

SUMMARY
Proper maintenance of mitochondrial activity is essential for metabolic homeostasis. Widespread phosphorylation of mitochondrial proteins may be an important element of this process; yet little is known about which enzymes control mitochondrial phosphorylation, or which phosphosites have functional impact. We investigate these issues by disrupting Ptc7p—a conserved but largely uncharacterized mitochondrial matrix PP2C-type phosphatase. Loss of Ptc7p causes respiratory growth defects concomitant with elevated phosphorylation of select matrix proteins. Among these, Δptc7 yeast exhibit an increase in phosphorylation of Cit1p—the canonical citrate synthase of the tricarboxylic acid cycle—that diminishes its activity. We find that phosphorylation of S462 can eliminate Cit1p enzymatic activity likely by disrupting its proper dimerization, and that Ptc7p-driven dephosphorylation rescues Cit1p activity. Collectively, our work connects Ptc7p to an essential TCA cycle function and to additional phosphorylation events that may affect mitochondrial activity inadvertently or in a regulatory manner.
Graphical abstract
INTRODUCTION
Nearly all cells rely on mitochondria for their core metabolic needs. As such, it is essential that cells are able to appropriately maintain the content, function, and activity of these organelles. Mitochondrial adaptation can involve transcriptional reprogramming, protease-based proteome remodeling, altered mitochondrial dynamics, and the use of post-translational modifications (PTMs) to regulate protein activity (Lin et al., 2005; Nunnari and Suomalainen, 2012; Pagliarini and Dixon, 2006; Quiros et al., 2015). The importance of PTMs in mitochondrial metabolism was recognized ~50 years ago with the discovery that reversible phosphorylation modulates the activity of pyruvate dehydrogenase (PDH) (Linn et al., 1969). More recently, roles for mitochondrial acylation (He et al., 2012), nitrosylation (Doulias et al., 2013), and redox modifications (Mailloux et al., 2014) have been identified, and a more exhaustive cataloging of protein phosphorylation sites has been achieved (Grimsrud et al., 2012). From this collective work, it is clear that PTMs are abundant, dynamic, and can affect enzymatic function.
Despite this progress, various aspects of PTM-driven regulation of mitochondrial function are unclear. First, it remains largely uncertain which mitochondrial PTMs—from among the thousands identified—might affect protein function, and which may instead be inconsequential. Second, the enzymes that regulate these modifications are largely unidentified or uncharacterized. To explore these issues, we focused on Ptc7p, a poorly characterized mitochondrial PP2C phosphatase conserved from yeast through humans. Our work connects this phosphatase to a select set of ~20 mitochondrial phosphoproteins from amongst thousands of observed phosphoisoforms that span distinct pathways. In particular, we demonstrate that Ptc7p-driven dephosphorylation of mitochondrial citrate synthase may be an important process for maintaining optimal mitochondrial activity.
RESULTS
Ptc7p phosphatase activity supports respiratory function
To further elucidate the nature of mitochondrial protein phosphorylation, we focused on identifying phosphatases that could directly dephosphorylate matrix phosphoproteins. The MitoCarta compendium includes 11 proteins with predicted PFAM phosphatase domains (Calvo et al., 2016; Pagliarini et al., 2008), and two additional dual specificity phosphatases have also been localized to mitochondria (Rardin et al., 2008). Among these, we prioritized PPTC7 for further study based on its matrix localization (Rhee et al., 2013), conservation through Saccharomyces cerevisiae (Ptc7p), and canonical PP2C protein phosphatase domain (Figure 1A).
Figure 1. Ptc7p phosphatase activity supports respiratory function.

(A) Summary table of mitochondrial phosphatases, analyzed for protein phosphatase domains, matrix localization, non-association with a protein complex (e.g. PDH), and presence of a yeast ortholog. PPTC7 (highlighted) is the only phosphatase to meet all four criteria.
(B) In vitro phosphatase activity assay of Ptc7p against pNPP with divalent cations Mn2+ and Mg2+ (mean ± SD, n=3).
(C) In vitro pNPP phosphatase activity assay of WT Ptc7p and two mutants predicted to disrupt metal binding (mean ± SD, n=3). Coomassie staining (below) demonstrates comparable protein concentration and purity.
(D) Maximum growth rate of WT and Δptc7 yeast in Ura− media containing 3% glycerol (G) (mean ± SD, n=4). Rescue strains express a plasmid containing ptc7 (WT: wild type ptc7; D109A: catalytically inactive mutant of ptc7).
(E) OCR of same cultures as in (D) grown in Ura− media containing 2% dextrose (D) (mean ± SD, n=3).
* p-value < 0.05; ** p-value < 0.01; *** p-value < 0.001; N.S., not significant.
See also Figure S1.
We expressed and purified recombinant Ptc7p from E. coli and found that it dephosphorylates para-nitrophenyl phosphate (pNPP) in vitro with kinetics consistent with known protein phosphatases (Figure S1A) (McAvoy and Nairn, 2010), and that it strongly prefers Mn2+ to Mg2+ for catalysis (Figure 1B). Mutation of the conserved aspartic acid residues that coordinate these metal ions completely abrogated activity (Figure 1C), consistent with the established catalytic mechanism of PP2C-type phosphatases (Das et al., 1996).
We next examined phenotypes related to mitochondrial function in Δptc7 yeast. We found no difference in growth between wild type (WT) and Δptc7 yeast in dextrose-based media (Figure S1B), but a marked decrease in Δptc7 growth rate in non-fermentable glycerol-based media (Figure 1D). This growth deficiency was rescued by expression of WT but not the catalytically inactive D109A Ptc7p mutant (Figure 1D). Consistently, Δptc7 yeast also exhibited a decreased oxygen consumption rate (OCR) that was likewise rescued only by WT Ptc7p (Figure 1E). In contrast to a recent report (Martin-Montalvo et al., 2013), we did not observe a defect in coenzyme Q (CoQ) levels in Δptc7 yeast (Figure S1C), and thus hypothesize that the growth impairment we observe is caused by distinct mitochondrial defects. Collectively, these results suggest that Ptc7p is an active protein phosphatase poised to dephosphorylate mitochondrial matrix proteins to promote mitochondrial function.
Quantitative phosphoproteomics identifies potential Ptc7p substrates
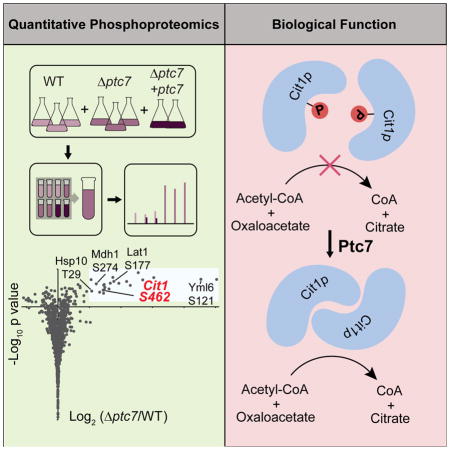
To identify potential substrates of Ptc7p, we performed quantitative phosphoproteomic analysis on WT, Δptc7, and Δptc7 + ptc7 (rescue) yeast strains (Figure 2A, Table S1, Table S2). Yeast were cultured in media containing 0.1% glucose and 3% glycerol to enable a short fermentation phase (allowing the growth of sufficient yeast for proteomics analysis), followed by a longer respiration phase during which the Δptc7 growth defect is evident (Figure S2A). Total proteomic and phosphoproteomic analyses were performed using 8-plex tandem mass tags (TMTs) for isobaric quantification, resulting in the quantification of 4271 proteins (886 mitochondrial) and 6647 phosphoisoforms (1182 mitochondrial) (Figure 2B).
Figure 2. Quantitative phosphoproteomics identifies potential Ptc7p substrates.

(A) Experimental workflow of phosphoproteomics. Peptides from three strains (WT, Δptc7, and Δptc7 + ptc7) were tagged with 8-plex TMT for isobaric quantification.
(B) Fold changes in mitochondrial phosphoisoform abundances (log2(Δptc7/WT), normalized to total protein abundance, n=3) versus significance (−log10(p-value)). Grey area indicates significance threshold (log2(Δptc7/WT) > 1 and p-value < 0.05). Five highlighted phosphoisoforms are prioritized candidate Ptc7p substrates. Inset table summarizes quantified total and mitochondrial proteins or phosphoisoforms; up arrow indicates significantly increased changes; down arrow indicates significantly decreased changes.
(C) Heat map of 19 mitochondrial phosphoisoforms whose abundances were significantly increased in Δptc7, and were restored to WT level or below in the rescue strain (Δptc7 + ptc7). * denotes mitochondrial matrix phosphosites conserved in higher eukaryotes.
Given that Ptc7p is an active protein phosphatase, we hypothesized that Δptc7 yeast would exhibit increased phosphorylation levels of its substrate(s). Indeed, the deletion of ptc7 caused a marked increase of 23 mitochondrial phosphoisoforms (Figure 2B). In contrast, only 7 non-mitochondrial phosphoisoforms (~0.1%) were significantly increased (Figures 2B, S2B). Furthermore, 19 of the 23 upregulated phosphoisoforms returned to WT levels (or below) when we rescued the Δptc7 strain with plasmid-based ptc7 expression (Figure 2C). Coupled with the general lack of proteome alterations in Δptc7 yeast (Figure S2C), these data suggest that these 19 phosphoproteins may be direct substrates of Ptc7p. None of these phosphorylation sites were associated with enzymes in CoQ biosynthesis, again suggesting our observed mitochondrial defect in Δptc7 yeast is distinct from this pathway.
To prioritize these likely Ptc7p substrates, we considered the submitochondrial localization of each protein and the conservation of each phosphosite. Five of the 19 phosphoproteins noted above—Yml6p, Lat1p, Cit1p, Mdh1p, and Hsp10p—share matrix localization with Ptc7p and have conserved phosphorylatable residues at the positions identified in our analyses (Figures S2D – S2H). Notably, three of these proteins (Lat1p, Cit1p, and Mdh1p) are enzymes associated with the pyruvate dehydrogenase complex/TCA cycle. From among these, we focused on Cit1p, the mitochondrial citrate synthase in yeast whose activity is generally not known to be affected by PTMs.
Phosphorylation of Cit1p at S462 disrupts enzyme function
The mitochondrial dysfunction of Δptc7 yeast (Figure 1) suggests that the phosphorylation of putative Ptc7p substrates likely diminishes their function. To test this hypothesis, we focused on phosphorylation of S462 on Cit1p (Figure S3A). We made non-phosphorylatable (S462A) and phosphomimetic (S462E) mutants of Cit1p and assessed their functional effects. First, we tested the ability of these mutants to rescue the growth of Δcit1 yeast on acetate-based media. Consistent with our hypothesis, S462E was unable to rescue the Δcit1 growth deficiency, while WT and S462A fully restored growth (Figure 3A). Second, we measured the activity of each construct from yeast lysate and found that Δcit1 yeast expressing S462E had no Cit1p activity, while lysate containing a S462A mutant had a slight increase in activity versus WT (Figure 3B). Further, expression of S462E failed to restore total cellular citrate levels in Δcit1 yeast (Figure S3B), and resulted in an altered metabolic profile more similar to Δcit1 yeast expressing empty vector (EV) than those expressing WT or S462A (Figure S3C). Finally, we purified recombinant WT and Cit1p mutants and measured their kinetics with oxaloacetate (OAA). Consistent with our in vivo data, the phosphomimetic S462E completely disrupted activity, while, surprisingly, S462A showed a significantly higher Vmax for OAA compared to WT (Figure 3C).
Figure 3. Phosphorylation of Cit1p at S462 disrupts enzyme function.

(A) Serial dilutions of WT and Δcit1 yeast expressing various plasmids grown on glucose- or acetate-containing Ura− plates.
(B) Citrate synthase activity of Δcit1 lysate expressing EV, WT, S462A, or S462E Cit1p (mean ± SD, n=3). Statistics are relative to WT activity (lane #2). Inset shows immunoblot against FLAG (Cit1p-FLAG), or actin (loading control).
(C) Kinetic curve of recombinant WT, S462A, or S462E Cit1p. Citrate synthase activity (μM/sec) is plotted versus concentration of OAA (μM). Inset shows comparable loading (Coomassie staining). Table shows calculated Vmax and Km for each Cit1p mutant.
(D) Citrate synthase activity of lysate from WT or Δptc7 (mean ± SD, n=4). Rescue strains express a plasmid containing ptc7 (WT: wild type ptc7; D109A: catalytically inactive mutant of ptc7).
(E) Citrate synthase activity of recombinant WT or phospho-S462 (pSer462) Cit1p treated with WT or D109A Ptc7p (mean ± SD, n=3).
(F) Coomassie staining of a PhosTag gel loaded with samples in (E).
* p-value < 0.05; ** p-value < 0.01; *** p-value < 0.001; N.S., not significant.
See also Figure S3.
To further assess whether Cit1p is a bona fide substrate of Ptc7p, we tested citrate synthase activity in Δptc7 yeast. Citrate synthase activity was decreased by ~40% in this strain, and was restored by WT Ptc7p, but not by the D109A mutant (Figure 3D). These data are consistent with our hypothesis that phosphorylation of Cit1p S462 can decrease its enzymatic activity, especially given that Cit1p protein levels are largely unchanged in Δptc7 yeast (Figure S3D). Ptc7 can be alternatively spliced to create mRNAs that encode Ptc7p isoforms that translocate to the nuclear envelope (the unspliced form) or to mitochondria (the spliced form) (Juneau et al., 2009). To ensure that the change in citrate synthase activity was due to the mitochondrial form of Ptc7p, we mutated endogenous ptc7 to disrupt its splicing. This strain (uns-ptc7), which produces only the non-mitochondrial form of Ptc7p, exhibited the same loss of citrate synthase activity as did Δptc7 yeast (Figure S3E, S3F).
To confirm the ability of Ptc7p to dephosphorylate Cit1p, we performed phosphatase assays with phospho-S462 Cit1p at the peptide and protein level. Ptc7p can efficiently dephosphorylate the S462 phosphopeptide and exhibits a more favorable Km for this substrate than for pNPP (Figure S3G). Ptc7p also robustly dephosphorylated recombinant Cit1p phosphorylated specifically at S462, which caused a band shift on a PhosTag gel and resulted in a marked increase in Cit1p catalytic activity (Figures 3E, 3F). Notably, Ptc7p did not affect the activity of unphosphorylated Cit1p, demonstrating that the differences in activity are due specifically to phosphorylation. These data confirm that Ptc7p can directly dephosphorylate Cit1p at S462 to enhance its catalytic activity.
Phosphorylation of Cit1p occurs at the dimer interface
We next explored how a single phosphorylation event could cause a dramatic decrease in Cit1p activity. As the region surrounding S462 is highly conserved (Figure S4A), we used the porcine (PDB:3ENJ) and chicken (PDB:5CSC) citrate synthase structures to investigate the potential effects of S462 phosphorylation. These structures have revealed that eukaryotic citrate synthases function as obligate homodimers, with each subunit contributing essential residues for activity (Figure S4B) (Larson et al., 2009; Liao et al., 1991). Interestingly, S462 lies at this dimer interface, where it produces inter-subunit contacts (Figure S4C) and packs tightly with the L89 side chain of the opposite monomer (Figure 4A). To assess the effects of disrupting S462, we mutated this residue in silico (Kulp et al., 2012) and found that neither phosphorylation of S462 nor the phosphomimetic substitution (S462E) is accommodated due to significant clashes that arise with L89, while the S462A substitution is readily accommodated (Figures 4B–E). Further, our analyses reveal that S462 is proximal to a salt-bridge between K461 and E277 on the opposite subunit (Figure S4D), suggesting that phospho-S462 may disrupt inter-subunit interaction through competition with E277 to destabilize this salt-bridge. Collectively, these data suggest that S462 likely mediates important interactions between citrate synthase monomers, leading us to hypothesize that phosphorylation of S462 may interfere with proper dimer formation or subunit orientation.
Figure 4. Phosphorylation of Cit1p occurs at the dimer interface.

(A–D) Structural interaction of residue 462 with L89 in the opposite chain with residue 462 modeled as (A) S (WT), (B) A, (C) pS (phospho-serine), and (D) E (based on structure PDB code 3ENJ).
(E) Energy of computed models of A, pS, and E mutants at position 462, compared to WT, with analysis of pig and chicken structures and a yeast homology model.
(F) Coomassie staining of native-PAGE loaded with recombinant WT (2x biological replicates), S462A, and S462E Cit1p.
(G) FLAG immunoblot of native- and SDS-PAGE resolved lysates of Δcit1 expressing EV, or c-terminal FLAG-tagged WT, S462A, or S462E Cit1p.
See also Figure S4.
To test this hypothesis, we determined the dimerization states of recombinant WT and mutant Cit1p purified from E. coli using native-PAGE. WT Cit1p from two separate purifications displayed a mixed proportion of dimeric and monomeric states, with the majority of Cit1p existing as a monomer (Figure 4F). Mutation of S462 disrupted this equilibrium: S462A purified almost exclusively as a dimer, while S462E was almost exclusively monomeric (Figure 4F). The increased proportion of the S462A dimer likely explains the increased Vmax we observe for this variant (Figure 3C). To confirm that the S462E mutation is sufficient to disrupt dimerization in vivo, we determined the native states of WT, S462A, and S462E mutants in yeast lysate. Consistent with our in vitro data, WT and S462A Cit1p formed dimers in vivo, whereas S462E was almost exclusively monomeric (Figure 4G). Collectively, these experiments highlight the importance of a single conserved non-catalytic residue (S462) in enabling Cit1p function, and support a model whereby the proper dimerization state—and thus activity—of Cit1p can be influenced by Ptc7p-dependent dephosphorylation in vivo.
DISCUSSION
Numerous recent investigations have revealed a vast landscape of mitochondrial PTMs associated with a diverse array of activities. From among these, select phosphorylation sites have already been implicated in apoptosis, oxidative phosphorylation, mitochondrial protein import, ketogenesis, and mitochondrial biogenesis (O’Rourke et al., 2011; Pagliarini and Dixon, 2006; Schmidt et al., 2011). Despite these findings, the nature and importance of most protein phosphorylation events in mitochondria remain unclear, and there is uncertainty regarding which enzymes are responsible for modulating the levels of this PTM.
To begin addressing these issues, we focused on Ptc7p, one of multiple likely protein phosphatases in the core mitochondrial proteome (Pagliarini et al., 2008; Rardin et al., 2008). We rationalized that disruption of Ptc7p would result in higher levels of the phosphosites that it dephosphorylates. Indeed, the knockout of ptc7 resulted in significantly elevated levels of just 23 mitochondrial phosphosites—less than 2% of those observed in mitochondria. Moreover, groups of these sites are on proteins are of related function. We focused on one of these proteins, Cit1p—the mitochondrial citrate synthase in S. cerevisiae—because of its central function in mitochondrial and cellular metabolism. We discovered that phosphorylation of S462 can inhibit Cit1p activity, likely through disruption of proper dimerization, and that Ptc7p can dephosphorylate Cit1p in vitro and in vivo to reverse this inhibition.
Further studies will be required to determine whether the phosphorylation of Cit1p and other Ptc7p-related targets is part of bona fide regulatory processes. Given the general absence of established kinases for these events, and the fact that mitochondrial enzymes can exhibit autophosphorylation in a potentially spurious manner (Phillips et al., 2011), it is possible that Ptc7p serves to reverse unintended and damaging protein phosphorylation. However, multiple lines of evidence suggest a more discrete regulatory process is also a possibility. First, it is curious that evolution has selected and maintained a phosphorylatable residue for Cit1p at its dimer interface (Figure S2H), given our demonstration that substitution of an alanine markedly enhanced its activity. This suggests that a balance between the monomeric and dimeric forms of Cit1p may play a role in its regulation, although this remains to be proven. Second, the thioredoxin-mediated redox regulation of citrate synthase in Arabidopsis and the regulation of the glyoxylate cycle Cit2p by ubiquitinylation in yeast suggest that post-translational modification of citrate synthase can be beneficial (Nakatsukasa et al., 2015; Schmidtmann et al., 2014). Finally, acute regulation of citrate synthase could have cellular benefits. In addition to its function in the TCA cycle, citrate acts allosterically to regulate glycolytic flux and fatty acid synthesis, is the source of extra-mitochondrial acetyl-CoA used for de novo fatty acid synthesis, and its overabundance can contribute to iron accumulation-induced mitochondrial dysfunction (Chen et al., 2002).
Beyond citrate synthase, our data suggest that additional mitochondrial proteins from diverse pathways and processes may likewise be affected by Ptc7p (Figure 2B) and contribute to the Δptc7 phenotype. Moreover, ptc7 is alternatively spliced in response to cellular nutrient status, with the protein isoform encoded by the spliced message translocating to the mitochondrion (Juneau et al., 2009). Collectively, these data suggest that Ptc7p may participate in a nutrient-responsive adaptive process aimed at fine tuning mitochondrial function. Determining the origin of these PTMs and their full effects on enzyme and organellar function will be critical toward understanding the broader role of PTMs in maintaining mitochondrial metabolic homeostasis.
EXPERIMENTAL PROCEDURES
Yeast Strains and Cultures
BY4741 yeast (Open Biosystems) were transformed with vector (p416-GPD) or ptc7-expressing plasmids. Knockout (Δptc7) and rescue (Δptc7 + ptc7) strains were generated by replacing ptc7 with His3MX in cells expressing vector or ptc7 plasmids, respectively. 48 hours after transformation, individual colonies were picked directly into uracil dropout (Ura−) media containing 0.1% glucose and 3% glycerol. Yeast were grown at 30°C for 16–24 hours to an OD ~1 before collection, and were snap frozen for proteomics analysis.
OCR and Growth Curve Analysis
OCR was analyzed using a Seahorse analyzer as previously described (He et al., 2013). Growth rates of yeast were calculated based on the logarithmic growth phase (ΔOD600/minute) in strains grown in 3% glycerol in a 96-well plate.
Ptc7p Recombinant Protein Purification and Phosphatase assay
N-terminally truncated ptc7 (Nd38) was expressed in RIPL E. coli and purified using methods previously described (Stefely et al., 2015). Ptc7p and associated mutants were assayed for phosphatase activity using pNPP absorbance at 402 nm (A402).
LC-MS/MS Proteomics
Yeast were lysed, digested and labeled with 8-plex TMT as previously described (Hebert et al., 2014) with modification. Phosphopeptides were enriched by immobilized metal affinity chromatography (IMAC). High pH reversed phase chromatography was used to fractionate both nonphospho- and phospho-fraction after IMAC. Nano LC-MS/MS analysis was performed on Thermo Orbitrap Fusion, and raw MS data searching was done by our custom software COMPASS with 1% false discovery rate (FDR) for both peptide and protein group.
Citrate Synthase Activity Assay
A citrate synthase enzyme assay was performed using 100 mM Tris (pH=7.4), 0.3 mM acetyl CoA, 0.1 mM 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB), and 0.5 mM OAA (all final concentrations). Cell lysate or recombinant Cit1p protein were added as source of enzyme. Absorbance at 412 nm (A412) was monitored for reaction rate calculation.
Cit1p Recombinant Protein Purification
N-terminally truncated cit1 (Nd48) was cloned into pGEX-6P-1, expressed in RIPL E. coli, and purified as described (Fuhs et al., 2015). Phosphoserine-incorporated Cit1p was generated using the Rinehart lab reagents for recombinant protein expression (Addgene). Briefly, Nd48-Cit1p with S462 mutated to a TAG codon as well as WT protein (both in pGEX) were co-expressed with SepOTSλ (a gift from Jesse Rinehart; Addgene # 68292) in C321.ΔA E. coli (a gift from Jesse Rinehart; Addgene # 68306), and purified to generate pSer462 Cit1p and WT Cit1p. Phospho-incorporation was confirmed using PhosTag acrylamide (Wako Chemical).
Molecular modeling
Molecular modeling of S462 variants (A, E and pS) was based on the structures of citrate synthase from pig heart (3ENJ) and chicken (5CSC). Side chains were optimized using conformers from the Energy-Based Conformer Library (Subramaniam and Senes, 2012). The calculations were performed with programs written with the Molecular Software Library (Kulp et al., 2012).
Native-PAGE
Samples were loaded on a 4–16% Native-PAGE Bis-Tris gel and run for 2 hours according to manufactural protocol (Life Technologies). Protein was visualized via western blotting or Coomassie staining.
Statistical Analysis
Statistical analysis was performed using Excel with a two-tailed Student’s t-test.
Further details for all procedures can be found in the Supplemental Experimental Procedures.
Supplementary Material
Acknowledgments
We thank Joshua J. Carson, Alex S. Hebert, and the Pagliarini laboratory for helpful discussions and technical support. This work was supported by NIH grants U01GM94622, R01DK098672, and R01GM115591 (to D.J.P.), P41GM108538 (to J.J.C. and D.J.P.), R01GM099752 (to A.S.), and R35GM118110 (to J.J.C.), an NSF grant CHE-1415910 (to A.S.), a United Mitochondrial Disease Foundation (UMDF) research grant (to N.M.N.), and an NLM grant 5T15LM007359 (to S.G.F.C.). We would like to give special thanks to the Emma Francis Dalton Research fund for their generous support of this project via the UMDF.
Footnotes
Supplemental Information includes supplemental experimental procedures, 4 supplemental figures, and 2 supplemental tables.
AUTHOR CONTRIBUTIONS
X.G., N.M.N., and D.J.P. conceived of the project and its design. X.G., N.M.N., A.S., and D.J.P. wrote the manuscript. All authors performed experiments and data analysis.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Calvo SE, Clauser KR, Mootha VK. MitoCarta2. 0: an updated inventory of mammalian mitochondrial proteins. Nucleic acids research. 2015:gkv1003. doi: 10.1093/nar/gkv1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo SE, Clauser KR, Mootha VK. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016;44:D1251–1257. doi: 10.1093/nar/gkv1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen OS, Hemenway S, Kaplan J. Genetic analysis of iron citrate toxicity in yeast: implications for mammalian iron homeostasis. Proc Natl Acad Sci U S A. 2002;99:16922–16927. doi: 10.1073/pnas.232392299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das AK, Helps NR, Cohen PT, Barford D. Crystal structure of the protein serine/threonine phosphatase 2C at 2.0 A resolution. EMBO J. 1996;15:6798–6809. [PMC free article] [PubMed] [Google Scholar]
- Doulias PT, Tenopoulou M, Greene JL, Raju K, Ischiropoulos H. Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Sci Signal. 2013;6:rs1. doi: 10.1126/scisignal.2003252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhs SR, Meisenhelder J, Aslanian A, Ma L, Zagorska A, Stankova M, Binnie A, Al-Obeidi F, Mauger J, Lemke G, et al. Monoclonal 1- and 3-Phosphohistidine Antibodies: New Tools to Study Histidine Phosphorylation. Cell. 2015;162:198–210. doi: 10.1016/j.cell.2015.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimsrud PA, Carson JJ, Hebert AS, Hubler SL, Niemi NM, Bailey DJ, Jochem A, Stapleton DS, Keller MP, Westphall MS, et al. A quantitative map of the liver mitochondrial phosphoproteome reveals posttranslational control of ketogenesis. Cell Metab. 2012;16:672–683. doi: 10.1016/j.cmet.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Newman JC, Wang MZ, Ho L, Verdin E. Mitochondrial sirtuins: regulators of protein acylation and metabolism. Trends Endocrinol Metab. 2012;23:467–476. doi: 10.1016/j.tem.2012.07.004. [DOI] [PubMed] [Google Scholar]
- He X, Zhu X, Wang X, Wang W, Dai Y, Yan Q. Nuclear modifier MTO2 modulates the aminoglycoside-sensitivity of mitochondrial 15S rRNA C1477G mutation in Saccharomyces cerevisiae. PLoS One. 2013;8:e81490. doi: 10.1371/journal.pone.0081490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert AS, Richards AL, Bailey DJ, Ulbrich A, Coughlin EE, Westphall MS, Coon JJ. The one hour yeast proteome. Molecular & Cellular Proteomics. 2014;13:339–347. doi: 10.1074/mcp.M113.034769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juneau K, Nislow C, Davis RW. Alternative splicing of PTC7 in Saccharomyces cerevisiae determines protein localization. Genetics. 2009;183:185–194. doi: 10.1534/genetics.109.105155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulp DW, Subramaniam S, Donald JE, Hannigan BT, Mueller BK, Grigoryan G, Senes A. Structural informatics, modeling, and design with an open-source Molecular Software Library (MSL) J Comput Chem. 2012;33:1645–1661. doi: 10.1002/jcc.22968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson SB, Day JS, Nguyen C, Cudney R, McPherson A. Structure of pig heart citrate synthase at 1.78 A resolution. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2009;65:430–434. doi: 10.1107/S1744309109008343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao DI, Karpusas M, Remington SJ. Crystal structure of an open conformation of citrate synthase from chicken heart at 2.8-A resolution. Biochemistry. 1991;30:6031–6036. doi: 10.1021/bi00238a029. [DOI] [PubMed] [Google Scholar]
- Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Linn TC, Pettit FH, Hucho F, Reed LJ. Alpha-keto acid dehydrogenase complexes. XI. Comparative studies of regulatory properties of the pyruvate dehydrogenase complexes from kidney, heart, and liver mitochondria. Proc Natl Acad Sci U S A. 1969;64:227–234. doi: 10.1073/pnas.64.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailloux RJ, Jin X, Willmore WG. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2014;2:123–139. doi: 10.1016/j.redox.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Montalvo A, Gonzalez-Mariscal I, Pomares-Viciana T, Padilla-Lopez S, Ballesteros M, Vazquez-Fonseca L, Gandolfo P, Brautigan DL, Navas P, Santos-Ocana C. The phosphatase Ptc7 induces coenzyme Q biosynthesis by activating the hydroxylase Coq7 in yeast. J Biol Chem. 2013;288:28126–28137. doi: 10.1074/jbc.M113.474494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAvoy T, Nairn AC. Serine/threonine protein phosphatase assays. Curr Protoc Mol Biol. 2010;Chapter 18(Unit 18):18. doi: 10.1002/0471142727.mb1818s92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsukasa K, Nishimura T, Byrne SD, Okamoto M, Takahashi-Nakaguchi A, Chibana H, Okumura F, Kamura T. The Ubiquitin Ligase SCF(Ucc1) Acts as a Metabolic Switch for the Glyoxylate Cycle. Mol Cell. 2015;59:22–34. doi: 10.1016/j.molcel.2015.04.013. [DOI] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke B, Van Eyk JE, Foster DB. Mitochondrial protein phosphorylation as a regulatory modality: implications for mitochondrial dysfunction in heart failure. Congest Heart Fail. 2011;17:269–282. doi: 10.1111/j.1751-7133.2011.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini DJ, Dixon JE. Mitochondrial modulation: reversible phosphorylation takes center stage? Trends Biochem Sci. 2006;31:26–34. doi: 10.1016/j.tibs.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Phillips D, Aponte AM, Covian R, Balaban RS. Intrinsic protein kinase activity in mitochondrial oxidative phosphorylation complexes. Biochemistry. 2011;50:2515–2529. doi: 10.1021/bi101434x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiros PM, Langer T, Lopez-Otin C. New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol. 2015;16:345–359. doi: 10.1038/nrm3984. [DOI] [PubMed] [Google Scholar]
- Rardin MJ, Wiley SE, Murphy AN, Pagliarini DJ, Dixon JE. Dual specificity phosphatases 18 and 21 target to opposing sides of the mitochondrial inner membrane. Journal of Biological Chemistry. 2008;283:15440–15450. doi: 10.1074/jbc.M709547200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, Ting AY. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013;339:1328–1331. doi: 10.1126/science.1230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt O, Harbauer AB, Rao S, Eyrich B, Zahedi RP, Stojanovski D, Schonfisch B, Guiard B, Sickmann A, Pfanner N, et al. Regulation of mitochondrial protein import by cytosolic kinases. Cell. 2011;144:227–239. doi: 10.1016/j.cell.2010.12.015. [DOI] [PubMed] [Google Scholar]
- Schmidtmann E, Konig AC, Orwat A, Leister D, Hartl M, Finkemeier I. Redox regulation of Arabidopsis mitochondrial citrate synthase. Mol Plant. 2014;7:156–169. doi: 10.1093/mp/sst144. [DOI] [PubMed] [Google Scholar]
- Stefely JA, Reidenbach AG, Ulbrich A, Oruganty K, Floyd BJ, Jochem A, Saunders JM, Johnson IE, Minogue CE, Wrobel RL, et al. Mitochondrial ADCK3 employs an atypical protein kinase-like fold to enable coenzyme Q biosynthesis. Mol Cell. 2015;57:83–94. doi: 10.1016/j.molcel.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam S, Senes A. An energy-based conformer library for side chain optimization: improved prediction and adjustable sampling. Proteins. 2012;80:2218–2234. doi: 10.1002/prot.24111. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.