receptor reserve in the β-arrestin assay as these cells express approximately 10 times the number of D1Rs versus the cells used in the cAMP assay. Moreover, cell background appears not to be a factor as similar results were observed in two different cell backgrounds (HEK293 and CHO). Importantly, consistent results were obtained in investigating agonist-induced D1R internalization, which is mediated by β-arrestin.60 The compounds that were inactive in recruiting β-arrestin to the D1R were inactive in promoting receptor internalization (with the exception of SKF38393, which promoted a small but significant amount of receptor internalization). These results suggest that, when administered to animals, these biased agonists will only stimulate G protein-mediated signaling by the D1R (primarily cAMP accumulation), while not promoting receptor internalization or β-arrestin-mediated signaling. In fact, such compounds will block dopamine signaling through β-arrestin or dopamine-induced receptor internalization.
Currently, little is known about D1R signaling through β-arrestin-mediated pathways; however, Urs et al.45 have shown that opioid-promoted locomotor activity in mice is regulated by a D1R/β-arrestin-2/pERK complex. Moreover, Fieblinger et al.62 have shown that the formation of D1R/pERK complexes is regulated by mGluR5 receptors and may be involved in dyskinesias. The development and use of signaling-biased agonists should now help in the identification of the physiologic and potentially therapeutic roles of downstream signaling pathways of the D1R.
Notably, among the G protein-biased agonists that we identified was SKF83959. This compound has been used extensively for investigations of the D1R and has a complicated pharmacological history (reviewed in ref 41). It has variously been reported as a D1R agonist or antagonist for stimulating cAMP accumulation but has also been described as a biased agonist for stimulating PLCβ activity and mobilizing Ca2+ (see ref 41 and references cited therein). This hypothesis has recently undergone revision to suggest that SKF83959 is a D1R–D2R dimer-selective agonist, where the D1R–D2R dimer is selectively linked to PLCβ activation and Ca2+ mobilization (ref 38 and references cited therein). We recently investigated this hypothesis and found that, while co-expression of both D1 and D2 receptors was required to observe a dopamine-stimulated Ca2+ response in HEK293 cells, this largely appeared to be due to downstream cross-talk pathways rather than D1R–D2R interactions.39 Surprisingly, we found that SKF83959 did not stimulate Ca2+ mobilization but rather functioned as an antagonist of this response. We also observed that SKF83959 was a somewhat promiscuous ligand that exhibited moderate affinity for a number of GPCRs.39 On the basis of these and other data, Mailman and colleagues40,41 have suggested that SKF83959 is not a PLCβ-biased agonist at the D1R, or a D1R–D2R dimer-selective agonist, but simply a partial D1R agonist that exhibits a number of behavioral effects caused by off-target activity at other GPCRs. In support of this hypothesis is the observation that in D1R knockout mice, “D1-selective” stimulation of PLCβ is still observed63 and that in adult rodents D1R–D2R dimers are either nonexistent or expressed at extremely low levels.64 We would now like to suggest an additional hypothesis, which is that the G protein-biased activity of SKF83959 may be partially responsible for some of the unexpected behavioral effects of this ligand in vivo when compared to nonbiased D1R agonists.
Our current results do differ somewhat from those of Mailman and colleagues in that they found SKF83959 was a partial agonist of both D1R-mediated cAMP accumulation and β-arrestin recruitment.40 An explanation for the difference in these results for β-arrestin recruitment is not immediately obvious; however, there are several observations that are important to note. First, we investigated a complete series of substituted benzazepines and identified several that exhibited the same signaling bias as SKF83959. Thus, our results are not limited to SKF83959. Second, our results with the G protein-biased agonists, including SKF83959, were corroborated using receptor internalization, which is mediated by β-arrestin,60 as the functional output. Third, it is of concern that in the study of Lee et al.40 the SKF83959-induced β-arrestin recruitment was only partially attenuated by a saturating concentration of the D1R antagonist SCH23390. Finally, Mailman and colleagues have previously published data57 suggesting that SKF83959 does not promote D1R internalization, or downregulation, defined as a loss of [3H]SCH23390 binding, which would be in agreement with our data showing that SKF83959 does not enhance β-arrestin recruitment or promote D1R internalization. Thus, taken together, we feel that all of the data support the notion that SKF83959 is a G protein-biased agonist at the D1R, although we do agree that SKF83959 is not biased for promoting D1R (or D1R–D2R dimer)-mediated PLCβ activation.
It was of interest to compare the activities of the biased compounds at both D1R- and D5R-mediated signaling outputs as these two receptors comprise the D1-like receptor subfamily. Surprisingly, despite the high degrees of structural similarity of these receptors,3 we observed significant differences in the functional activities of the D1R-biased compounds. Notably, all these compounds were able to promote the recruitment of β-arrestin to the D5R in contrast to their inability to recruit β-arrestin to the D1R. For the most part, the D1R-biased compounds were partial agonists on both D5R-mediated cAMP accumulation and β-arrestin recruitment. Strikingly, however, SKF83959 was devoid of activity on D5R-mediated cAMP accumulation despite it being a potent but low-efficacy partial agonist of D5R–β-arrestin recruitment. These data suggest that SKF83959 is actually a β-arrestin-biased partial agonist at the D5R, thus adding to its pharmacological complexity. Given the high degree of structural similary of the D1R and D5R, the small differences in structure may provide a starting point for identifying a structural basis for the biased signaling properties of the D1R and D5R.
Close inspection of the chemical structures of the biased and nonbiased substituted benzazepines did not reveal an obvious structure–activity relationship (SAR) for biased signaling activity at the D1R. Notably, however, the compounds used in this study were simply those that were commercially available rather than ones that were synthesized to interrogate the SAR for functional selectivity at the D1R. Future research will be directed at examining the chemical basis for the G protein signaling bias of the identified benzazepines, and determining if any of them are more D1R-selective than SKF83959. Such data might provide for the rational design of signaling-biased D1R agonists.
A surprising finding from this study was the G protein signaling bias of apomorphine at the D1R. This compound is frequently used as a dopaminergic agonist in behavioral studies and, in fact, is an FDA-approved drug for the acute treatment of Parkinson’s disease. It will thus be of interest to further explore the apomorphine scaffold for developing compounds with biased signaling activity at the D1R and other dopamine receptors.
In summary, we have identified the first G protein signaling-biased agonists for the D1R. While D1R-selective agonists have shown great potential for the treatment of various neuro-psychiatric disorders, such as Parkinson’s disease and impaired cognition, significant side effects such as hypotension and tolerance have precluded the approval of a number of candidate compounds. Our results suggest that a possible way forward for identifying improved drugs is to develop functionally selective D1R compounds, such as G protein-biased agonists that will exhibit high clinical efficacy but will have fewer side effects, such as drug-induced tolerance.
METHODS
Materials
D1R and D5R stably transfected cell lines were made in house by stably transfecting HEK293T cells with the human D1R or were purchased from either DiscoveRx Corp. (Fremont, CA) (D1R-U2OS cells and D1R-CHOK1 cells) or Codex Biosolutions (Gaithersburg, MD) (D1R and D5R HEK293 cells), as indicated. Cell-plating (CP) media and all components of the PathHunter and HitHunter detection kits were purchased from DiscoveRx Corp. Cell culture media and reagents were purchased from Mediatech/Cellgro (Manassas, VA). Cell culture flasks and assay plates were purchased from ThermoFisher Scientific (Waltham, MA). SKF83959 and SKF83822 were purchased from Tocris Bioscience/RD Systems (Minneapolis, MN). All other compounds and reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise specified.
cAMP Accumulation Assay
Assays were performed on cells stably expressing the human D1R or D5R, including D1R-CHOK1 cells (made in house), D1R-HEK293 cells (Codex Biosolutions), and D5R-HEK293 cells (Codex Biosolutions), as indicated. HEK293 cell lines were maintained in Dulbecco’s modified Eagle’s medium, supplemented with 10% fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin, 1 mM sodium pyruvate, 10 μg/mL gentamycin, and 250 μg/mL G418. CHOK1 cells were maintained in Ham’s F12 supplemented with 10% fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin, 20 mM HEPES, 500 μg/mL hygromycin, and 400 μg/mL G418. Cells were incubated at 37 °C, 5% CO2, and 90% humidity. Cells were seeded in 384-well black, clear-bottom plates at a density of 5000 cells/well at 10 μL/well. After incubation for 18–24 h at 37 °C, 5% CO2, and 90% humidity, the medium was removed and replaced with 5 μL of PBS/well. Cells were then treated with 2.5 μL of varying concentrations of compound diluted in PBS containing 25 μM 4-(3-butoxy-4-methoxybenzyl)-imidazolin-2-one (Ro 20-1724), 1 μM propranolol, and 0.2 mM sodium metabisulfite and incubated for 60 min at 37 °C, 5% CO2, and 90% humidity. cAMP was measured using the DiscoveRx HitHunter kit, according to the manufacturer’s recommendations. Briefly, an antibody and a working solution were added to each well according to the manufacturer’s protocol and incubated in the dark at room temperature for 60 min. Following incubation, EA reagent was added to the plates and luminescence (RLU) was measured (FDSS μCell, Hamamatsu Photonics K. K., Bridgewater, NJ) following a 3 h incubation in the dark at room temperature. Data are represented as a percentage of the control maximal dopamine-stimulated cAMP signal.
β-Arrestin Recruitment Assay
D1R and D5R agonist-mediated recruitment of β-arrestin-2 was assessed using the DiscoveRx PathHunter complementation assay, as previously described by our laboratory.27,65,66 Briefly, CHO-K1 cells stably expressing the D1R or D5R were seeded in cell plating (CP) media (DiscoveRx Corp.) at a density of 2625 cells/well in 384-well black, clear-bottom plates. Following incubation for 18–24 h, the cells were treated with the indicated concentrations of compound in PBS buffer containing 0.2 mM sodium metabisulfite and incubated at 37 °C for 90 min. DiscoveRx reagent was added to cells according to the manufacturer’s protocol followed by a 60 min incubation in the dark at room temperature. Luminescence was measured on a Hamamatsu FDSS μ-Cell reader for 5 s (Hamamatsu), and data were collected using the FDSS software. Data were collected as relative luminescence units (RLUs) and normalized to a percentage of the control luminescence seen with a maximal concentration of dopamine, with zero percent being RLUs produced in the absence of any compound. The Hill coefficients of the concentration response curves did not significantly differ from unity.
Internalization Assay
Agonist-mediated D1R internalization was assessed using the PathHunter Total GPCR Internalization Assay System (DiscoveRx Corp.), as deployed for other GPCRs,61 which utilizes a U2OS cell line stably expressing the D1R tagged with a Prolink tag, and an enzyme acceptor tag fused to an endosomal marker protein. Trafficking of the tagged receptor to the endosomes results in complementation of the two enzyme fragments and a subsequent chemiluminescent signal. The assay was conducted according to the manufacturer’s recommendation. Briefly, cells were seeded in cell plating (CP5) media (DiscoveRx) at a density of 2250 cells/well in 384-well black, clear-bottom plates. Following incubation for 24 h, cells were treated with varying concentrations of compound in PBS containing 0.2 mM sodium metabisulfite and incubated at 37 °C for 180 min. Reagent was then added to cells according to the manufacturer’s protocol followed by incubation in the dark at room temperature for 60 min. Luminescence was then measured on a Hamamatsu FDSS μ-cell reader for 8–12 s (Hamamatsu), and data were collected using the FDSS software. Data are represented as a percentage of the maximal internalization produced with dopamine, with zero percent represented by luminescence seen in the absence of any compound.
Radioligand Binding Assays
Radioligand competition binding assays were conducted with slight modifications as previously described by our laboratory.39 HEK293 cells stably transfected with human D1R (Codex Biosolutions) were dissociated from plates using EBSS-, and intact cells were collected by centrifugation at 900g for 10 min. Cells were resuspended and lysed using 5 mM Tris-HCl and 5 mM MgCl2 (pH 7.4) at 4 °C. The cell lysate was centrifuged at 30000g for 30 min, and the membrane faction was resuspended in EBSS with calcium at pH 7.4. Cell membranes (100 μL, containing ~8 μg of protein for D2-like receptor assays or ~10 μg of protein for D1-like receptor assays) were incubated for 90 min at room temperature with either [3H]SCH23390 (D1R and D5R) or [3H]methylspiperone (D2R–D4R) in a final reaction volume of 250 μL. Nonspecific binding was assessed in the presence of 4 μM (+)-butaclamol. Bound ligand was separated from free by filtration through a PerkinElmer Unifilter-96 GF/C 96-well microplate using the PerkinElmer Unifilter-96 Harvester, followed by washing three times, 1 mL per well, with ice-cold assay buffer. After samples had dried, 50 μL of liquid scintillation cocktail (MicroScint PS, PerkinElmer, Waltham, MA) was added to each well and plates were sealed and analyzed on a PerkinElmer Topcount NXT instrument.
Statistical Analysis
Data are expressed as a percentage of control values for each individual experiment. Nonlinear regression analyses were conducted using GraphPad Prism version 5.01 (GraphPad Software, Inc., La Jolla, CA). Results are expressed as means ± SEM. IC50 and Emax values were calculated from individual concentration responses and then averaged to generate means and SEM values.
Supplementary Material
Table 4.
Stimulation of D5R Receptor Signaling by Biased D1R Agonists
| compound | cAMP accumulationa
|
β-arrestin recruitmenta
|
||
|---|---|---|---|---|
| EC50 (nM) | Emax (% of DA) | EC50 (nM) | Emax (% of DA) | |
| dopamine | 184.7 ± 57 | 100 | 13.6 ± 2.4 | 100 |
| SKF38393 | 40.6 ± 3.9 | 77.4 ± 7.3 | 23.7 ± 5.8 | 42.9 ± 1.9 |
| SKF83959 | >10000 | <5 | 3.7 ± 2 | 20.3 ± 1 |
| SKF82957 | 0.17 ± 0.3 | 37.7 ± 2.6 | 0.82 ± 0.3 | 30.9 ± 1.1 |
| SKF77434 | 80.8 ± 68.4 | 69.2 ± 7.3 | 29.9 ± 5.1 | 34.7 ± 1 |
| SKF75670 | 0.019 ± 0.003 | 53.8 ± 8.5 | 5.5 ± 0.2 | 31.7 ± 0.6 |
EC50 and Emax values were obtained from nonlinear regression analysis of individual dose–response curves for cAMP accumulation or β-arrestin recruitment as shown in Figure 6. Comparison to the control maximal response exhibited by dopamine (DA) derives the percent response (Emax) of a test compound.
Acknowledgments
Funding
This project was funded by the Intramural Research Program of the National Institute of Neurological Disorders and Stroke (NINDS), in the National Institutes of Health (NIH).
We thank Mr. Trevor Doyle, Ms. Preetha Nandi, and Dr. Kadee Luderman for excellent technical assistance and control binding experiments.
ABBREVIATIONS
- D1R
D1 dopamine receptor
- D2R
D2 dopamine receptor
- D3R
D3 dopamine receptor
- D4R
D4 dopamine receptor
- D5R
D5 dopamine receptor
- EC50
50% excitatory concentration
- IC50
50% inhibitory concentration
- SEM
standard error of the mean
Footnotes
Author Contributions
J.L.C. conducted experiments, performed data analysis, participated in research design, and participated in writing the manuscript. R.B.F. performed data analysis, participated in research design, and contributed to writing the manuscript. D.R.S. oversaw the research project, contributed to research design, and contributed to writing the manuscript.
The authors declare no competing financial interest.
CHO cells stably expressing the D1R were assayed for agonist stimulation of cAMP accumulation. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Rankin ML, Hazelwood LA, Free RB, Namkung Y, Rex EB, Roof RA, Sibley DR. Molecular pharmacology of the dopamine receptors. In: Iversen LL, Iversen SD, Dunnett SB, Bjorklund A, editors. Dopamine Handbook. Oxford University Press; New York: 2009. pp. 63–87. [Google Scholar]
- 2.Kapur S, Mamo D. Half a century of antipsychotics and still a central role for dopamine D2 receptors. Prog Neuro-Psychopharmacol Biol Psychiatry. 2003;27:1081–1090. doi: 10.1016/j.pnpbp.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 3.Sibley DR, Monsma FJ., Jr Molecular biology of dopamine receptors. Trends Pharmacol Sci. 1992;13:61–69. doi: 10.1016/0165-6147(92)90025-2. [DOI] [PubMed] [Google Scholar]
- 4.Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- 5.Swinney DC. Successful drug discovery. Curr Top Med Chem. 2006;6:403–404. doi: 10.2174/156802606776743110. [DOI] [PubMed] [Google Scholar]
- 6.Walters RW, Shukla AK, Kovacs JJ, Violin JD, DeWire SM, Lam CM, Chen JR, Muehlbauer MJ, Whalen EJ, Lefkowitz RJ. β-Arrestin1 mediates nicotinic acid-induced flushing, but not its antilipolytic effect, in mice. J Clin Invest. 2009;119:1312–1321. doi: 10.1172/JCI36806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobilka BK, Deupi X. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol Sci. 2007;28:397–406. doi: 10.1016/j.tips.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 8.Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, Miller KJ, Spedding M, Mailman RB. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- 9.Wess J, Han SJ, Kim SK, Jacobson KA, Li JH. Conformational changes involved in G-protein-coupled-receptor activation. Trends Pharmacol Sci. 2008;29:616–625. doi: 10.1016/j.tips.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whalen EJ, Rajagopal S, Lefkowitz RJ. Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol Med. 2011;17:126–139. doi: 10.1016/j.molmed.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kenakin T. Collateral efficacy in drug discovery: Taking advantage of the good (allosteric) nature of 7TM receptors. Trends Pharmacol Sci. 2007;28:407–415. doi: 10.1016/j.tips.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 12.Kenakin T. Functional selectivity through protean and biased agonism: Who steers the ship? Mol Pharmacol. 2007;72:1393–1401. doi: 10.1124/mol.107.040352. [DOI] [PubMed] [Google Scholar]
- 13.Kenakin TP. Pharmacological onomastics: What’s in a name? Br J Pharmacol. 2008;153:432–438. doi: 10.1038/sj.bjp.0707407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mailman RB. GPCR functional selectivity has therapeutic impact. Trends Pharmacol Sci. 2007;28:390–396. doi: 10.1016/j.tips.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abdul-Ridha A, Lopez L, Keov P, Thal DM, Mistry SN, Sexton PM, Lane JR, Canals M, Christopoulos A. Molecular determinants of allosteric modulation at the M1 muscarinic acetylcholine receptor. J Biol Chem. 2014;289:6067–6079. doi: 10.1074/jbc.M113.539080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dror RO, Green HF, Valant C, Borhani DW, Valcourt JR, Pan AC, Arlow DH, Canals M, Lane JR, Rahmani R, Baell JB, Sexton PM, Christopoulos A, Shaw DE. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature. 2013;503:295–299. doi: 10.1038/nature12595. [DOI] [PubMed] [Google Scholar]
- 17.Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hubner H, Pardon E, Valant C, Sexton PM, Christopoulos A, Felder CC, Gmeiner P, Steyaert J, Weis WI, Garcia KC, Wess J, Kobilka BK. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 2013;504:101–106. doi: 10.1038/nature12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu JJ, Horst R, Katritch V, Stevens RC, Wuthrich K. Biased signaling pathways in β2-adrenergic receptor characterized by 19F-NMR. Science. 2012;335:1106–1110. doi: 10.1126/science.1215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wacker D, Wang C, Katritch V, Han GW, Huang XP, Vardy E, McCorvy JD, Jiang Y, Chu M, Siu FY, Liu W, Xu HE, Cherezov V, Roth BL, Stevens RC. Structural features for functional selectivity at serotonin receptors. Science. 2013;340:615–619. doi: 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kilts JD, Connery HS, Arrington EG, Lewis MM, Lawler CP, Oxford GS, O’Malley KL, Todd RD, Blake BL, Nichols DE, Mailman RB. Functional selectivity of dopamine receptor agonists. II Actions of dihydrexidine in D2L receptor-transfected MN9D cells and pituitary lactotrophs. J Pharmacol Exp Ther. 2002;301:1179–1189. doi: 10.1124/jpet.301.3.1179. [DOI] [PubMed] [Google Scholar]
- 21.Mottola DM, Kilts JD, Lewis MM, Connery HS, Walker QD, Jones SR, Booth RG, Hyslop DK, Piercey M, Wightman RM, Lawler CP, Nichols DE, Mailman RB. Functional selectivity of dopamine receptor agonists. I Selective activation of postsynaptic dopamine D2 receptors linked to adenylate cyclase. J Pharmacol Exp Ther. 2002;301:1166–1178. doi: 10.1124/jpet.301.3.1166. [DOI] [PubMed] [Google Scholar]
- 22.Gay EA, Urban JD, Nichols DE, Oxford GS, Mailman RB. Functional selectivity of D2 receptor ligands in a Chinese hamster ovary hD2L cell line: Evidence for induction of ligand-specific receptor states. Mol Pharmacol. 2004;66:97–105. doi: 10.1124/mol.66.1.97. [DOI] [PubMed] [Google Scholar]
- 23.Lane JR, Powney B, Wise A, Rees S, Milligan G. Protean agonism at the dopamine D2 receptor: (S)-3-(3-Hydroxyphenyl)-N-propylpiperidine is an agonist for activation of Go1 but an antagonist/inverse agonist for Gi1, Gi2, and Gi3. Mol Pharmacol. 2007;71:1349–1359. doi: 10.1124/mol.106.032722. [DOI] [PubMed] [Google Scholar]
- 24.Lane JR, Powney B, Wise A, Rees S, Milligan G. G protein coupling and ligand selectivity of the D2L and D3 dopamine receptors. J Pharmacol Exp Ther. 2008;325:319–330. doi: 10.1124/jpet.107.134296. [DOI] [PubMed] [Google Scholar]
- 25.Beaulieu JM, Gainetdinov RR, Caron MG. The Akt-GSK-3 signaling cascade in the actions of dopamine. Trends Pharmacol Sci. 2007;28:166–172. doi: 10.1016/j.tips.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 26.Beaulieu JM, Gainetdinov RR, Caron MG. Akt/GSK3 signaling in the action of psychotropic drugs. Annu Rev Pharmacol Toxicol. 2009;49:327–347. doi: 10.1146/annurev.pharmtox.011008.145634. [DOI] [PubMed] [Google Scholar]
- 27.Free RB, Chun LS, Moritz AE, Miller BN, Doyle TB, Conroy JL, Padron A, Meade JA, Xiao J, Hu X, Dulcey AE, Han Y, Duan L, Titus S, Bryant-Genevier M, Barnaeva E, Ferrer M, Javitch JA, Beuming T, Shi L, Southall NT, Marugan JJ, Sibley DR. Discovery and characterization of a G protein-biased agonist that inhibits β-arrestin recruitment to the D2 dopamine receptor. Mol Pharmacol. 2014;86:96–105. doi: 10.1124/mol.113.090563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allen JA, Yost JM, Setola V, Chen X, Sassano MF, Chen M, Peterson S, Yadav PN, Huang XP, Feng B, Jensen NH, Che X, Bai X, Frye SV, Wetsel WC, Caron MG, Javitch JA, Roth BL, Jin J. Discovery of β-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc Natl Acad Sci USA. 2011;108:18488–18493. doi: 10.1073/pnas.1104807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen X, Sassano MF, Zheng L, Setola V, Chen M, Bai X, Frye SV, Wetsel WC, Roth BL, Jin J. Structure-functional selectivity relationship studies of β-arrestin-biased dopamine D(2) receptor agonists. J Med Chem. 2012;55:7141–7153. doi: 10.1021/jm300603y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Undie AS, Friedman E. Stimulation of a dopamine D1 receptor enhances inositol phosphates formation in rat brain. J Pharmacol Exp Ther. 1990;253:987–992. [PubMed] [Google Scholar]
- 31.Undie AS, Friedman E. Selective dopaminergic mechanism of dopamine and SKF38393 stimulation of inositol phosphate formation in rat brain. Eur J Pharmacol. 1992;226:297–302. doi: 10.1016/0922-4106(92)90046-x. [DOI] [PubMed] [Google Scholar]
- 32.Downes RP, Waddington JL. Grooming and vacuous chewing induced by SK&F 83959, an agonist of dopamine ‘D1-like’ receptors that inhibits dopamine-sensitive adenylyl cyclase. Eur J Pharmacol. 1993;234:135–136. doi: 10.1016/0014-2999(93)90718-w. [DOI] [PubMed] [Google Scholar]
- 33.Deveney AM, Waddington JL. Pharmacological characterization of behavioural responses to SK&F 83959 in relation to ‘D1-like’ dopamine receptors not linked to adenylyl cyclase. Br J Pharmacol. 1995;116:2120–2126. doi: 10.1111/j.1476-5381.1995.tb16420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Panchalingam S, Undie AS. SKF83959 exhibits biochemical agonism by stimulating [35S]GTPγS binding and phosphoinositide hydrolysis in rat and monkey brain. Neuro-pharmacology. 2001;40:826–837. doi: 10.1016/s0028-3908(01)00011-9. [DOI] [PubMed] [Google Scholar]
- 35.Jin LQ, Goswami S, Cai G, Zhen X, Friedman E. SKF83959 selectively regulates phosphatidylinositol-linked D1 dopamine receptors in rat brain. J Neurochem. 2003;85:378–386. doi: 10.1046/j.1471-4159.2003.01698.x. [DOI] [PubMed] [Google Scholar]
- 36.Lee SP, So CH, Rashid AJ, Varghese G, Cheng R, Lanca AJ, O’Dowd BF, George SR. Dopamine D1 and D2 receptor co-activation generates a novel phospholipase C-mediated calcium signal. J Biol Chem. 2004;279:35671–35678. doi: 10.1074/jbc.M401923200. [DOI] [PubMed] [Google Scholar]
- 37.Rashid AJ, So CH, Kong MM, Furtak T, El-Ghundi M, Cheng R, O’Dowd BF, George SR. D1–D2 dopamine receptor heterooligomers with unique pharmacology are coupled to rapid activation of Gq/11 in the striatum. Proc Natl Acad Sci USA. 2007;104:654–659. doi: 10.1073/pnas.0604049104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perreault ML, Shen MY, Fan T, George SR. Regulation of c-fos expression by the dopamine D1–D2 receptor heteromer. Neuroscience. 2014;285C:194–203. doi: 10.1016/j.neuroscience.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chun LS, Free RB, Doyle TB, Huang XP, Rankin ML, Sibley DR. D1–D2 dopamine receptor synergy promotes calcium signaling via multiple mechanisms. Mol Pharmacol. 2013;84:190–200. doi: 10.1124/mol.113.085175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee SM, Kant A, Blake D, Murthy V, Boyd K, Wyrick SJ, Mailman RB. SKF-83959 is not a highly-biased functionally selective D1 dopamine receptor ligand with activity at phospholipase C. Neuropharmacology. 2014;86:145–154. doi: 10.1016/j.neuropharm.2014.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee SM, Yang Y, Mailman RB. Dopamine D1 receptor signaling: Does GαQ-phospholipase C actually play a role? J Pharmacol Exp Ther. 2014;351:9–17. doi: 10.1124/jpet.114.214411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Andringa G, Stoof JC, Cools AR. Sub-chronic administration of the dopamine D(1) antagonist SKF 83959 in bilaterally MPTP-treated rhesus monkeys: Stable therapeutic effects and wearing-off dyskinesia. Psychopharmacology. 1999;146:328–334. doi: 10.1007/s002130051124. [DOI] [PubMed] [Google Scholar]
- 43.Fang X, Guo L, Jia J, Jin GZ, Zhao B, Zheng YY, Li JQ, Zhang A, Zhen XC. SKF83959 is a novel triple reuptake inhibitor that elicits anti-depressant activity. Acta Pharmacol Sin. 2013;34:1149–1155. doi: 10.1038/aps.2013.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo L, Zhao J, Jin G, Zhao B, Wang G, Zhang A, Zhen X. SKF83959 is a potent allosteric modulator of sigma-1 receptor. Mol Pharmacol. 2013;83:577–586. doi: 10.1124/mol.112.083840. [DOI] [PubMed] [Google Scholar]
- 45.Urs NM, Daigle TL, Caron MG. A dopamine D1 receptor-dependent β-arrestin signaling complex potentially regulates morphine-induced psychomotor activation but not reward in mice. Neuropsychopharmacology. 2011;36:551–558. doi: 10.1038/npp.2010.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mailman RB, Nichols DE. Dopamine D1 receptor agonists as antiparkinson drugs. Trends Pharmacol Sci. 1998;19:255–256. doi: 10.1016/s0165-6147(98)01219-x. [DOI] [PubMed] [Google Scholar]
- 47.Arnsten AF, Cai JX, Murphy BL, Goldman-Rakic PS. Dopamine D1 receptor mechanisms in the cognitive performance of young adult and aged monkeys. Psychopharmacology. 1994;116:143–151. doi: 10.1007/BF02245056. [DOI] [PubMed] [Google Scholar]
- 48.Goldman-Rakic PS. Working memory dysfunction in schizophrenia. J Neuropsychiatry Clin Neurosci. 1994;6:348–357. doi: 10.1176/jnp.6.4.348. [DOI] [PubMed] [Google Scholar]
- 49.Nichols DE. Dopamine Receptor Subtype-Selective Drugs: D1-Like Receptors. In: Neve KA, editor. The Dopamine Receptors. 2. Humana Press; Totowa, NJ: 2009. pp. 75–100. [Google Scholar]
- 50.Weed MR, Vanover KE, Woolverton WL. Reinforcing effect of the D1 dopamine agonist SKF 81297 in rhesus monkeys. Psychopharmacology. 1993;113:51–52. doi: 10.1007/BF02244333. [DOI] [PubMed] [Google Scholar]
- 51.Undie AS, Weinstock J, Sarau HM, Friedman E. Evidence for a distinct D1-like dopamine receptor that couples to activation of phosphoinositide metabolism in brain. J Neurochem. 1994;62:2045–2048. doi: 10.1046/j.1471-4159.1994.62052045.x. [DOI] [PubMed] [Google Scholar]
- 52.Peacock L, Gerlach J. Aberrant behavioral effects of a dopamine D1 receptor antagonist and agonist in monkeys: Evidence of uncharted dopamine D1 receptor actions. Biol Psychiatry. 2001;50:501–509. doi: 10.1016/s0006-3223(01)01189-1. [DOI] [PubMed] [Google Scholar]
- 53.Chausmer AL, Katz JL. Comparison of interactions of D1-like agonists, SKF 81297, SKF 82958 and A-77636, with cocaine: Locomotor activity and drug discrimination studies in rodents. Psychopharmacology. 2002;159:145–153. doi: 10.1007/s002130100896. [DOI] [PubMed] [Google Scholar]
- 54.Nichols AJ, Ruffolo RR, Jr, Brooks DP. The pharmacology of fenoldopam. Am J Hypertens. 1990;3:116S–119S. doi: 10.1093/ajh/3.6.116s. [DOI] [PubMed] [Google Scholar]
- 55.Arnt J, Hyttel J, Sanchez C. Partial and full dopamine D1 receptor agonists in mice and rats: Relation between behavioural effects and stimulation of adenylate cyclase activity in vitro. Eur J Pharmacol. 1992;213:259–267. doi: 10.1016/0014-2999(92)90690-6. [DOI] [PubMed] [Google Scholar]
- 56.Lewis MM, Watts VJ, Lawler CP, Nichols DE, Mailman RB. Homologous desensitization of the D1A dopamine receptor: Efficacy in causing desensitization dissociates from both receptor occupancy and functional potency. J Pharmacol Exp Ther. 1998;286:345–353. [PubMed] [Google Scholar]
- 57.Ryman-Rasmussen JP, Nichols DE, Mailman RB. Differential activation of adenylate cyclase and receptor internalization by novel dopamine D1 receptor agonists. Mol Pharmacol. 2005;68:1039–1048. doi: 10.1124/mol.105.012153. [DOI] [PubMed] [Google Scholar]
- 58.Glick SD, Carlson JN, Baird JL, Maisonneuve IM, Bullock AE. Basal and amphetamine-induced asymmetries in striatal dopamine release and metabolism: Bilateral in vivo micro-dialysis in normal rats. Brain Res. 1988;473:161–164. doi: 10.1016/0006-8993(88)90329-0. [DOI] [PubMed] [Google Scholar]
- 59.Schulz DW, Stanford EJ, Wyrick SW, Mailman RB. Binding of [3H]SCH23390 in rat brain: Regional distribution and effects of assay conditions and GTP suggest interactions at a D1-like dopamine receptor. J Neurochem. 1985;45:1601–1611. doi: 10.1111/j.1471-4159.1985.tb07233.x. [DOI] [PubMed] [Google Scholar]
- 60.Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. Differential affinities of visual arrestin, β arrestin1, and β arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem. 2000;275:17201–17210. doi: 10.1074/jbc.M910348199. [DOI] [PubMed] [Google Scholar]
- 61.DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, Chen XT, Pitis PM, Gotchev D, Yuan C, Koblish M, Lark MW, Violin JD. A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther. 2013;344:708–717. doi: 10.1124/jpet.112.201616. [DOI] [PubMed] [Google Scholar]
- 62.Fieblinger T, Sebastianutto I, Alcacer C, Bimpisidis Z, Maslava N, Sandberg S, Engblom D, Cenci MA. Mechanisms of dopamine D1 receptor-mediated ERK1/2 activation in the parkinsonian striatum and their modulation by metabotropic glutamate receptor type 5. J Neurosci. 2014;34:4728–4740. doi: 10.1523/JNEUROSCI.2702-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Friedman E, Jin LQ, Cai GP, Hollon TR, Drago J, Sibley DR, Wang HY. D1-like dopaminergic activation of phosphoinositide hydrolysis is independent of D1A dopamine receptors: Evidence from D1A knockout mice. Mol Pharmacol. 1997;51:6–11. doi: 10.1124/mol.51.1.6. [DOI] [PubMed] [Google Scholar]
- 64.Frederick AL, Yano H, Trifilieff P, Vishwasrao HD, Biezonski D, Meszaros J, Sibley DR, Kellendonk C, Sonntag KC, Graham DL, Colbran RJ, Stanwood GD, Javitch JA. Evidence against dopamine D1/D2 receptor heteromers. Mol Psychiatry. 2015 doi: 10.1038/mp.2014.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Banala AK, Levy BA, Khatri SS, Furman CA, Roof RA, Mishra Y, Griffin SA, Sibley DR, Luedtke RR, Newman AH. N-(3-Fluoro-4-(4-(2-methoxy or 2,3-dichlorophenyl)-piperazine-1-yl)butyl)arylcarboxamides as selective dopamine D3 receptor ligands: Critical role of the carboxamide linker for D3 receptor selectivity. J Med Chem. 2011;54:3581–3594. doi: 10.1021/jm200288r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bergman J, Roof RA, Furman CA, Conroy JL, Mello NK, Sibley DR, Skolnick P. Modification of cocaine self-administration by buspirone (buspar(R)): Potential involvement of D3 and D4 dopamine receptors. Int J Neuropsychopharmacol. 2013;16:445–458. doi: 10.1017/S1461145712000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.