

SUMMARY
N6-methyladenosine (m6A) represents the most prevalent internal modification in mammalian mRNAs. Despite its functional importance in various fundamental bioprocesses, the studies of m6A in cancer have been limited. Here we show that FTO, as an m6A demethylase, plays a critical oncogenic role in acute myeloid leukemia (AML). FTO is highly expressed in AMLs with t(11q23)/MLL-rearrangements, t(15;17)/PML-RARA, FLT3-ITD and/or NPM1 mutations. FTO enhances leukemic oncogene-mediated cell transformation and leukemogenesis, and inhibits all-trans-retinoic acid (ATRA)-induced AML cell differentiation, through regulating expression of targets such as ASB2 and RARA by reducing m6A levels in these mRNA transcripts. Collectively, our study demonstrates the functional importance of the m6A methylation and the corresponding proteins in cancer, and provides profound insights into leukemogensis and drug response.
eTOC Blurb
Li et al. show that FTO, an N6-methyladenosine (m6A) demethylase, is highly expressed in subtypes of AML, promotes leukemogenesis, and inhibits all-trans-retinoic acid-induced leukemia cell differentiation. FTO exerts its oncogenic role by regulating mRNA targets such as ASB2 and RARA by reducing their m6A levels.
INSTRUCTION
N6-methyladenosine (m6A) is the most abundant internal modification in messenger RNA (mRNA) mainly occurring at consensus motif of G[G>A]m6AC[U>A>C] (Fu et al., 2014; Meyer and Jaffrey, 2014; Niu et al., 2013; Yue et al., 2015). Although m6A was first discovered in 1970s (Desrosiers et al., 1974; Perry and Kelley, 1974), the lack of technologies to study RNA modifications limited research on m6A and the field had not advanced for several decades. The identification of the fat mass and obesity-associated protein (FTO) as the first RNA demethylase (Jia et al., 2011) revived RNA methylation research, suggesting that m6A is a reversible and dynamic RNA modification that may impact biological regulation analogous to the well-studied reversible DNA and histone modifications (Jia et al., 2013). In 2012, two groups reported transcriptome-wide approaches for m6A RNA immunoprecipitation followed by next-generation sequencing (termed as m6A-seq or MeRIP-seq) and detected m6A peaks in more than 7,000 mRNA transcripts and hundreds of long non-coding RNAs (lncRNAs) in both human and mouse cells, with many of the m6A peaks conserved between humans and mice (Dominissini et al., 2012; Meyer et al., 2012). Thus, these studies suggest m6A methylation in mRNAs is a prevalent modification that likely possesses functional importance.
Recent studies have shown that m6A modification in mRNAs or non-coding RNAs plays critical roles in tissue development, stem cell self-renewal and differentiation, control of heat shock response, and circadian clock controlling, as well as in RNA fate and functions such as mRNA stability, splicing, transport, localization and translation, primary microRNA processing, and RNA-protein interactions (Alarcon et al., 2015; Chen et al., 2015; Dominissini et al., 2012; Geula et al., 2015; Liu et al., 2015; Meyer et al., 2015; Meyer et al., 2012; Wang et al., 2014a; Wang et al., 2015; Wang et al., 2014b; Zhao et al., 2014; Zheng et al., 2013; Zhou et al., 2015). FTO and ALKBH5, the second RNA demethylase identified in 2013 (Zheng et al., 2013), both belong to the AlkB family and catalyze m6A demethylation in a Fe(II)- and α-ketoglutarate-dependent manner, and are referred to as m6A ‘erasers’ (Fu et al., 2014; Yue et al., 2015). METTL3 and METTL14 were identified as m6A ‘writers’ that form a heterodimer with support of WTAP to catalyze m6A methylation (Bokar et al., 1997; Liu et al., 2014; Ping et al., 2014; Wang et al., 2014b). YTHDF1/2/3 were identified as m6A ‘readers’ that preferentially bind to RNA that contains m6A at the G[G>A] m6ACU consensus sequence and lead to different biological consequence (Dominissini et al., 2012; Wang et al., 2014a; Wang et al., 2015), such as inducing RNA decay (Wang et al., 2014a) and promoting mRNA translation (Wang et al., 2015).
FTO was known to be robustly associated with increased body mass and obesity in humans (Dina et al., 2007; Frayling et al., 2007; Scuteri et al., 2007). Animal model studies showed that Fto deficiency protected from obesity and caused growth retardation (Fischer et al., 2009; Gao et al., 2010), while overexpression of Fto led to increased food intake and obesity (Church et al., 2010). In humans, loss-of-function mutations in FTO also caused severe growth retardation and multiple malformations that resulted in premature death (Boissel et al., 2009). As the first identified RNA demethylase that regulates the demethylation of target mRNAs, FTO has been reported to regulate dopaminergic signaling in brain (Hess et al., 2013), and also regulate mRNA splicing of adipogenetic regulatory factors and thus play a critical role in adipogenesis (Ben-Haim et al., 2015; Zhao et al., 2014). However, the impact of FTO, especially as a RNA demethylase, in cancer development and progression has yet to be investigated.
Acute myeloid leukemia (AML) is one of the most common and fatal forms of hematopoietic malignancies with distinct genetic (e.g., t(11q23)/MLL-rearranged, inv(16), t(8;21), and t(15;17)) and molecular (e.g., FLT3-ITD and NPM1 mutations) abnormalities and variable response to treatment (Chen et al., 2010; Dohner et al., 2015; Marcucci et al., 2005). With standard chemotherapies, only 35%–40% of younger (aged <60) and 5%–15% of older (aged ≥60) patients with AML survive over 5 years (Dohner et al., 2015). Thus, to develop effective targeted therapies to treat AML is an urgent and significant unmet medical need, which relies on better understanding of the molecular mechanisms underlying the pathogenesis and drug response of AML.
In the present study, we sought to determine the biological function of FTO in the pathogenesis and drug response of AMLs and also investigate the underlying molecular mechanism through identification of critical mRNA targets of FTO.
RESULTS
FTO is highly expressed in certain subtypes of AML
In analysis of our in-house microarray dataset of 100 human AML with t(11q23)/MLL-rearranged, t(8;21), inv(16) or t(15;17) and 9 normal control samples, we found that FTO was expressed at a significantly higher level in MLL-rearranged AML than in both normal controls (p=0.04) and non-MLL-rearranged AML samples (p=0.002); amongst non-MLL-rearranged AML, FTO is expressed at a significantly higher level in t(15;17) AML, also called acute promyelocytic leukemia (APL), than in t(8;21) and inv(16) AMLs (Figure 1A). Our qPCR assay also showed that FTO is expressed at a significantly higher level in CD34+ bone marrow (BM) cells isolated from primary MLL-rearranged AML patients relative to normal CD34+ BM cells isolated from healthy donors (Figure 1B). In contrast, ALKBH5, another m6A demethylase (Zheng et al., 2013), was not significantly up-regulated in MLL-rearranged AML relative to normal controls (Figures S1A and S1B), although a very recent study suggests involvement of ALKBH5 in breast cancer cell proliferation in vitro (Zhang et al., 2016).
Figure 1. FTO is highly expressed in certain AML subtypes.
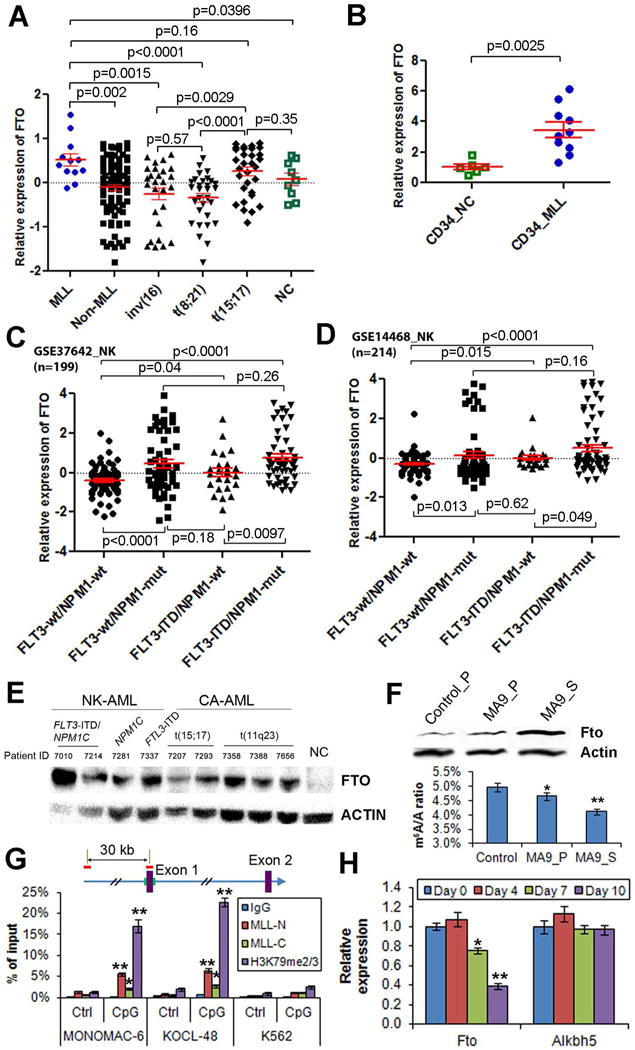
(A) Comparison of FTO expression between human primary AML cases with MLL rearrangements/t(11q23) (MLL) and those without MLL rearrangements (non-MLL), or AML cases with inv(16), t(8;21) or t(15;17), or normal controls (NC). The expression values were detected by Affymetrix exon arrays (Huang et al., 2013). The expression values were log2-transformed and mean centered. (B) qPCR analysis of FTO expression in human CD34+ AML BM cells isolated from 10 primary MLL-rearranged AML patients (CD34_MLL) and normal CD34+ BM cells isolated from 6 healthy donors (CD34_NC). The average expression level of FTO in the CD34_NC samples was set as 1. (C,D) The expression patterns of FTO across cytogenetically normal (or normal karyotype; NK) AMLs within GSE37642 set (n=562) and GSE14468 set (n=518) AML datasets. (E) Western blot assay of FTO expression in human primary AML specimens with different fusion genes or mutant oncogenes and a healthy donor sample (NC). Mononuclear cells isolated from primary AML patients and the healthy donor were used for the assay. (F) The protein level of Fto (upper panel) and m6A level (lower panel; by QQQ-MS) in the representative samples of the control group (from a primary BMT recipient) or MA9 leukemic group (one each from primary and secondary BMT recipients). (G) ChIP-qPCR assays of the enrichment of MLL-N (i.e., MLL N-terminal, representing both wild-type MLL and MLL-fusion proteins), MLL-C (i.e., MLL C-terminal, representing wild-type MLL only), and H3K79me2/3 at the promoter region of FTO (CpG site) and a distal upstream region (Control site) in MONOMAC-6, KOCL-48 and K562 cells. IgG was used as a negative control. (H) qPCR analysis of Fto and Alkbh5 expression in mouse MLL-ENL-ERtm cells after withdrawal of 4-OHT. *, p<0.05; **, p<0.01; t-test. See also Figure S1.
Consistently, in analysis of two large-cohort AML datasets, including GSE37642 set (n=562) and GSE14468 set (n=518), we observed that FTO is expressed at a significantly higher level in t(11q23) and t(15;17) AMLs compared to other subtypes of cytogenetically abnormal (CA) AMLs including inv(16) and t(8;21) AMLs (Figures S1C and S1D). The expression level of FTO in normal karyotype (NK) AMLs is comparable to that in t(11q23) and t(15;17) AMLs (Figures S1C and S1D). Within NK-AMLs, FTO is expressed at a significantly higher level in AML with FLT3-ITD and/or NPM1 mutations (i.e., NPM1c) (especially that with both) compared to those without (Figures 1C and 1D). We also observed that FTO was aberrantly up-regulated at the protein level in human primary AML specimens of the above subtypes (Figure 1E).
Expression of FTO can be upregulated by the relevant leukemic oncogenes
We then conducted both Western blot and qPCR assays of mouse BM progenitor cell samples transformed by MLL-AF9 (the most common form of MLL-fusions (Chen et al., 2010)), PML-RARA (fusion gene of t(15;17)), and FLT3-ITD/NPM1-mutant, showing that FTO can be upregulated by the oncogenes at both protein and RNA levels (Figure S1E). Moreover, Fto, but not Alkbh5, was also significantly up-regulated by MLL-fusion in mouse leukemic BM cells collected from primary BM transplantation (BMT) recipient mice, relative to normal control cells; notably, the up-regulation of Fto was further enhanced after transplantation of the primary MLL-AF9 leukemic BM cells into secondary recipients (Figures 1F, S1F and S1G). Fto overxpression is accompanied with mRNA m6A level decrease in the samples (Figure 1F).
We also used MLL-rearranged leukemia as a model to further investigate whether FTO is directly up-regulated by the oncogenic proteins. Through chromatin immunoprecipitation (ChIP) assays, we found that MLL (see MLL-C binding) and particularly MLL-fusion proteins (see the portion of MLL-N binding exceeding that of MLL-C) are enriched at the CpG area (CpG sites), but not the distal upstream site (Control site), of FTO locus in human MONOMAC-6 (a MLL-AF9 AML line) and KOCL-48 (a MLL-AF4 AML line) cells; the locus also shows a significant enrichment of H3K79 methylation (H3K79me2/3), a mark of active transcription (Bernt et al., 2011; Okada et al., 2005) (Figure 1G). No such significant enrichments were observed in a control cell line, K562 (a human erythroleukemic cell line) (Figure 1G). Thus, our data suggest that FTO is likely a direct target of MLL fusions. In addition, we observed that Fto (but not Alkbh5) expression was significantly (p<0.01) down-regulated in MLL-ENL-ERtm mouse myeloid cells carrying tamoxifen-inducible MLL-ENL (Zeisig et al., 2004) when expression of MLL-ENL was depleted after withdrawal of 4-Hydroxy-tamoxifen (4-OHT) (Figure 1H), indicating that Fto expression in MLL-ENL-ERtm cells relies on the presence of MLL-ENL.
FTO promotes cell proliferation/transformation and suppresses apoptosis in vitro
Both gain- and loss-of-function studies were performed to investigate the pathological role of FTO in AML. As shown in Figures 2A–F, lentivirally transduced wild-type FTO, but not FTO mutant (carrying two point mutations, H231A and D233A, which disrupt the enzymatic activity of FTO (Jia et al., 2011; Lin et al., 2014)), promoted cell growth/proliferation and viability, while decreasing apoptosis and the global mRNA m6A level, in two MLL-rearranged AML cell lines (MONOMAC-6 and MV4-11); the opposite is true when endogenous expression of FTO was knocked down by FTO shRNAs. Similar phenomena were observed when FTO was overexpressed by a retroviral construct or knocked down by siRNAs (Figures S2A–D). In contrast, while forced expression of FTO exhibited moderate (though significant) effects, FTO knockdown exhibited no significant effects on cell growth/proliferation, growth or apoptosis in K562 AML cells (a control line) (Figures S2E–I); thus, FTO is less functionally essential in K562 cells than in MLL-rearranged AML cells, likely due to its much lower endogenous expression in K562 cells (Figure S2J). We next performed mouse BM colony-forming/replating assays (CFAs) to investigate the function of FTO in MLL-AF9-mediated cell transformation. As expected, co-overexpression of FTO, but not FTO-Mut, significantly increased colony numbers after replating, compared to the MLL-AF9 alone group; knockdown expression of Fto led to the opposite (Figures 2G and S2K).
Figure 2. Biological effects of forced expression or knockdown of FTO/Fto expression in MLL-rearranged AML.
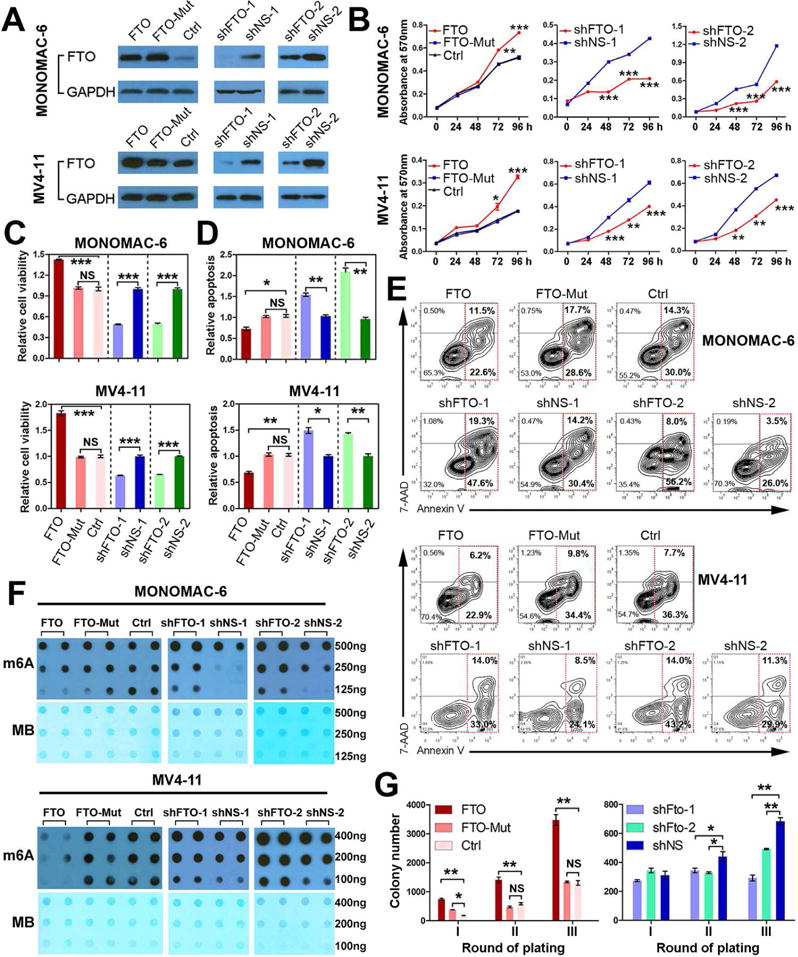
(A) Western Blotting confirmation of forced expression and knockdown of FTO by lentiviral constructs in MONOMAC-6 and MV4-11 cells. FTO, pMIRNA1-FTO; FTO-Mut, pMIRNA1-FTO-Mut; Ctrl, control vector (empty pMIRNA1); shFTO-1, pLKO.1-shFTO-1; shNS-1, pLKO.1-shNS-1; shFTO-2, pGFP-C-shLenti-shFTO-2; shNS-2, pGFP-C-shLenti-shNS-2. (B–E) Effects of forced expression or knockdown of FTO expression on cell growth/proliferation (B), viability (C,D), and apoptosis (E) in MONOMAC-6 and MV4-11 cells. h, hour. (F) m6A dot blot assays of MONOMAC-6 and MV4-11 cells with or without forced expression or knockdown of FTO. MB, methylene blue staining (as loading control). (G) Effects of forced expression of FTO or FTO-Mut (with the above pMIRNA1 constructs) or knockdown of Fto expression (with pGFP-V-RS-Fto shRNAs) on colony-forming/replating capacity of mouse normal BM progenitor cells transduced by MSCVneo-MLL-AF9 (MA9). Colony cells were replated every 7 days. *, p<0.05; **, p<0.01; ***, p<0.001; NS, not significant (p>0.05); t-test. See also Figure S2.
Similar patterns were observed in the PML-RARA/t(15;17) and FTL3-ITD/NPM1 leukemic cell models when both gain- and loss-of-function studies of FTO/Fto were conducted (see Figures 3 and S3), demonstrating the oncogenic role of FTO in these AML subtypes.
Figure 3. Effects of forced expression or knockdown of FTO/Fto in PML-RARA and FLT3-ITD/NPM1-mutant AML.
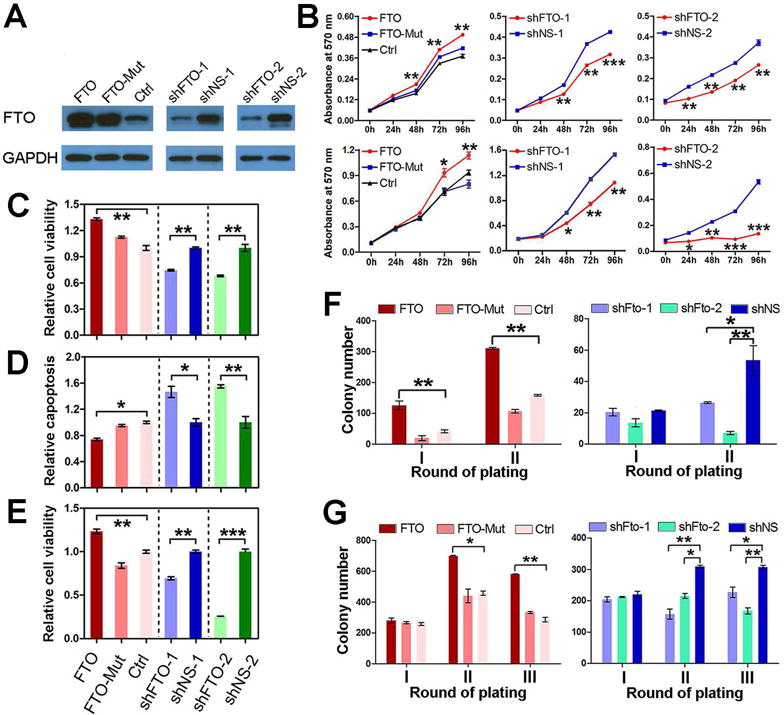
(A) Confirmation of overexpression and knockdown of FTO by Western blotting in PL-21/t(15;17) AML cells. (B) Effects of forced expression or knockdown of FTO expression on cell growth/proliferation in PL-21 (upper panels) and NB4/t(15;17) (lower panels) AML cells. (C–E) Effects of forced expression or knockdown of FTO expression on cell viability in PL-21 (C) and NB4 (E), and apoptosis in PL-21 (D) cells. (F, G) Effects of forced expression of FTO or FTO-Mut, and knockdown of Fto expression on colony-forming/replating capacity of mouse BM progenitor cells carrying PML-RARA (F) or FLT3-ITD/NPM1-mutant (G). Colony cells were replated every 7 days. *, p<0.05; **, p<0.01; ***, p<0.001. See also Figure S3.
Fto significantly enhances leukemogenesis in vivo
To investigate the role of Fto in vivo, we conducted mouse BM transplantation (BMT) assays. We found that forced expression of Fto significantly (p<0.05; log-rank test) accelerated MLL-AF9-induced leukemogenesis in recipient mice (Figures 4A and S4), and this pattern is repeatable (Figure 4B). Notably, forced expression of Fto significantly increased c-Kit+ immature blast cell proportion (Figure 4C) and decreased global m6A level in leukemic BM cells (Figure 4D). Conversely, the knockdown of Fto by shFto-1 or shFto-2 significantly delayed MLL-AF9-mediated leukemogenesis in mice (Figure 4E). Similarly, MLL-AF9-tranduced BM progenitor cells from Fto+/− mice (Gao et al., 2010) caused leukemia in recipient mice significantly slower than did MLL-AF9-tranduced BM progenitor cells from wild-type (Fto+/+) mice (Figure 4F), and were associated with a decrease in c-Kit+ cell proportion (Figure 4G) and an increase in m6A abundance in leukemic BM cells (Figure 4H).
Figure 4. The role of Fto in leukemogenesis mediated by MLL-AF9.
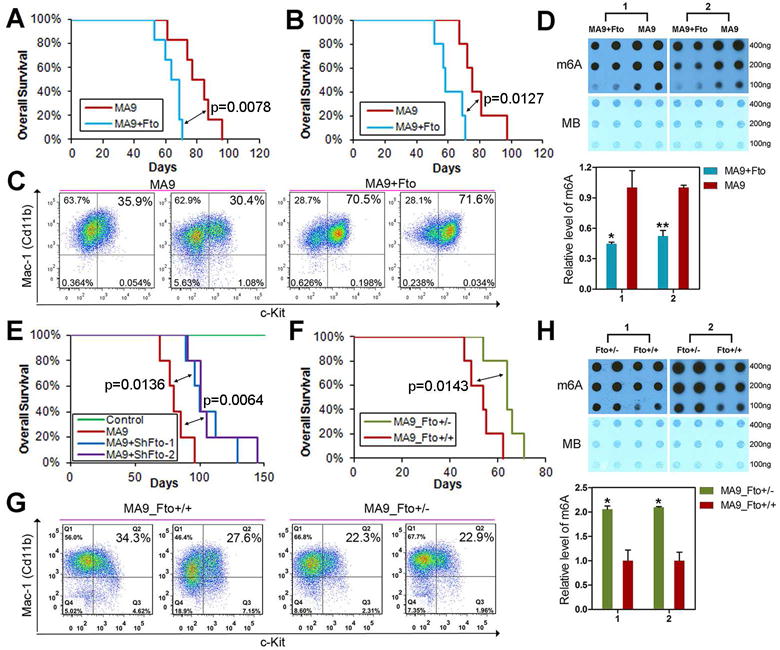
(A,B) Effect of forced expression of Fto on MLL-AF9 (MA9)-induced leukemogenesis. Kaplan-Meier curves are shown for two cohorts of transplanted mice including MSCVneo-MA9+MSCV-PIG (MA9) and MSCVneo-MA9+MSCV-PIG-Fto (MA9+Fto) from two independent BM transplantation (BMT) assays. Six mice per group in plot A and 5 mice per group in plot B. The p values were calculated by log-rank test. (C,D) Flow cytometry analysis of Mac-1+ and c-Kit+ cell populations (C) and m6A dot blot analysis (D) in BM cells of the representative leukemic mice from the BMT assay shown in Figure 4A. (E) Effect of depleted expression of Fto by shRNAs on MA9-induced leukemogenesis. Kaplan-Meier curves are shown for four cohorts of transplanted mice including MSCVneo+MSCV-PIG (Control), MSCVneo-MA9+MSCV-PIG (MA9), MSCVneo-MA9+pGFP-V-RS-shFto-1 (MA9+shFto-1) and MSCVneo-MA9+pGFP-V-RS-shFto-2 (MA9+shFto-2). Five mice were studied per group. (F) Effect of depleted expression of Fto by genetic knockout (heterozygous) on MA9-induced leukemogenesis. Kaplan-Meier curves are shown for two cohorts of recipient mice transplanted with MSCVneo-MA9 transduced wild-type donor cells (MA9_Fto+/+) and MSCVneo-MLL-AF9 transduced Fto+/− donor cells (MA9_Fto+/−). Five mice were studied per group. (G, H) Flow cytometry analysis of Mac-1+ and c-Kit+ cell populations (G) and m6A dot blot analysis (H) in BM cells of the representative leukemic mice from the BMT assay shown in Figure 4F. *, p<0.05; **, p<0.01; t-test. See also Figure S4.
Transcriptome-wide m6A-seq and RNA-seq assays to identify potential targets of FTO
To identify potential mRNA targets of FTO whose m6A levels are reduced by FTO in AML cells, we retrovirally transduced MSCV-PIG-FTO (i.e., FTO) or MSCV-PIG (i.e., Ctrl/Control) into human MONOMAC-6 AML cells and then selected individual stable clones under selection of puromycin (0.5ug/ml). Two FTO-overexpressing (namely FTO_1 and _2) and two control (namely Ctrl_1 and _2) stable cell lines were selected for transcriptome-wide m6A-sequencing (m6A-seq) and RNA-sequencing (RNA-seq) assays. The FTO-overexpressing stable lines exhibit a 4–5 fold increase in FTO protein level (Figure 5A) and a noticeable decrease in m6A level (Figures 5B and 5C), compared to the control lines. As shown in Table S1, 17.7 million – 83.1 million reads were generated from each m6A-seq or RNA-seq (also serving as the input control of the corresponding m6A-seq) library. A total of 13,278 m6A peaks were identified by both MACS (Zhang et al., 2008) and exomePeak (Meng et al., 2013) methods from at least two of the four m6A-seq libraries (Figure S5A). Consistent with previous studies (Chen et al., 2015; Dominissini et al., 2012; Meyer et al., 2012), the most common m6A motif GGAC is significantly enriched in our 13,278 m6A peaks (Figure S5B); the m6A peaks are mostly located in exons (>87%; Figure S5C) and are especially enriched in the vicinity of the stop codon (Figures S5D and S5E).
Figure 5. Identification of potential targets of FTO in AML via transcriptome-wide m6A-seq and RNA-seq assays.
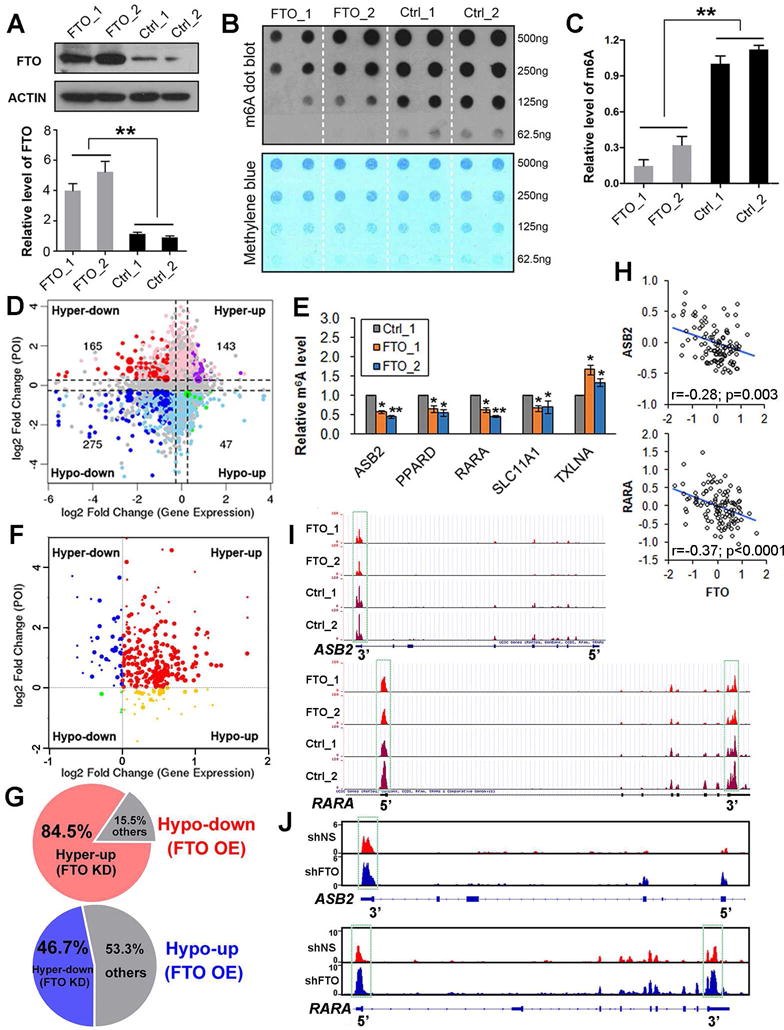
(A) Western blot assay of FTO expression in human MONOMAC-6 AML cell lines with or without forced expression of FTO, including FTO_1/2 and Ctrl_1/2 cell lines. Upper panel shows the image and the lower panel shows the relative quantitative information of FTO expression at the protein level in different AML cell lines. (B,C) The m6A dot blot assay (B) and relative quantitative information (C) of global m6A abundance in transcriptomes of the above four cell lines. (D) Distribution of genes with a significant change in both m6A level and overall transcript (i.e., expression) level in FTO-overexpressing (FTO_1 and FTO_2) compared to control (Ctrl_1 and Ctrl_2) MONOMAC-6 AML cells. (E) Gene-specific m6A qPCR validation of m6A level changes of five representative m6A-Hypo genes in MONOMAC-6 cells. (F) Distribution of the FTO-induced hypo-genes (including the hypo-down and hypo-up groups shown in Figure 5D) in FTO-knockdown MA9/FLT3-ITD AML cells relative to the control AML cells. (G) Up to 84.5% Hypo-down genes in FTO-overexpressing (FTO OE) MONOMAC-6 AML cells display Hyper-up pattern in FTO knockdown (FTO KD) MA9/FLT3-ITD AML cells; 46.7% Hypo-up genes in the FTO OE cells display Hyper-down pattern in the FTO KD cells. (H) Correlation of expression between FTO and ASB2 or RARA across the 109 (100 AML and 9 normal control) samples shown in Figure 1A. (I, J) The m6A abundances in ASB2 and RARA mRNA transcripts in FTO-overexpressing (FTO_1 and FTO_2) and control (Ctrl_1 and Ctrl_2) MONOMAC-6 AML cells (I), and in FTO-knockdown (shFTO-1) and control (shNS) MA9/FLT3-ITD AML cells (J), as detected by m6A-seq. The m6A peaks shown in the green rectangles are those have a significant reduced abundance (p<0.005; fold change>1.2) in FTO_1/2 than in Ctrl_1/2 cells. *, p<0.05; **, p<0.01; t-test. See also Figure S5 and Tables S1–3.
Potential target genes of FTO tend to be negatively regulated by FTO
We next compared the abundance (normalized to input) of the 13,278 m6A peaks between FTO-overexpressing cells (i.e., FTO_1/2) and the control cells (i.e., Ctrl_1/2). A total of 2,785 and 3,180 m6A peaks showed a significant decrease and increase (p<0.005; fold change ≥1.2), respectively, in abundance, in FTO_1/2 cells relative to Ctrl_1/2 cells, and thereby they were termed as hypo- and hyper-methylated m6A peaks, respectively (Figure S5F). Through analysis of the RNA-seq data, we identified 322 hypo-methylated m6A peaks whose mRNA transcripts were significantly (p<0.005; fold change ≥1.2) down-regulated (275; Hypo-down) or up-regulated (47; Hypo-up) in FTO_1/2 cells relative to Ctrl_1/2 cells (Figure 5D). Notably, 85% of the 322 hypo-methylated m6A peaks are associated with down-regulated mRNA transcripts in FTO-overexpressing cells, significantly more frequent (p<0.0001; χ2-test) than the rate (54%) in the 308 hyper-methylated m6A peaks (Figure 5D). Thus, mRNA transcripts carrying hypo-methylated m6A peaks, which are likely potential targets of FTO since FTO is an m6A demethylase, tend to be down-regulated in FTO-overexpressing AML cells.
Through searching the Molecular Signature Database (MSigDB) of GSEA (Subramanian et al., 2005), we found that the Hypo-up transcripts were significantly enriched with target genes of SOX2, NANOG and LEF1, key transcription factors for embryonic stem cell pluripotency or WNT signaling activation (Figure S5G). Hypo-down transcripts were significantly enriched with genes involving the interferon signaling and immune system; interestingly, the most enriched genes are a set of potential direct target genes of PML-RARA, a fusion protein resulting from t(15;17) in APL (Figure S5G). The enriched genes sets in Hyper-up or Hyper-down transcripts are shown in Figure S5H. We next investigated the correlation between FTO and the above genes in expression across four large cohorts of AML patient datasets (see Table S2). We found that 8 Hypo-down (i.e., ASB2, KCNG1, PPARD, RAB17, RARA, SLC11A1, SLCO4A1, and TBC1D9) and 3 Hypo-up (C21orf59, MZF1, and TXLNA) genes exhibited a significantly negative and positive correlation, respectively, with FTO in expression across all the four datasets (Table S2). We next conducted gene-specific m6A qPCR assays for five such m6A-Hypo genes (ASB2, PPARD, RARA, SLC11A1, and TXLNA) and confirmed the m6A-level decrease in 4 out of the 5 genes (80%; Figure 5E), demonstrating the reliability of our transcriptome-wide m6A-seq data.
We then conducted m6A-seq and RNA-seq of MA9/FLT3-ITD leukemic cells (i.e., MLL-AF9 plus FLT3-ITD-transformed human CD34+ cord blood cells (Wunderlich et al., 2013)) with or without FTO-shRNA knockdown. Remarkably, more than 90% m6A-Hypo transcripts identified from FTO-overexpressing MONOMAC-6 cells turned into m6A-Hyper in FTO-knockdown MA9/FLT3-ITD cells (Figure 5F), with approximately 85% of the Hypo-down and 47% of Hypo-up transcripts became Hyper-up and Hyper-down, respectively (see Figures 5F and 5G), which might be genuine targets of FTO. Overall, the vast majority of potential targets of FTO are likely negatively regulated by FTO. Eight out of the 11 m6A-Hypo genes listed in Table S2 showed expected patterns in the m6A and RNA level changes in FTO-knockdown cells (Table S3).
ASB2 and RARA are functionally important target genes of FTO in AML
Interestingly, amongst the genes listed in Tables S2 and S3, ASB2 and RARA have been reported to be up-regulated during normal hematopoiesis and in ATRA-induced differentiation of leukemia cells and function as key regulators during the processes (Glasow et al., 2005; Guibal et al., 2002; Kohroki et al., 2001; Sakamoto et al., 2014; Wang et al., 2012; Zhu et al., 2001). Notably, ASB2 can also degrade MLL during hematopoietic differentiation via ubiquitination (Wang et al., 2012). Consistent with their down-regulation in FTO-overexpressing AML cells, ASB2 and RARA exhibit a significant inverse correlation with FTO in expression across all four independent AML cohorts (see Table S2 and Figure 5H). Our m6A-seq data indicates that FTO targets the 3′UTR of ASB2 and both 3′UTR and 5′UTR of RARA transcripts; FTO overexpression and knockdown causes a significant decrease and increase, respectively, in the m6A level of the UTR(s) (Figures 5I and 5J). We then focused on these two FTO potential targets for further studies.
As expected, we showed that in both MONOMAC-6 and NB4/t(15;17) AML cells, forced expression of wild-type FTO, but not mutant FTO, substantially reduced expression of ASB2 and RARA, while increasing expression of MLL, a negative downstream target of ASB2 (Wang et al., 2012) (Figures 6A and S6A). The opposite phenomena were observed when we knocked down endogenous expression of FTO (Figures 6B, 6C, and S6B). Forced expression or depletion of FTO affected expression of RARA, but not that of other RAR genes such as RARG (Figure S6C). In addition, we observed similar changes in RARA and ASB2 expression after overexpression or knockdown of FTO in nuclear and cytoplasm (Figure S6D).
Figure 6. ASB2 and RARA are two critical target genes of FTO in AML.
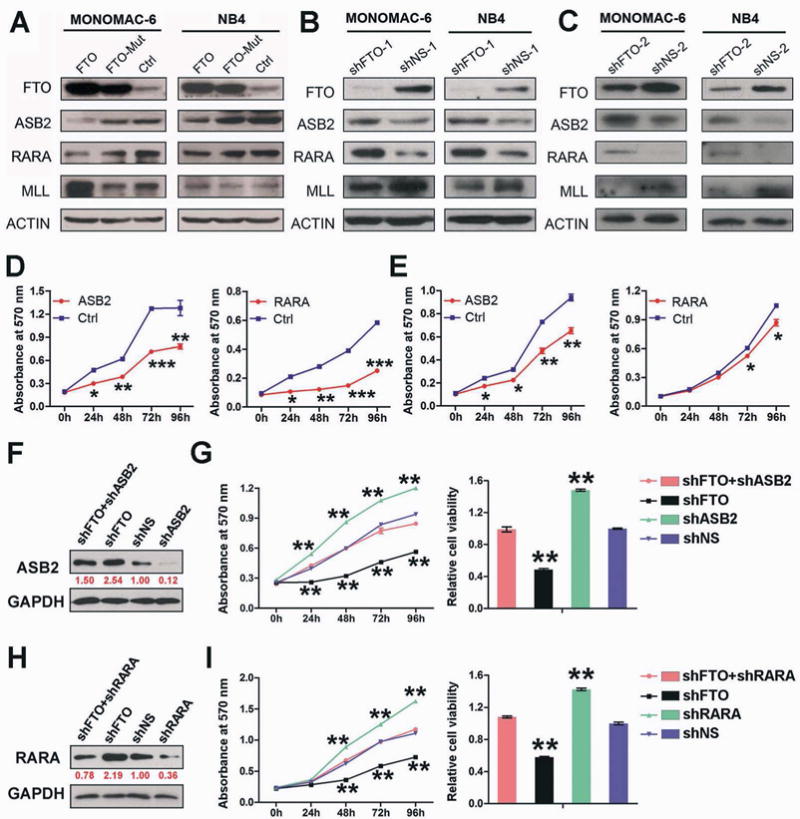
(A–C) Western blot assays of FTO, ASB2, RARA and MLL in MONOMAC-6 or NB4 AML cells with lentivirally transduced FTO (pmiRNA1-FTO), FTO mutant (H231A and D233A; pmiRNA1-FTO-Mut) or control (Ctrl; pmiRNA1) construct (A), as well as shFTO-1/shNS-1 (B) and shFTO-2/shNS-2 (C). ACTIN was used as the endogenous control protein for loading control. (D, E) Effects of forced expression of ASB2 and RARA on cell growth/proliferation in MONOMAC-6 (D) and NB4 (E) AML cells. (F) Western blot assays of ASB2 in MONOMAC-6 cells transduced with shFTO+shASB2 (pLKO.1-shFTO-1+ pTRIPZ-shASB2), shFTO (pLKO.1-shFTO-1 + pTRIPZ), shNS (pLKO.1-shNS + pTRIPZ) or shASB2 (pLKO.1-shNS-1 + pTRIPZ-shASB2). (G) Effects of FTO and/or ASB2 knockdown on cell growth/proliferation (left panel) and viability (right panel) in MONOMAC-6 cells. (H) Western blot assays of RARA in MONOMAC-6 AML cells transduced with shFTO+shRARA (pLKO.1-shFTO-1+ shRARA), shFTO (pLKO.1-shFTO-1+ shNS), shNS (pLKO.1-shNS + shNS) or shRARA (pLKO.1-shNS-1 + shRARA). (I) Effects of FTO and/or RARA knockdown on cell growth/proliferation (left panel) and viability (right panel) in MONOMAC-6 cells. *, p<0.05; **, p<0.01; ***, p<0.001; t-test. See also Figure S6.
We then cloned ASB2 and RARA coding regions (CDS) or shRNAs into lentiviral vectors and investigated their functions in AML cells. As expected, forced expression of either ASB2 or RARA largely recapitulated the phenotypes caused by FTO knockdown in both MONOMAC-6 and NB4 cells (Figures 6D–E vs. Figures 2B and 3B). Moreover, the effects of FTO overexpression can be largely rescued by forced expression of RARA or ASB2 (Figures S6E–J). Conversely, knockdown of ASB2 or RARA significantly enhanced AML cell growth and viability, which mimics the effect of FTO overexpression, and was sufficient to rescue the inhibitory effect of FTO knockdown on AML cell growth and viability (Figures 6F–I). Collectively, our data demonstrate that ASB2 and RARA are functionally important targets of FTO.
FTO-mediated regulation of ASB2 and RARA depends on its m6A demethylase activity and the m6A modifications in the target mRNA transcripts
To elucidate the molecular mechanism underlying FTO-mediated regulation of expression of its targets such as ASB2 and RARA, we first validated effects of FTO expression changes on the m6A levels in target mRNA transcripts. Using gene-specific m6A qPCR assays, we demonstrated that forced expression and knockdown of FTO reduced and increased, respectively, the m6A levels on ASB2 and RARA mRNA transcripts (Figure 7A and data not shown). More importantly, to assess the requirement of the target mRNA m6A modifications for FTO-mediated gene regulation, we conducted Luciferase reporter and mutagenesis assays (Figure 7B). As expected, compared with mutant FTO or empty vector, ectopically expressed wild-type FTO substantially reduced luciferase activity of the individual reporter constructs bearing wild-type ASB2 3′UTR, RARA 3′UTR or RARA 5′UTR that has intact m6A sites, while mutations in the m6A sites abrogated the inhibition (Figure 7B). To validate whether the effects observed above are related to m6A modifications, we conducted gene-specific m6A qPCR of the cloned wild-type and mutant 3′UTR fragments of ASB2 or RARA in HEK-293T cells that were used for the Luciferase reporter assays. As expected, while the wild-type 3′UTR fragments have high abundance of m6A modifications, the mutant 3′UTR fragments contain no or minimal level of m6A modifications; co-tansfected wild-type FTO, but not FTO mutant, significantly reduced the m6A abundance on the wild-type 3′UTR fragments of ASB2 or RARA in HEK-293T cells (see Figure 7C). Together, our data demonstrate that FTO-mediated gene regulation relies on its demethylase activity and m6A modifications in the target mRNA transcripts.
Figure 7. FTO-mediated regulation of expression of ASB2 and RARA relies on its m6A demethylase activity and the m6A modifications in target mRNAs.
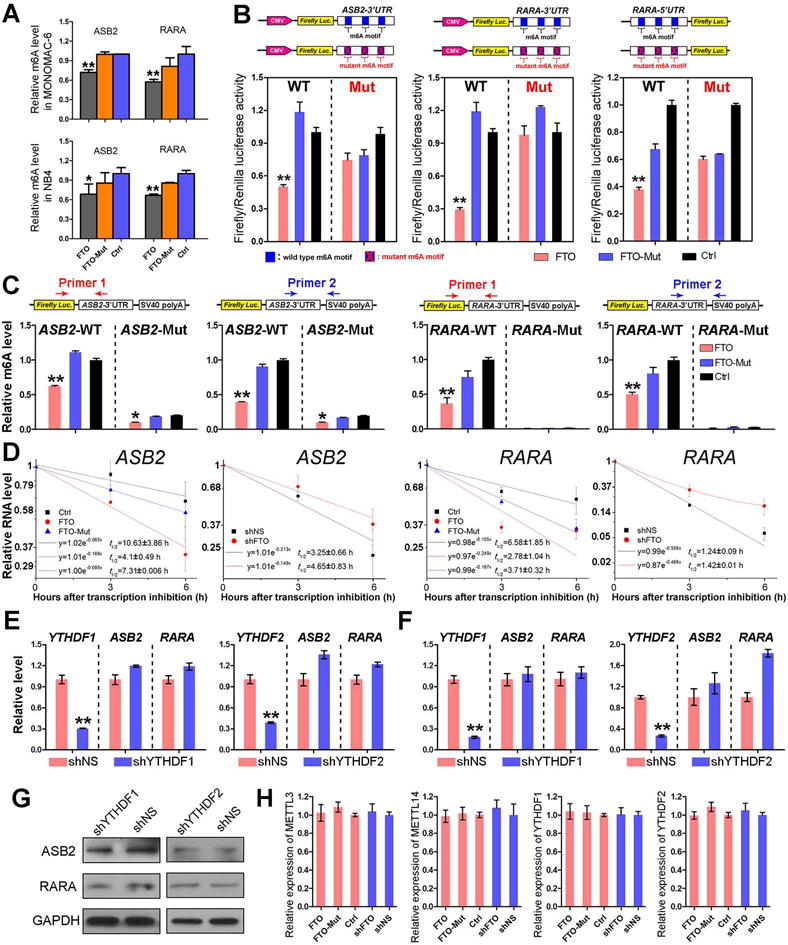
(A) Gene-specific m6A qPCR analysis of m6A level in mRNA transcripts of each gene in MONOMAC-6 and NB4 cells transduced with FTO (FTO), FTO mutant (FTO-Mut) or control vector (Ctrl). (B) Relative luciferase activity of pMIR-REPORT-ASB2-3′UTR (left panel), pMIR-REPORT-RARA-3′UTR (middle panel) and pGL3-basic-RARA-5′UTR (right panel) with either wild-type or mutant (A-to-T mutation) m6A sites after co-transfection with FTO (FTO), FTO mutant (FTO-Mut) or control vector (Ctrl) into HEK-293T cell. Firefly luciferase activity was measured and normalized to Renilla luciferase activity. (C) Luciferase reporter assay related gene-specific m6A qPCR analysis of m6A levels in exogenous mRNA transcripts of Firefly Luc-ASB2 3′UTR or Firefly Luc-RARA 3′UTR in HEK-293T cells. For each luciferase reporter construct, we designed two different pairs of primers crossing the inserted ASB2 or RARA 3′UTR fragment and pMIR-Report vector fragment. Primer 1 covers the joint of Firefly Luc and ASB2-3′UTR or RARA-3′UTR. Primer 2 covers the joint of ASB2-3′UTR or RARA-3′UTR and SV40 poly A. (D) The mRNA half-life (t1/2) of ASB2 or RARA in NB4 cells transduced with FTO (FTO), FTO mutant (FTO-Mut) or control vector (Ctrl), or with depleted expression of FTO (shFTO-1) or not (shNS-1). (E, F) Relative expression of ASB2 and RARA after knockdown of m6A readers, YTHDF1 or YTHDF2, in MONOMAC-6 (E) and NB4 (F) cells. (G) Western blot assay of ASB2 and RARA expression after depletion of m6A readers in MONOMAC-6 cells. (H) Relative expression of m6A writers and readers with forced or depleted expression of FTO in MONOMAC-6 cells. *, p<0.05; **, p<0.01; t-test. See also Figure S7.
A set of m6A-modified transcripts could be recognized by YTHDF2, which exhibits a shorter half-life than non-methylated ones (Wang et al., 2014a). Thus, it is surprising that most of the FTO potential targets we identified tend to be negatively regulated by FTO in AML cells (Figures 5D, 5F and 5G). To analyze the effect of m6A level decrease on the stability of potential target transcripts of FTO, we conducted RNA stability assays (Wang et al., 2014a) and showed that forced expression and knockdown of FTO shortened and prolonged, respectively, the half-life of ASB2 and RARA mRNA transcripts in AML cells (Figures 7D, S7A and S7B). Thus, FTO-induced repression of ASB2 and RARA expression is at least in part due to the decreased stability of ASB2 and RARA mRNA transcripts upon FTO-mediated decrease of m6A level in their mRNA transcripts. Consistent with the effect of FTO overexpression, we found that knockdown of METTL3 and especially of METTL14, two m6A writers, also resulted in the down-regulation of ASB2 and RARA levels (Figure S7C).
Our results suggest that alternative m6A reading processes may exist which recognize m6A sites in FTO target transcripts and promote their stability. YTHDF1 and YTHDF2 are two well-established m6A readers with each recognizing a few thousands of methylated transcripts in mammalian cells (Wang et al., 2014a; Wang et al., 2015). Nonetheless, knockdown of either of them did not lead to a significant reduction at the mRNA levels of ASB2 or RARA (Figures 7E and 7F), indicating that YTHDF1 and YTHDF2 are unlikely the readers that can promote the stability of ASB2 and RARA mRNAs. While YTHDF1 knockdown resulted in a minor decrease in the protein levels of ASB2 and RARA, YTHDF2 knockdown showed no effect on their protein levels at all (Figure 7G). Furthermore, neither forced expression nor knockdown of FTO affected expression of METTL3, METTL14, YTHDF1 or YTHDF2 (Figure 7H). Thus, the effects of FTO overexpression or knockdown on ASB2 and RARA mRNA stability are unlikely due to the changes in the above m6A writers or readers. The reader(s) that promotes the mRNA stability of FTO target transcripts (e.g., ASB2 and RARA) has yet to be identified.
The FTO⊣ASB2/RARA axis contributes to the response of APL cells to ATRA treatment
While ATRA-based differentiation therapy has transformed APL from a highly fatal disease to a highly curable one (Huang et al., 1988; Wang and Chen, 2008), it is still important to better understand the underlying molecular mechanism(s). As shown in Figure 8A, upon ATRA treatment, FTO is significantly down-regulated in NB4 APL cells, associated with a significant up-regulation of RARA and especially ASB2. To investigate the potential role of FTO in ATRA-induced APL cell differentiation, we compared FTO-overexpressing NB4 cells and control NB4 cells on their response to ATRA treatment through flow cytometry assays. As ATRA can induce APL cells towards granulocytic and monocytic differentiation (Arteaga et al., 2013; Carlesso et al., 1999; Gocek et al., 2014), anti-CD11b (a granulocytic differentiation marker) and anti-CD14 (a monocytic differentiation marker) antibodies were used in the flow cytometric assays. As shown in Figure 8B, forced expression of FTO, but not FTO mutant, noticeably increased the undifferentiated NB4 cell population (i.e., CD11b−/CD14− cells) after 48 hours of 500 nM ATRA treatment. We then conducted a loss-of-function study, and found that depletion of FTO expression by two individual different shRNAs could substantially enhanced ATRA-induced cell differentiation, resulting in a striking decrease in undifferentiated population of NB4 cells after 48 hours of treatment with a lower concentration of ATRA (100 nM) (Figure 8C). Figure 8D shows a summary of the results from three independent experiments. Consistent with the phenotype caused by FTO knockdown, forced expression of either RARA or ASB2 could also substantially enhance NB4 cell differentiation (Figures 8E and 8F). Collectively, FTO inhibits ATRA-induced differentiation of APL cells likely through post-transcriptionally repressing expression/function of RARA and ASB2, and such function of FTO relies on its m6A demethylase activity.
Figure 8. The potential role of the FTO⊣RARA/ASB2 axis in ATRA-induced NB4 cell differentiation and a schematic model of FTO signaling in AML.
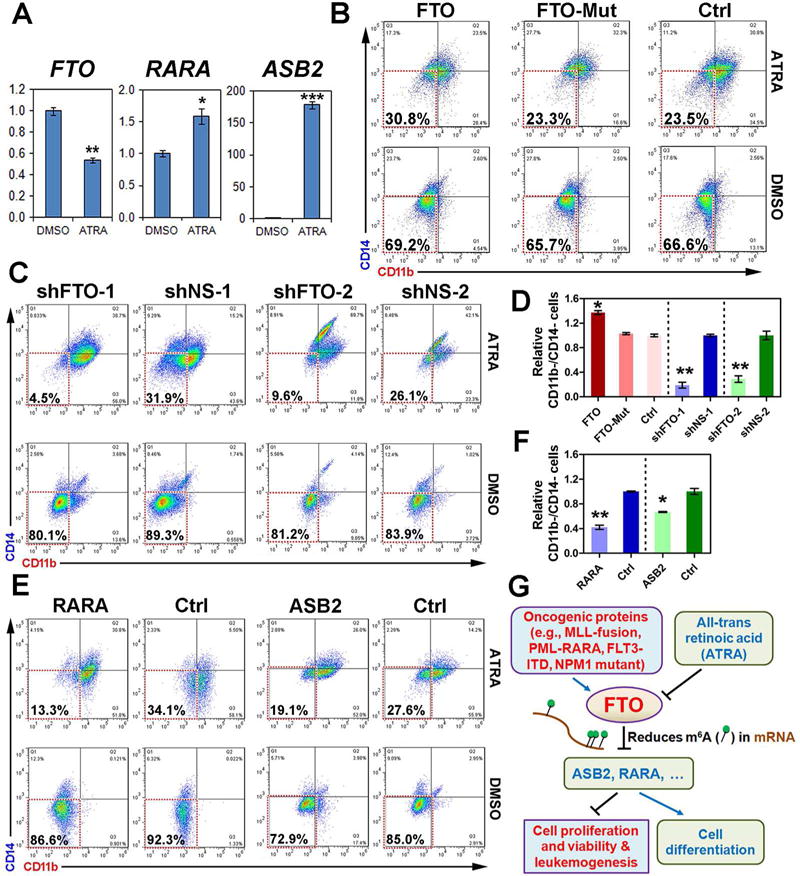
(A) Expressional changes of FTO, RARA, and ASB2 in NB4 cells 48 hours post treatment with 100 nM ATRA as detected by qPCR. *, p<0.05; **, p<0.01; ***, p<0.001; t-test. (B) Flow cytometric analyses of NB4 cells transduced with FTO (FTO), FTO mutant (FTO-Mut) or control vector (Ctrl) 48 hours post treatment with 500 nM ATRA or DMSO. (C) Flow cytometric analyses of NB4 cells with knockdown of FTO (shFTO-1 or shFTO-2) 48 hours post treatment with 100 nM ATRA or DMSO. (D) The summary of results from three independent experiments with those shown in Figure 8B or 8C as representatives. (E, F) The representative (E) or summary (F; from triplicates) results of flow cytometric analyses of NB4 cells with forced expression of RARA or ASB2 after exposed to 100 nM ATRA or DMSO for 48 hours. (G) The schematic model of the role and underlying mechanism of FTO in leukemogenesis and ATRA-induced differentiation of leukemic cells.
DISCUSSION
In the present study, we demonstrate that FTO, an obesity risk-associated gene and the first m6A eraser identified, plays a critical oncogenic role in hematopoietic cell transformation and leukemogenesis. Briefly, FTO expression can be up-regulated by certain oncogenic proteins (e.g., MLL-fusion proteins, PML-RARA, FLT3-ITD and NPM1 mutant) and thereby FTO is aberrantly up-regulated in certain subtypes of AMLs, e.g., t(11q23)/MLL-rearranged, t(15;17), FLT3-ITD and/or NPM1-mutanted AMLs. In vitro, we show that forced expression of FTO significantly enhances the viability and proliferation/growth of human AML cells, while inhibiting the apoptosis of the cells, and also significantly enhances leukemic oncogenes-mediated transformation/immortalization of normal hematopoietic stem/progenitor cells; the opposite is true when expression of FTO is knocked down. In vivo, we show that forced and depleted expression of Fto significantly promotes and inhibits, respectively, MLL-fusion-mediated leukemogenesis in mice. Our transcriptome-wide m6A-seq assay and the subsequent validation and functional studies suggest that ASB2 and RARA are two critical target genes of FTO. As an m6A RNA demethylase, FTO reduces the m6A levels of ASB2 and RARA mainly at untranslated regions (UTRs), which in turn leads to the down-regulation of these two genes at the RNA level and especially at the protein level. Mechanistically, we demonstrate that the biological function of FTO relies on its m6A demethylase activity as mutations in the FTO catalytic domain sufficiently abrogate the function of FTO. Moreover, our luciferase reporter/mutagenesis assays indicate that the m6A sites in the UTRs of its critical target genes such as ASB2 and RARA are essential for FTO to post-transcriptionally regulate their expression. Our data and previous studies (Glasow et al., 2005; Guibal et al., 2002; Kohroki et al., 2001; Zhu et al., 2001) demonstrate the anti-leukemic effects of ASB2 and RARA. Thus, the FTO⊣ASB2/RARA axis likely plays a critical role in the pathogenesis of AMLs. Furthermore, we provide evidence that this axis likely also plays an essential role in mediating the response of leukemic cells to ATRA treatment. A schematic model summarizing our discoveries is shown in Figure 8G.
Epidemiology studies demonstrate a strong association between FTO single nucleotide polymorphisms (SNPs) or overweight/obesity and the risk of various types of cancers, such as breast, prostate, kidney and pancreatic cancers, as well as hematopoietic malignancies including myeloma, lymphoma and leukemia (Castillo et al., 2012; Li et al., 2012a; Soderberg et al., 2009). Therefore, it is possible that increased expression of FTO, caused by its obesity-associated SNPs (Berulava and Horsthemke, 2010; Church et al., 2010), may contribute (to some extent) to the higher risk of individuals with overweight and obesity in developing various types of cancers. If so, FTO may play an oncogenic role also in other cancers, besides leukemia.
Previous studies suggest that mRNA transcripts with m6A modifications tend to be less stable (Schwartz et al., 2014; Wang et al., 2014a), largely due to the relocation of such mRNAs by YTHDF2 to RNA decay sites (Wang et al., 2014a). Surprisingly, herein we show that over 80% of FTO potential targets tend to be negatively regulated by FTO in AML cells. We show that forced and depleted expression of FTO substantially shortens and prolongs, respectively, the half life of its critical targets such as ASB2 and RARA, suggesting that FTO-mediated repression of ASB2 and RARA expression is at least in part due to the decreased stability of ASB2 and RARA mRNA transcripts. Our data further suggest that additional reading process may exist which controls the stability of FTO target transcripts. Neither YTHDF2 nor YTHDF1 target all m6A sites in mammalian cells. Other reading processes have been suggested (Liu et al., 2015). Although certain m6A-sites on ASB2 or RARA could be affected by YTHDF2 or YTHDF1, our data indicate that the FTO-targeted sites exhibit effects on mRNA distinct from these known reading processes. It will be very interesting to uncover such an alternative reading process in the future.
Interestingly, all the subtypes of AMLs with high levels of endogenous FTO expression, such as those carrying t(11q23), t(15;17), NPM1 mutations, and/or FLT3-ITD, are more sensitive to ATRA than the other AML subtypes (Dos Santos et al., 2013; El Hajj et al., 2015; Hu et al., 2009; Huang et al., 1988; Iijima et al., 2004; Jiang et al., 2016; Lo-Coco et al., 2013; Martelli et al., 2015; Niitsu et al., 2001; Schlenk et al., 2009). It is possible that the survival/proliferation of these subtypes of AML cells relies more on the FTO signaling, and thus they are more responsive to ATRA treatment, as ATRA can release the expression/function of ASB2 and RARA, two negative targets of FTO, and thereby trigger cell differentiation.
In summary, here we provide compelling in vitro and in vivo evidence demonstrating that FTO, an m6A demethylase, plays a critical oncogenic role in cell transformation and leukemogenesis as well as in ATRA-mediated differentiation of leukemic cells, through reducing m6A levels in mRNA transcripts of its critical target genes such as ASB2 and RARA and thereby triggering corresponding signaling cascades. Our study highlights the functional importance of the m6A modification machinery in cancer, and provides profound insights into the molecular mechanisms underlying tumorigenesis by revealing a previously unrecognized mechanism of gene regulation in cancer. In addition, given the functional importance of FTO in leukemogenesis and drug response, targeting FTO signaling by selective inhibitors may represent a promising therapeutic strategy to treat leukemia, especially in combination of ATRA treatment. As FTO has also been implicated in other types of cancers, our discoveries may have a broad impact in cancer biology and cancer therapy.
EXPERIMENTAL PROCEDURES
Leukemic patient samples
The leukemia patient samples were obtained at the time of diagnosis or relapse and with informed consent at the University of Chicago Hospital (UCH) or other collaborative hospitals, and were approved by the institutional review board of the institutes/hospitals.
The care and maintenance of animals
It was approved by Institutional Animal Care and Use Committee (IACUC) of the University of Chicago or University of Cincinnati.
Cell Culture and Transfection, Chromatin Immunoprecipitation (ChIP), qPCR, In vitro Functional Study Assays, and in vivo Bone Marrow Transplantation Studies
These assays were performed as we did previously (Huang et al., 2013; Jiang et al., 2012; Li et al., 2012b) with some modifications.
Global m6A Quantitative Assays, m6A-seq, RNA-seq, Gene-Specific m6A qPCR and RNA Stability Assays
These assays were conducted as described previously (Dominissini et al., 2013; Jia et al., 2011; Liu et al., 2014; Wang et al., 2014a) with some modifications (see Supplemental Information for details).
Supplementary Material
SIGNIFICANCE.
The identification of FTO as the first N6-methyladenosine RNA demethylase and the high prevalence of m6A methylation demonstrated in mammalian mRNA transcriptomes have spurred immense interest in the function of m6A modifications in post-transcriptional regulation. However, little is known about the function of FTO or m6A modifications in cancer. Here we show that FTO functions as an oncogene that promotes leukemic oncogene-mediated cell transformation and leukemogenesis, and inhibits all-trans-retinoic acid (ATRA)-mediated leukemia cell differentiation. Mechanistically, FTO exerts its oncogenic role as an m6A demethylase by targeting a set of critical transcripts such as ASB2 and RARA. Thus, our study reveals a previously unrecognized mechanism of gene regulation in tumorigenesis and highlights functional importance of FTO and m6A modification in cancer.
HIGHLIGHTS.
-
➢
FTO is highly expressed in certain subtypes of AMLs such as MLL-rearranged AML
-
➢
FTO promotes leukemic oncogene-mediated cell transformation and leukemogenesis
-
➢
FTO targets a set of genes (e.g. ASB2 and RARA) in AML as an m6A RNA demethylase
-
➢
The FTO⊣ASB2/RARA axis mediates AML cell growth and (ATRA-induced) differentiation
Acknowledgments
We thank Dr. Michelle M. Le Beau for providing primary AML patient samples and James Mulloy for providing the MA9/FLT3-ITD AML cell line. This work was supported in part by the National Institutes of Health (NIH) R01 Grants CA178454 (J.C.), CA182528 (J.C.) and GM071440 (C.H.), Leukemia & Lymphoma Society (LLS) Special Fellowship (Z.L.), LLS Translational Research Grant (J.C.), American Cancer Society (ACS) Research Scholar grant (J.C.), ACS-IL Research Scholar grant (Z.L.), Gabrielle’s Angel Foundation for Cancer Research (J.C., Z.L., X.J. and H.H.), China Scholarship Council (CSC) visiting scholar (X.W.), and Foundation of Innovation Team for Basic and Clinical Research of Zhejiang Province (Grant 2011R50015) (J.J.). S.N. is an HHMI Fellow of the Damon Runyon Cancer Research Foundation (DRG-2215-15). C.H. is an investigator of the Howard Hughes Medical Institute (HHMI).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ACCESSION NUMBERS
The microarray, m6A-seq and RNA-seq data have been deposited in the Gene expression Ominibus (GEO) repository with the accession numbers GSE34184, GSE30285, GSE76414, GSE84944 and GSE85008.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, Figures and Tables.
AUTHOR CONTRIBUTIONS
Z.L. and J.C. conceived the project. Z.L., R.S., C.H. and J.C. designed the research and supervised experiments conducted in the laboratories. Z.L., H.W., R.S., X.W., Z.Z., C.L., H.H., S.N., L.D., C.H., X.Q., L.T., G.M.H., Y.W., H.H., X.W., P.C., S.G., S.A., Y.L., S.L., J.S., X.J., J.C. performed experiments and/or data analyses; Z.L., H.H., M.B.N., R.A.L., X.J., P.Z., J.J., C.H., and J.C. contributed reagents/analytic tools and/or grant support; Z.L., H.W., R.S., X.W., Z.Z., C.H., and J.C. wrote the paper. All authors discussed the results and commented on the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- Alarcon CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. N6-methyladenosine marks primary microRNAs for processing. Nature. 2015;519:482–485. doi: 10.1038/nature14281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arteaga MF, Mikesch JH, Qiu J, Christensen J, Helin K, Kogan SC, Dong S, So CW. The histone demethylase PHF8 governs retinoic acid response in acute promyelocytic leukemia. Cancer Cell. 2013;23:376–389. doi: 10.1016/j.ccr.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Haim MS, Moshitch-Moshkovitz S, Rechavi G. FTO: linking m6A demethylation to adipogenesis. Cell research. 2015;25:3–4. doi: 10.1038/cr.2014.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, Feng Z, Punt N, Daigle A, Bullinger L, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20:66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berulava T, Horsthemke B. The obesity-associated SNPs in intron 1 of the FTO gene affect primary transcript levels. Eur J Hum Genet. 2010;18:1054–1056. doi: 10.1038/ejhg.2010.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissel S, Reish O, Proulx K, Kawagoe-Takaki H, Sedgwick B, Yeo GS, Meyre D, Golzio C, Molinari F, Kadhom N, et al. Loss-of-function mutation in the dioxygenase-encoding FTO gene causes severe growth retardation and multiple malformations. Am J Hum Genet. 2009;85:106–111. doi: 10.1016/j.ajhg.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. Rna. 1997;3:1233–1247. [PMC free article] [PubMed] [Google Scholar]
- Carlesso N, Aster JC, Sklar J, Scadden DT. Notch1-induced delay of human hematopoietic progenitor cell differentiation is associated with altered cell cycle kinetics. Blood. 1999;93:838–848. [PubMed] [Google Scholar]
- Castillo JJ, Mull N, Reagan JL, Nemr S, Mitri J. Increased incidence of non-Hodgkin lymphoma, leukemia, and myeloma in patients with diabetes mellitus type 2: a meta-analysis of observational studies. Blood. 2012;119:4845–4850. doi: 10.1182/blood-2011-06-362830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Odenike O, Rowley JD. Leukaemogenesis: more than mutant genes. Nat Rev Cancer. 2010;10:23–36. doi: 10.1038/nrc2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Hao YJ, Zhang Y, Li MM, Wang M, Han W, Wu Y, Lv Y, Hao J, Wang L, et al. m(6)A RNA methylation is regulated by microRNAs and promotes reprogramming to pluripotency. Cell Stem Cell. 2015;16:289–301. doi: 10.1016/j.stem.2015.01.016. [DOI] [PubMed] [Google Scholar]
- Church C, Moir L, McMurray F, Girard C, Banks GT, Teboul L, Wells S, Bruning JC, Nolan PM, Ashcroft FM, Cox RD. Overexpression of Fto leads to increased food intake and results in obesity. Nat Genet. 2010;42:1086–1092. doi: 10.1038/ng.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci USA. 1974;71:3971–3975. doi: 10.1073/pnas.71.10.3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dina C, Meyre D, Gallina S, Durand E, Korner A, Jacobson P, Carlsson LM, Kiess W, Vatin V, Lecoeur C, et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet. 2007;39:724–726. doi: 10.1038/ng2048. [DOI] [PubMed] [Google Scholar]
- Dohner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med. 2015;373:1136–1152. doi: 10.1056/NEJMra1406184. [DOI] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Salmon-Divon M, Amariglio N, Rechavi G. Transcriptome-wide mapping of N(6)-methyladenosine by m(6)A-seq based on immunocapturing and massively parallel sequencing. Nat Protoc. 2013;8:176–189. doi: 10.1038/nprot.2012.148. [DOI] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- Dos Santos GA, Kats L, Pandolfi PP. Synergy against PML-RARa: targeting transcription, proteolysis, differentiation, and self-renewal in acute promyelocytic leukemia. J Exp Med. 2013;210:2793–2802. doi: 10.1084/jem.20131121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hajj H, Dassouki Z, Berthier C, Raffoux E, Ades L, Legrand O, Hleihel R, Sahin U, Tawil N, Salameh A, et al. Retinoic acid and arsenic trioxide trigger degradation of mutated NPM1, resulting in apoptosis of AML cells. Blood. 2015;125:3447–3454. doi: 10.1182/blood-2014-11-612416. [DOI] [PubMed] [Google Scholar]
- Fischer J, Koch L, Emmerling C, Vierkotten J, Peters T, Bruning JC, Ruther U. Inactivation of the Fto gene protects from obesity. Nature. 2009;458:894–898. doi: 10.1038/nature07848. [DOI] [PubMed] [Google Scholar]
- Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, Perry JR, Elliott KS, Lango H, Rayner NW, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–894. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat Rev Genet. 2014;15:293–306. doi: 10.1038/nrg3724. [DOI] [PubMed] [Google Scholar]
- Gao X, Shin YH, Li M, Wang F, Tong Q, Zhang P. The fat mass and obesity associated gene FTO functions in the brain to regulate postnatal growth in mice. PLoS ONE. 2010;5:e14005. doi: 10.1371/journal.pone.0014005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, et al. Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science. 2015;347:1002–1006. doi: 10.1126/science.1261417. [DOI] [PubMed] [Google Scholar]
- Glasow A, Prodromou N, Xu K, von Lindern M, Zelent A. Retinoids and myelomonocytic growth factors cooperatively activate RARA and induce human myeloid leukemia cell differentiation via MAP kinase pathways. Blood. 2005;105:341–349. doi: 10.1182/blood-2004-03-1074. [DOI] [PubMed] [Google Scholar]
- Gocek E, Marchwicka A, Bujko K, Marcinkowska E. NADPH-cytochrome P450 reductase is regulated by all-trans retinoic acid and by 1,25-dihydroxyvitamin D3 in human acute myeloid leukemia cells. PLoS ONE. 2014;9:e91752. doi: 10.1371/journal.pone.0091752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guibal FC, Moog-Lutz C, Smolewski P, Di Gioia Y, Darzynkiewicz Z, Lutz PG, Cayre YE. ASB-2 inhibits growth and promotes commitment in myeloid leukemia cells. J Biol Chem. 2002;277:218–224. doi: 10.1074/jbc.M108476200. [DOI] [PubMed] [Google Scholar]
- Hess ME, Hess S, Meyer KD, Verhagen LA, Koch L, Bronneke HS, Dietrich MO, Jordan SD, Saletore Y, Elemento O, et al. The fat mass and obesity associated gene (Fto) regulates activity of the dopaminergic midbrain circuitry. Nat Neurosci. 2013;16:1042–1048. doi: 10.1038/nn.3449. [DOI] [PubMed] [Google Scholar]
- Hu J, Liu YF, Wu CF, Xu F, Shen ZX, Zhu YM, Li JM, Tang W, Zhao WL, Wu W, et al. Long-term efficacy and safety of all-trans retinoic acid/arsenic trioxide-based therapy in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci USA. 2009;106:3342–3347. doi: 10.1073/pnas.0813280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Jiang X, Li Z, Li Y, Song CX, He C, Sun M, Chen P, Gurbuxani S, Wang J, et al. TET1 plays an essential oncogenic role in MLL-rearranged leukemia. Proc Natl Acad Sci USA. 2013;110:11994–11999. doi: 10.1073/pnas.1310656110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ME, Ye YC, Chen SR, Chai JR, Lu JX, Zhoa L, Gu LJ, Wang ZY. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood. 1988;72:567–572. [PubMed] [Google Scholar]
- Iijima K, Honma Y, Niitsu N. Granulocytic differentiation of leukemic cells with t(9;11)(p22;q23) induced by all-trans-retinoic acid. Leukemia & lymphoma. 2004;45:1017–1024. doi: 10.1080/1042819031000163887. [DOI] [PubMed] [Google Scholar]
- Jia G, Fu Y, He C. Reversible RNA adenosine methylation in biological regulation. Trends Genet. 2013;29:108–115. doi: 10.1016/j.tig.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, He C. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Hu C, Arnovitz S, Bugno J, Yu M, Zuo Z, Chen P, Huang H, Ulrich B, Gurbuxani S, et al. miR-22 has a potent anti-tumor role with therapeutic potential in acute myeloid leukemia. Nat Commun. 2016;7:11452. doi: 10.1038/ncomms11452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Huang H, Li Z, Li Y, Wang X, Gurbuxani S, Chen P, He C, You D, Zhang S, et al. Blockade of miR-150 maturation by MLL-fusion/MYC/LIN-28 is required for MLL-associated leukemia. Cancer Cell. 2012;22:524–535. doi: 10.1016/j.ccr.2012.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohroki J, Fujita S, Itoh N, Yamada Y, Imai H, Yumoto N, Nakanishi T, Tanaka K. ATRA-regulated Asb-2 gene induced in differentiation of HL-60 leukemia cells. FEBS Lett. 2001;505:223–228. doi: 10.1016/s0014-5793(01)02829-0. [DOI] [PubMed] [Google Scholar]
- Li G, Chen Q, Wang L, Ke D, Yuan Z. Association between FTO gene polymorphism and cancer risk: evidence from 16,277 cases and 31,153 controls. Tumour Biol. 2012a;33:1237–1243. doi: 10.1007/s13277-012-0372-9. [DOI] [PubMed] [Google Scholar]
- Li Z, Huang H, Chen P, He M, Li Y, Arnovitz S, Jiang X, He C, Hyjek E, Zhang J, et al. miR-196b directly targets both HOXA9/MEIS1 oncogenes and FAS tumour suppressor in MLL-rearranged leukaemia. Nat Commun. 2012b;2:688. doi: 10.1038/ncomms1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Hales CM, Garber K, Jin P. Fat mass and obesity-associated (FTO) protein interacts with CaMKII and modulates the activity of CREB signaling pathway. Hum Mol Genet. 2014;23:3299–3306. doi: 10.1093/hmg/ddu043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–95. doi: 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560–564. doi: 10.1038/nature14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo-Coco F, Avvisati G, Vignetti M, Thiede C, Orlando SM, Iacobelli S, Ferrara F, Fazi P, Cicconi L, Di Bona E, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369:111–121. doi: 10.1056/NEJMoa1300874. [DOI] [PubMed] [Google Scholar]
- Marcucci G, Mrozek K, Bloomfield CD. Molecular heterogeneity and prognostic biomarkers in adults with acute myeloid leukemia and normal cytogenetics. Curr Opin Hematol. 2005;12:68–75. doi: 10.1097/01.moh.0000149608.29685.d1. [DOI] [PubMed] [Google Scholar]
- Martelli MP, Gionfriddo I, Mezzasoma F, Milano F, Pierangeli S, Mulas F, Pacini R, Tabarrini A, Pettirossi V, Rossi R, et al. Arsenic trioxide and all-trans retinoic acid target NPM1 mutant oncoprotein levels and induce apoptosis in NPM1-mutated AML cells. Blood. 2015;125:3455–3465. doi: 10.1182/blood-2014-11-611459. [DOI] [PubMed] [Google Scholar]
- Meng J, Cui X, Rao MK, Chen Y, Huang Y. Exome-based analysis for RNA epigenome sequencing data. Bioinformatics. 2013;29:1565–1567. doi: 10.1093/bioinformatics/btt171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Jaffrey SR. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nature reviews Molecular cell biology. 2014;15:313–326. doi: 10.1038/nrm3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian SB, Jaffrey SR. 5′ UTR m(6)A Promotes Cap-Independent Translation. Cell. 2015;163:999–1010. doi: 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niitsu N, Hayashi Y, Sugita K, Honma Y. Sensitization by 5-aza-2′-deoxycytidine of leukaemia cells with MLL abnormalities to induction of differentiation by all-trans retinoic acid and 1alpha,25-dihydroxyvitamin D3. Br J Haematol. 2001;112:315–326. doi: 10.1046/j.1365-2141.2001.02523.x. [DOI] [PubMed] [Google Scholar]
- Niu Y, Zhao X, Wu YS, Li MM, Wang XJ, Yang YG. N6-methyl-adenosine (m6A) in RNA: an old modification with a novel epigenetic function. Genomics Proteomics Bioinformatics. 2013;11:8–17. doi: 10.1016/j.gpb.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, Su L, Xu G, Zhang Y. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121:167–178. doi: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- Perry RP, Kelley DE. Existence of methylated messenger RNA in mouse L cells. Cell. 1974;1:37–42. [Google Scholar]
- Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24:177–189. doi: 10.1038/cr.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K, Imamura T, Yano M, Yoshida H, Fujiki A, Hirashima Y, Hosoi H. Sensitivity of MLL-rearranged AML cells to all-trans retinoic acid is associated with the level of H3K4me2 in the RARalpha promoter region. Blood Cancer J. 2014;4:e205. doi: 10.1038/bcj.2014.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlenk RF, Dohner K, Kneba M, Gotze K, Hartmann F, Del Valle F, Kirchen H, Koller E, Fischer JT, Bullinger L, et al. Gene mutations and response to treatment with all-trans retinoic acid in elderly patients with acute myeloid leukemia. Results from the AMLSG Trial AML HD98B. Haematologica. 2009;94:54–60. doi: 10.3324/haematol.13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, Mertins P, Ter-Ovanesyan D, Habib N, Cacchiarelli D, et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 2014;8:284–296. doi: 10.1016/j.celrep.2014.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scuteri A, Sanna S, Chen WM, Uda M, Albai G, Strait J, Najjar S, Nagaraja R, Orru M, Usala G, et al. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet. 2007;3:e115. doi: 10.1371/journal.pgen.0030115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderberg KC, Kaprio J, Verkasalo PK, Pukkala E, Koskenvuo M, Lundqvist E, Feychting M. Overweight, obesity and risk of haematological malignancies: a cohort study of Swedish and Finnish twins. Eur J Cancer. 2009;45:1232–1238. doi: 10.1016/j.ejca.2008.11.004. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Muntean AG, Hess JL. ECSASB2 mediates MLL degradation during hematopoietic differentiation. Blood. 2012;119:1151–1161. doi: 10.1182/blood-2011-06-362079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014a;505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, He C. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell. 2015;161:1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol. 2014b;16:191–198. doi: 10.1038/ncb2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood. 2008;111:2505–2515. doi: 10.1182/blood-2007-07-102798. [DOI] [PubMed] [Google Scholar]
- Wunderlich M, Mizukawa B, Chou FS, Sexton C, Shrestha M, Saunthararajah Y, Mulloy JC. AML cells are differentially sensitive to chemotherapy treatment in a human xenograft model. Blood. 2013;121:e90–97. doi: 10.1182/blood-2012-10-464677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Liu J, He C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 2015;29:1343–1355. doi: 10.1101/gad.262766.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisig BB, Milne T, Garcia-Cuellar MP, Schreiner S, Martin ME, Fuchs U, Borkhardt A, Chanda SK, Walker J, Soden R, et al. Hoxa9 and Meis1 are key targets for MLL-ENL-mediated cellular immortalization. Mol Cell Biol. 2004;24:617–628. doi: 10.1128/MCB.24.2.617-628.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, He X, Semenza GL. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A. 2016;113:E2047–2056. doi: 10.1073/pnas.1602883113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Yang Y, Sun BF, Shi Y, Yang X, Xiao W, Hao YJ, Ping XL, Chen YS, Wang WJ, et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014;24:1403–1419. doi: 10.1038/cr.2014.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, Vagbo CB, Shi Y, Wang WL, Song SH, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18–29. doi: 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, Qian SB. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature. 2015;526:591–594. doi: 10.1038/nature15377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Heyworth CM, Glasow A, Huang QH, Petrie K, Lanotte M, Benoit G, Gallagher R, Waxman S, Enver T, Zelent A. Lineage restriction of the RARalpha gene expression in myeloid differentiation. Blood. 2001;98:2563–2567. doi: 10.1182/blood.v98.8.2563. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.